

Abstract
Mutation studies previously showed that the lentivirus lytic peptide (LLP2) sequence of the cytoplasmic C-terminal tail of the HIV-1 gp41 envelope protein inhibited viral-initiated T-cell death and T-cell syncytium formation, at which time in the HIV life cycle the gp41 protein is embedded in the T-cell membrane. In striking contrast, the mutants did not affect virion infectivity, during which time the gp41 protein is embedded in the HIV envelope membrane. To examine the role of LLP2/membrane interactions, we applied synchrotron x-radiation to determine structure of hydrated membranes. We focused on WT LLP2 peptide (+3 charge) and MX2 mutant (−1 charge) with membrane mimics for the T-cell and the HIV-1 membranes. To investigate the influence of electrostatics, cholesterol content, and peptide palmitoylation, we also studied three other LLP2 variants and HIV-1 mimics without negatively charged lipids or cholesterol as well as extracted HIV-1 lipids. All LLP2 peptides bound strongly to T-cell membrane mimics, as indicated by changes in membrane structure and bending. In contrast, none of the weakly bound LLP2 variants changed the HIV-1 membrane mimic structure or properties. This correlates well with, and provides a biophysical basis for, previously published results that reported lack of a mutant effect in HIV virion infectivity in contrast to an inhibitory effect in T-cell syncytium formation. It shows that interaction of LLP2 with the T-cell membrane modulates biological function.
Introduction
The HIV-1 envelope glycoprotein (Env) is synthesized as a 160 kDa precursor, gp160, and then proteolytically cleaved into gp120 and gp41, which remain noncovalently associated in a trimer arrangement. HIV infection begins when gp120 contacts the CD4 receptor on the T-cell host, which is followed by contact with a second chemokine receptor, and then fusion of the HIV virion and T-cell membranes, catalyzed by the gp41 fusion protein. The gp41 fusion protein contains a long (150 AA) cytoplasmic C-terminal tail (CTT) that is a common feature of lentiviruses but is significantly longer than the CTT of other retroviruses. Three conserved amphipathic, primarily α-helical segments in the CTT referred to as lentiviral lytic peptides (LLP1, LLP2, and LLP3) have been shown to bind and perturb membranes (1–5). LLP2 is highly cationic with a preferential incorporation of arginine residues and it possesses a large hydrophobic moment (6). To understand the function of the CTT, deletions of the entire CTT have been carried out with different results depending upon cell type. For so-called permissive cell types (HeLa, COS, 293T, MT-4, and M8166), deletion has little effect on Env incorporation into virions (7,8). However, the majority of T-cell lines (including CED(12D-7), Jurkat, MT-2, H9, and SupT1) are nonpermissive in that CTT deletion results in a 10-fold reduction in Env incorporation and a complete blockage of viral replication (9). Murakami and Freed found the block to be at the early stage of Env incorporation (7).
Less drastic changes in the CTT have been carried out using rational site-directed mutations. Kalia et al. (10), for instance, produced a mutant MX2 of LLP2 where two highly conserved arginine residues (R3 and R21) were replaced with negatively charged glutamic acid, thus decreasing the net charge from +3 to −1. In virions derived from 293T cells, MX2 mutation showed no loss of Env expression or infectivity (10). These events occur when the gp41 protein is embedded in the HIV envelope membrane. In contrast, when the gp41 protein is in the T-cell membrane, both viral-initiated cell death and syncytium formation (cell-cell fusion) were greatly decreased in H9 cells by the MX2 mutant (10); this was not a case of permissive versus nonpermissive cell lines, because this inhibition was also found in permissive 293T cells using a luciferase expression fusion assay (10). In addition to these functional variations, it was demonstrated that the MX2 Env expressed at the T-cell surface displayed significant differences in reactivity with monoclonal antibodies compared to wild-type (WT) Env proteins (11). Changes in antigenicity of the MX2 mutant Env correlated with neutralization resistance of the mutant virus, which defined the LLP2 domain as a critical determinant of Env structure and antigenicity (11). However, a mechanism for the alteration of cell-cell but not virus-cell functionality by the MX2 mutant was not evident from the mutational studies. Indeed, a priori it would have seemed that the CTT should not affect Env activity because it is on the opposite side of the membrane.
