

Abstract
A 41-year-old woman had meaningful functional improvement following reinstitution of a low phenylalanine diet. She was diagnosed at birth with phenylketonuria and followed strict dietary adherence till the age of 16. Thereafter the diet was discontinued. She subsequently presented with subacute profound visual loss, cognitive dysfunction and paraparesis such that she was bed bound requiring full nursing care. Following dietary intervention there was meaningful improvement such that she was no longer demented and while her vision remains poor she is now independent for activities of daily living. This case report suggests that consideration of reimplementation of dietary intervention is warranted even after a prolonged period of time.
Background
Phenylketonuria (PKU) is an autosomal recessive inborn error of metabolism causing accumulation of phenylalanine resulting in cognitive, behavioural and pyramidal dysfunction. The prevalence of PKU is 1/10 000 live births in European countries. Treatment is lifelong adherence to a low phenylalanine diet.1 Improvement outcomes after introducing dietary restrictions in adulthood or in the reintroduction after prolonged periods of non-compliance are largely limited to subjective changes in behaviour and well-being.2–4 Despite the recognition of this disorder and its treatment for over half a century, there are a few case reports to date addressing the changes in motor function in adults who are non-compliant with PKU diet.5 6 This case report provides further evidence for the beneficial effects of reintroducing a low phenylalanine diet in an untreated adult with neurological complications in late adulthood.
Case presentation
A 41-year-old medically retired nurse was admitted to hospital with a 6 month history of declining mobility, generalised apathy, behavioural disturbance and a gradual deterioration in her vision. She was wheel-chair bound and incontinent of urine with excoriated skin upon admission.
She had a background of PKU, diagnosed at birth and had been treated with a low phenyalanine diet from birth until the age of 16 years. At that stage she opted to discontinue her diet upon medical advice and social convenience. At the age of 38 years she developed progressive visual loss to 6/60 bilaterally over a 6-month period necessitating early retirement from her nursing career. Initial investigations showed visual evoked potentials were delayed with small amplitude bilaterally. MRI of brain demonstrated extensive diffuse white matter changes. The patient then defaulted from follow-up until her current admission. Her functional decline meant her husband was acting as her full-time carer upon admission.
On examination she was orientated but unable to follow 1 stage commands consistently. She required a hoist for transfer and was bed bound. Visual acuity was reduced to finger counting bilaterally with bilateral pallor of the optic discs. There was spasticity of all four limbs (Ashworth scale 3 bilaterally) with sustained clonus in the lower limbs and pyramidal weakness grade 3/5. She was hyper-reflexic and the plantar responses were extensor. Sensation was normal.
Investigations
Initial investigations demonstrated a low folate (1.6 µg/L, normal range 3.1–17.5 µg/L) and borderline low vitamin B12 (211 ng/L, normal range 200–940 ng/L). Her tyrosine was 56 µmol/L with an elevated phenylalanine 1564 µmol/L (120–480 µmol/L), consistent with untreated PKU. MRI brain (figures 1 and 2) showed extensive abnormal T2 high signal affecting grey matter and subcortical white matter within the frontal and parietal lobes with some atrophy.
Figure 1.
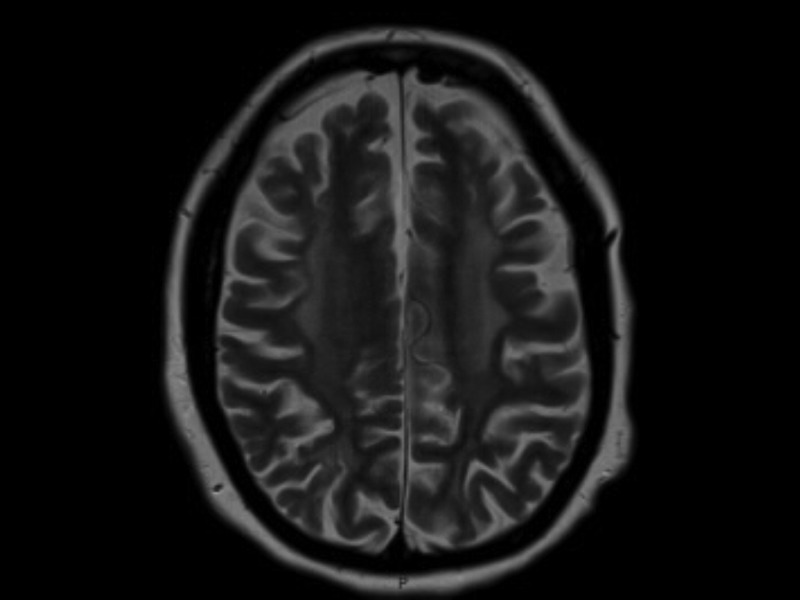
Axial T2-weighted MRI showing diffuse high signal change throughout the white matter with associated cortical atrophy.
Figure 2.