In the current work, we have investigated LLP2 variants in lipid bilayers to learn how changes in structure and properties correlate with biological function. We used our pioneering method of x-ray diffuse scattering (12–15) from oriented, fully hydrated peptide/lipid mixtures that provides both structural and bending flexibility data. As model HIV-1 and T-cell membranes, we used membrane mimics composed of 10 lipids and cholesterol based on previous determinations (16,17), as well as total lipids extracted from HIV membranes. Two additional HIV membrane mimics lacking negatively charged lipids (HIV-neg) or cholesterol (HIV-chol) were also investigated to probe the importance of negatively charged lipids and cholesterol in the HIV virion membrane. The primary peptides were WT and MX2 (WT mutant). We also studied CracWT (WT with Crac motif), CracWTpal (CracWT with palmitoylated cysteine), and CracMX2pal (MX2 mutant with Crac and palmitoylated cysteine). These peptides were specifically designed to investigate the role of the charge change from +3 in the WT to −1 in the MX2 mutant (10), as well as the effect of a Crac, or cholesterol-binding motif (18) immediately preceding the LLP2 sequence. Crac motifs were first discovered in the benzodiazepine receptor (19) and have a sequence that binds cholesterol (18,20). The peptide variants also tested the role of the palmitoylated cysteine, which precedes the Crac motif (21,22). Our data reveal that all peptide variants interact strongly with the T-cell membrane mimic, as observed by changes in structure and bending modulus, whereas the peptides only interacted weakly with the HIV membrane mimic or with the HIV extracted lipid membrane. Our work provides a structural basis for the effects seen in mutation studies (10), namely that LLP2 is a modulator of biological activity in T-cell membranes and not in HIV envelope membranes. It suggests that the T-cell membrane mediates inside-outside signaling by LLP2 peptides to the ectodomain of the Env protein.
Materials and Methods
Samples
Purified lipids were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. Membrane mimic mixtures were prepared by first dissolving lyophilized lipids in chloroform and then mixing these stock solutions in proportions following (16,17); (17) used H9 cells for the HIV and T-cell lipidome analysis with conditions that preserved the phosphoinositides, but because the cholesterol content was not clearly stated, we also used (16), which used 293T cells. Table 1 shows our lipid compositions, where PO indicates palmitoyloleoyl. Chemical structures of lipids are shown in Fig. S1 in the Supporting Material.
Table 1.
Lipid composition of membrane mimics
| Lipid | Mole percentage |
|||
|---|---|---|---|---|
| HIV | HIV-chol | HIV-neg | T-cell | |
| C 16 Ceramide | 0.8 | 1.5 | 1.0 | 1.1 |
| Soy Phosphoinositol (PI) | 1.3 | 2.3 | 0 | 6.3 |
| Brain PI(4)P | 1.3 | 2.3 | 0 | 0.9 |
| Brain PI(4,5)P2 | 1.3 | 2.3 | 0 | 1.1 |
| 12:0 diHydroSphingomyelin (diHSM) | 1.7 | 3.1 | 1.8 | 2.7 |
| PO-Phosphoethanolamine (POPE) | 4.2 | 7.6 | 4.7 | 6.3 |
| PO-Phosphoserine (POPS) | 7.5 | 13.7 | 0 | 9.1 |
| Plasmalogen PE | 7.5 | 13.7 | 8.5 | 10.9 |
| Egg Sphingomyeline (Egg SM) | 14.2 | 26.0 | 16.0 | 16.3 |
| PO-PhosphoCholine (POPC) | 15.1 | 27.5 | 17.0 | 15.4 |
| Cholesterol (Chol) | 45.2 | 0 | 51.0 | 29.9 |
Membrane mimics are HIV, HIV-chol (HIV without cholesterol), HIV-neg (HIV without negatively charged lipids), and T-cell. PO stands for palmitoyloleoyl.
HIV-1 virions were procured from the Biological Products Core Laboratory, AIDS and Cancer Virus Program, Frederick, MD, where the virus was attenuated by 1 mM Aldrithiol-2. HIV-1 NL4-3 was produced on host cell line SUPT1-CCR5 CL.30, Lot P4166. 113 × 0.25 ml aliquots (total capsid = 5.7 mg and total protein = 52.3 mg) were sent to our BSL-2+ facility at the University of Pittsburgh, where we carried out the Bligh/Dyer (23) lipid extraction. The recovered bottom lipid phase was washed with authentic upper phase according to the procedure. 33 mg of pure HIV lipids were collected from this preparation and divided into 4 mg aliquots for x-ray sample preparation. Organic solvents were removed by vacuum drying at room temperature.
Peptides were purchased from the Peptide Synthesis Facility (University of Pittsburgh, Pittsburgh, PA). Mass spectroscopy revealed >95% purity. Lentivirus lytic peptide-2 (LLP2 peptides corresponding to residues #768–788 in the gp160 protein in HXB2) variants were: WT H2N-YHRLRDLLLIVTRIVELLGRR-COOH, MX2 mutant H2N-YHELRDLLLIVTRIVELLGRE-COOH (10), (#764–788) CracWTpal H2N-C(pal)-LFLYHRLRDLLLIVTRIVELLGRR-COOH (24), CracWT H2N-GLFLYHRLRDLLLIVTRIVELLGRRCOOH, and CracMX2pal H2N-C(pal)-LFLYHELRDLLLIVTRIVELLGRECOOH. The peptide structures are shown lined up in Fig. S1. A low synthetic yield of CracMX2pal prevented using it in all experiments. Peptides and lipids were dissolved in HPLC chloroform and mixed together in molar ratios 20:1 or 40:1 (lipid/peptide). 4 mg lipid/peptide mixture in HPLC chloroform/trifluoroethanol (TFE) (1:1 v:v) was plated onto silicon wafers (15 × 30 × 1 mm) via the rock and roll method (25) to produce ∼1800 well-aligned bilayers, and solvents were removed by evaporation. Before x-ray exposure, samples were prehydrated through the vapor in polypropylene hydration chambers at 37°C for 1–3 h directly before hydrating in the thick-walled x-ray hydration chamber (13) for 0.5–1 h. Preequilibration allows sufficient time for equilibrium binding of peptides with membrane mimics and also reduces mosaic spread.
Volume determination
Volumes of lipid mixtures with and without peptides in fully hydrated multilamellar vesicles were determined at 37 ± 0.01°C using an Anton-Paar USA DMA5000M (Ashland, VA) vibrating tube densimeter (26).
X-ray scattering and liquid crystal analysis. Low-angle x-ray scattering (LAXS)
LAXS data from oriented fluid phase lipid mixtures at 37°C were obtained at the Cornell High Energy Synchrotron Source (CHESS) with the scattering geometry shown in Fig. S2 using previously described methods (12,15). The analysis of diffuse LAXS from oriented stacks of fluctuating fluid bilayers has been previously described (13). Typical beam width was 0.25 mm and beam height was 1–1.2 mm. Typical wavelength was 1.1775 Å with a total beam intensity of ∼1–5 × 1011 photons/s/mm2. Typical sample-to-charge-coupled device distance was 376 mm LAXS and 159 mm wide-angle x-ray scattering (WAXS), calibrated using a silver behenate standard with D-spacing 58.4 Å. Temperature was controlled at 37°C with a Julabo F25 (Allentown, PA).
Structural analysis
The x-ray |F(qz)| data were fit to the Fourier transform of a model of the electron density profile with components using a scattering density profile (SDP) procedure (27). The SDP procedure guarantees an important relation between the molecular area A and the zeroth-order x-ray form factor F(0) (28):
where V is the measured volume of the lipid/peptide mixture, n = nL + npepf/(1-f) is the total, nL is the lipid and npep is the peptide number of electrons, f is the mole fraction of peptide/peptide + lipid and ρW = 0.3323e/Å3 is the electron density of water at 37°C. Informed by our volume measurements, we constrained the ratio of peptide volume/lipid volume. We also constrained the Gaussian sigma widths of the lipid headgroup peaks to 2–5 Å, the methyl trough Gaussian sigma to 3–5 Å (corresponding to full width at half-maximum of 7–12 Å), and the width of the hydrocarbon interface to 2.4 Å. The peptide width was initially not constrained. The difference in distance DH1 between the maximum in the electron density profile and the Gibbs dividing surface for the hydrocarbon region, was loosely constrained to 4.95 Å, and the difference in distance DH2 between the phosphatidylcholine (PC) and carbonyl-glycerol (CG) Gaussians was constrained to 3.7 Å. Otherwise, the locations of the components remained free to provide estimates for the head-head distance and the position of the peptide in the membrane mimic as previously described (29). The smallest χ2 in the SDP model fit determined the peptide position. Estimated uncertainties in these positions are addressed in Fig. S3.