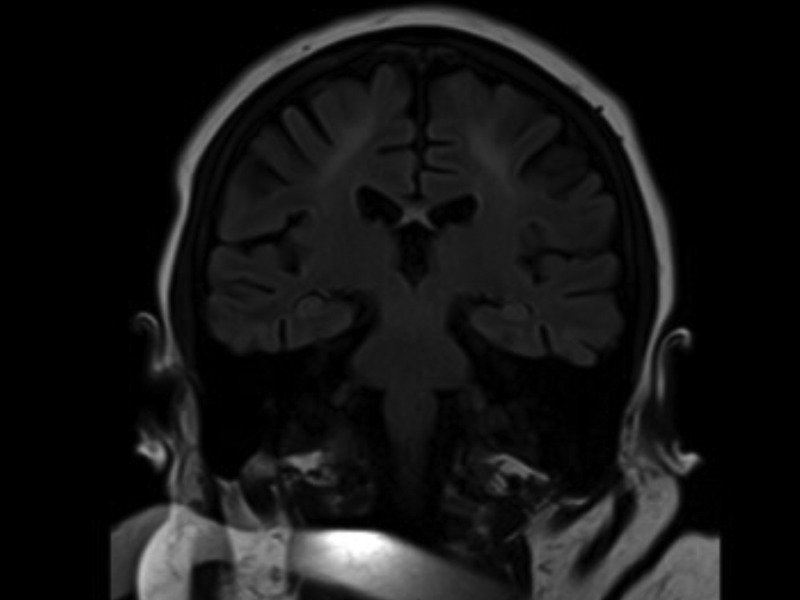
Coronal fluid attenuation inversion recovery sequence demonstrating high signal change on the parietal lobes.
The following tests were either normal or negative: HIV, antinuclear body, extractable nuclear antigen, antineutrophil cytoplasmic antibody plasma viscosity, syphilis antibody, vitamin B6, full blood count, liver function tests, thyroid function tests, protein electrophoresis and urine Bence-Jones protein, chest X-ray, pelvis and hip X-ray.
Differential diagnosis
Multiple sclerosis
The clinical course atypical and the imaging features were not in keeping with those of the Barkoff criteria.
HIV
Serology was negative.
Neuromyelitis optica (NMO)
This was considered and indeed the clinical presentation of bilateral optic neuropathy and spastic paraparesis afflicted the areas typical of NMO. The subacute evolution and lack of signal change in the spinal cord on MRI were atypical. Also, the antibody profile was negative. We accept that this may occur in 40% of patients with NMO but are confident this is not the aetiology in our case.
Subacute combined degeneration of the cord
We accept that a low serum folate may contribute to the spinal cord disturbance but in our case the dorsal columns were spared. Also there was no evidence of a peripheral neuropathy.
Human T-lymphotropic virus 1 causes a tropical spastic paraparesis.
Treatment
A low phenylalanine diet was started. She was given folate and vitamin B12 replacement and symptomatic treatment for spasticity.
Outcome and follow-up
Following normalisation of her phenylalanine (121 µmol/L) she made sustained improvements in cognitive function, affect and behaviour. She was now able to complete an Addenbrooke's cognitive examination—revised (ACE-r) scoring 53/84 (she was unable to complete visual sections). Two months later she was able to walk 40 m independently with a Zimmer frame, climb stairs and was no longer incontinent of urine. Her vision improved to 1/36 bilaterally. There was less spasticity (now Ashworth scale 1 bilaterally) with residual weakness confined to the hip flexors (4/5).
Discussion
This case demonstrates improvement in cognitive function, behaviour, visual and motor function after restarting of PKU diet in a 41-year-old patient, who last adhered to PKU diet until the age of 16. She was able to sustain normal functional state, working as a nurse until 3 years ago. There are only eight case reports to date on adult PKU non-compliance with diet leading motor deficits and these reports focus only on early adulthood or late adolescence, reporting quadraparesis, paraparesis, tremor, epilepsy, ataxia as presenting features.5 6 The duration of adherence to PKU diet during first 16 years of life appears to indicative of likely complications in adulthood.6 There is only one case report of late adulthood PKU diagnosis, reported as ‘Musty odour, mental retardation and spastic paraplegia revealing phenylketonuria in adulthood’,7 unfortunately the full case report is not available. Notably no previous case report has stated visual problems as sequelae of non-compliance with PKU diet. Given the accepted variable adherence to PKU diet in adult PKU population, the rare neurological complication of non-adherence to PKU diet and improved outcome upon reinstituting a low phenylalanine diet should be considered.
Learning points.
Adult patients with phenylketonuria who do not adhere to a low phenylalanine diet may develop poor cognitive function and motor deficits. There is scope for improvement on reinstitution of the dietary intervention plus rehabilitation.
Late neurological deterioration is possible despite relatively normal function for a substantial period of time.
Footnotes
Contributors: MSA is the reviewer of the manuscript and he helped BW with initial collecting and findings. JOR reviewed the final manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010;2013:1417–27 [DOI] [PubMed] [Google Scholar]
- 2.Gassio R, Campistol J, Vilaseca MA, et al. Do adult patients with phenylketonuria improve their quality of life after introduction/resumption of a phenylalanine-restricted diet? Acta Paediatrica 2003;2013:1474–8 [PubMed] [Google Scholar]
- 3.Channon S, Goodman G, Zlotowitz S, et al. Effects of dietary management of phenylketonuria on long-term cognitive outcome. Arch Dis Child 2007;2013:213–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.González MJ, Gutiérrez AP, Gassió R, et al. Neurological complications and behavioral problems in patients with phenylketonuria in a follow-up unit. Mol Genet Metab 2011;2013(Suppl):S73–9 [DOI] [PubMed] [Google Scholar]
- 5.Soyupeka F, Koyuncuoglub H, Cindasc A, et al. The rehabilitation programme of an adult phenylketonuria with upper motor neuron involvement. J Back Musculoskelet Rehabil 2009;2013:49–53 [DOI] [PubMed] [Google Scholar]
- 6.Thomson AJ, Smith I, Brenton D, et al. Neurological deterioration in young adults with phenylketonuria. Lancet 1990;2013:602–5 [DOI] [PubMed] [Google Scholar]
- 7.Jousserand G, Antoine JC, Camdessanché JP. Musty odour, mental retardation, and spastic paraplegia revealing phenylketonuria in adulthood. J Neurol 2010;2013:302–4 [DOI] [PubMed] [Google Scholar]