WAXS
The analysis of WAXS data has been previously described (14,30). Briefly, the orientational order parameter, Sxray, is similar to an NMR order parameter and is obtained as described in Fig. S4.
Circular dichroism spectroscopy
Samples for CD were prepared by spreading 0.3 mg peptide/lipid mixture in chloroform/TFE (1:1) onto one inside face of a quartz cuvette to make a thin film and solvents were removed under vacuum. 100 μl milli-Q water was introduced and hydration occurred through the vapor in sealed cuvettes at room temperature for 24 h. Our samples were purposely misoriented during spreading onto the cuvette side to minimize orientation effects on CD spectra (31,32), and two cuvette orientations, vertical and inverted, were used to sample different parts of the hydrated film. 8–10 scans were collected with a Jasco 715 on each sample from 260 to 180 nm at 100 nm/minute and averaged. CD data in mean residue ellipiticity were analyzed using the CDSSTR function in DichroWEB (33) with protein basis set #4 (34). Uncertainties were estimated by averaging multiple CDSSTR fits to the data and taking the average and standard deviation. We also attempted to use the more conventional method of incorporating LLP2 peptides into small unilamellar vesicles, but this was not successful with the LLP2 peptide variants. We therefore used another peptide, α-synuclein (αS) with both the small unilamellar vesicle method and our thin film method and determined that both methods agreed to within 15% for the helical content. We also measured the CD of peptides suspended in 3 ml water (0.008 mg/ml) with no lipid.
Results
LAXS
Oriented stacks of ∼1800 membrane mimics were hydrated through the vapor and synchrotron LAXS was obtained as shown in Fig. 1, a and b, for the T-cell and HIV membrane mimics, respectively. Comparison shows that the diffuse patterns are quite different; most notably, the HIV membrane mimic is missing the second diffuse scattering lobe compared to the T-cell membrane mimic. This requires that these membrane mimics have different quantitative structures (see Fig. S5).
Figure 1.
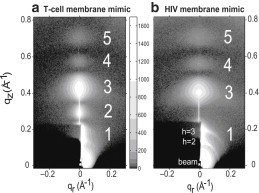
Contour plots of LAXS intensity (white is most intense) at 37°C where z is the direction of the membrane normal and r is the in-plane direction. (a) T-cell lipid membrane, (b) HIV lipid membrane. Diffuse scattering lobes (1–5) are identified with large, white numbers. The dark rectangle in the lower left corner is due to molybdenum sheets that attenuate the beam and the h = 1–3 diffraction peaks. The unattenuated arcs visible in (b) emanating from the h = 2 and h = 3 peaks are due to imperfect alignment.
Structurally, raw diffuse scattering, as in Fig. 1, is quantified by the bilayer form factor F(qz), which is the Fourier transform of the electron density profile along the bilayer normal, perpendicular to the membrane mimic surface. Fig. 2 shows F(qz) obtained for the T-cell and HIV membrane mimics with and without the LLP2 variants; the raw data from which these form factors are derived are shown in Fig. S6. Fig. 2 a clearly shows that the T-cell mimic form factors are affected by the addition of these peptides. A relative decrease in intensity occurs in the second lobe (near qz = 0.3 Å−1) and a relative increase occurs in the 4th and 5th lobes (near qz = 0.6 and 0.7 Å−1). These changes in form factors in the T-cell membrane mimic samples prove that the peptides are binding to the T-cell membrane mimic and affecting its structure. Form factor results were similar for the 20:1 samples.
Figure 2.
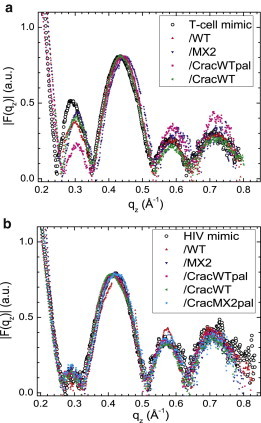
Form factors of T-cell and HIV lipid membranes with and without LLP2 peptides listed in the legends. T-cell lipid membrane samples (a) and HIV lipid membrane samples (b) at 40:1 lipid/peptide mole ratio. Data were normalized in the third lobe (qz ∼ 0.43 Å−1).
Table 2 summarizes the net charges and lamellar repeat D-spacings for all the samples. Our experimental setup allows us to change the hydration level, which then changes D. For the well-hydrated samples reported here these changes in D do not affect the F(qz) in Fig. 2. However, the D values are informative regarding electrostatic charge. Stacks of bilayers with sufficient net charge swell to very large values of D; this is often called membrane unbinding (35), not to be confused with unbinding of peptides from membranes. As an infinite D-spacing would disorient the sample, an osmotic pressure was imposed to limit the actual D-spacing of samples that would unbind. A U is written next to the osmotically imposed actual D-spacing in Table 2 for those samples that were in the process of unbinding. The T-cell membrane mimic has a net charge of −0.2e/lipid and the HIV membrane mimic has a net charge of −0.15e/lipid; both membranes unbind. Adding the negatively charged MX2 mutants increases the negative charges so those membrane mimics continue to unbind. Adding WT LLP2 with three positive charges to T-cell membrane mimics changes the net charge/lipid to −0.125 for 40:1 (unbound) and −0.05 for 20:1 (bound); for HIV samples the net charge/lipid is −0.075 for 40:1 and 0 for 20:1 (both bound). Results for the other WT modifications, except CracWT(20:1), suggest that the charge threshold for unbinding occurs for net charge between −0.075 and −0.125 e/lipid.
Table 2.
Net charge and lamellar D-spacings
| Sample |
Net charge (e) |
D-spacing (Å) |
||
|---|---|---|---|---|
| Lipid/peptide mole ratio | 20:1 | 40:1 | 20:1 | 40:1 |
| T-cell membrane mimic | −0.20 | 92, U | ||
| /WT | −0.05 | −0.125 | 69 | 75, U |
| /MX2 | −0.25 | −0.23 | 86, U | 122, U |
| /CracWTpal | −0.05 | −0.125 | 68 | 100, U |
| /CracWT | −0.05 | −0.125 | 113, U | 143, U |
| HIV membrane mimic | −0.15 | 119, U | ||
| /WT | 0 | −0.075 | 64 | 66 |
| /MX2 | −0.20 | −0.175 | 73, U | 75, U |
| /CracWTpal | 0 | −0.075 | 66 | 69 |
| /CracWT | 0 | −0.075 | 65 | 67 |
| /CracMX2pal | −0.20 | −0.175 | 69, U | 76, U |
Samples are T-cell mimics with WT (wild-type), MX2 (WT mutant), CracWTpal (WT with a cholesterol-binding Crac motif and palmitoylated cysteine) and CracWT (WT with a Crac motif), and HIV mimic with WT, MX2, CracWTpal, CracWT, and CracMX2pal (MX2 with a Crac motif and palmitoylated cysteine). U indicates unbinding of the D-spacing.
In contrast to the T-cell, the HIV form factors in Fig. 2 b exhibit negligible differences for any of the LLP2 analogs compared to the HIV membrane mimic. Similar to the HIV mimic samples, the HIV extracted lipid samples are also missing the second diffuse lobe, and this pattern is not affected by the peptides as shown by the example in Fig. S6. This surprising result at first suggested that the peptides had not been incorporated into our samples. However, that would have resulted in membrane unbinding for all the HIV samples, which is contrary to the membrane binding (finite D spacing) that occurs when the WT peptides were added (see Table 2). Furthermore, it appears that the peptides, at least those positively charged, remain weakly bound to the bilayer to reduce its net negative charge and bring about the finite D spacing. Note that although the peptide would change the electron density of the water space, our subsequent analysis indicates that such changes are consistent with the negligible differences in |F(qz)| in Fig. 2 b.
Electron density profiles
To estimate where the peptides reside within the T-cell membrane mimic and to verify our observation that the peptides do not enter the HIV membrane mimic, the SDP model fitting program was used (27). The SDP program fits the Fourier transform of a real space electron density ρ(z) model (27) to the F(qz) data. Fig. 3 a shows an example of the goodness of the fit to the x-ray form factor data. Fig. 3 b shows the component groups in the T-cell samples. As shown, the WT peptide resides in the T-cell headgroup region, whereas MX2 localizes further into the hydrocarbon interior. At 40:1 (lipid/peptide mole ratio) both peptides thin the T-cell membrane mimic, with MX2 causing the greatest (3 Å) thinning. Electron density profiles for the 20:1 T-cell samples showed similar thinning and peptide locations. For the HIV membrane mimics, the peptides only weakly associated with this mimic and the lipid structures were unchanged, consistent with unchanged form factors in Fig. 2 b; LLP2 has an electron density close to water, so there is insufficient contrast to localize its position or width. The electron density profiles for the 20:1 and 40:1 HIV samples were similarly unchanged.
Figure 3.
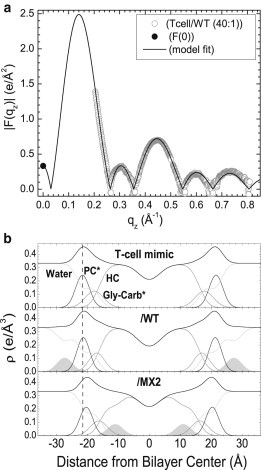
(a) Model fit to F(qz) obtained from x-ray data and F(0) obtained from volumetric data for T-cell/WT. Modeled total and component electron density profiles for T-cell samples (b) for 40:1 lipid/peptide. The component groups are water, PC∗ (phosphocholine∗), Gly-Carb∗ (glycerol plus carbonyls∗), HC (lipid hydrocarbon chains plus cholesterol), and LLP2 peptides (shaded). ∗Indicates a weighted average group (see Materials and Methods). Vertical dotted line facilitates comparison of membrane thickness.
Bending modulus KC
Table 3 shows the values of the bending modulus, KC, for three different membrane mimics: T-cell, HIV, and HIV-neg, and for the HIV extracted lipid membrane. The energy required to bend a membrane a fixed amount is proportional to KC (36). Estimated errors in KC (±0.5) were obtained from data taken at different lateral positions ( Fig. S2) on the same sample.
Table 3.
Bending and orientational results
| Sample | T-cell mimic |
Sxray | HIV mimic |
Sxray | HIV-neg mimic |
Sxray | HIV extracted |
|---|---|---|---|---|---|---|---|
| KC | KC | KC | KC | ||||
| Membrane | 7.1 | 0.71 | 3.5 | 0.80 | 3.0 | 0.80 | 2.6 |
| /WT(40:1) | 5.4 | 0.72 | 3.5 | 0.79 | 3.0 | 0.78 | 2.9 |
| /WT(20:1) | 3.2 | 0.73 | 2.3 | 0.78 | – | 0.81 | – |
| /MX2(40:1) | 2.2 | 0.67 | 2.9 | 0.77 | 2.9 | 0.79 | a |
| /MX2(20:1) | 1.7 | 0.72 | 3.8 | 0.80 | 2.8 | 0.77 | – |
| /CracWTpal(40:1) | 6.4 | 0.74 | 4.2 | 0.77 | 3.8 | 0.78 | 3.7 |
| /CracWTpal(20:1) | 5.3 | 0.78 | 3.7 | 0.78 | 2.5 | 0.79 | – |
| /CracWT(40:1) | 8.2 | 0.70 | 3.2 | 0.77 | 3.7 | 0.72 | 1.9 |
| /CracWT(20:1) | 9.6 | 0.64 | 3.0 | 0.73 | 3.4 | 0.72 | – |
| /CracMX2pal(40:1) | – | – | – | – | 2.5 | 0.79 | 2.5 |
| /CracMX2pal(20:1) | – | – | 2.4 | 0.81 | 2.6 | 0.76 | – |
Membrane mimics and samples are as in Table 2, but also include HIV-neg (HIV mimic without negatively charged lipids) and HIV extracted lipid membranes. KC units are ×10−20 J.
This sample did not reach full hydration.
The T-cell membrane mimic had a KC value near 7 × 10−20 J (Table 3), which is typical of commonly studied lipids (37). The fact that the KC values in the T-cell samples were sensitive to these peptides and their concentrations is further confirmation that these peptides strongly bound into the T-cell membrane mimic. When WT was added, KC decreased considerably, and it decreased even more when MX2 was added. Adding Crac to WT increased KC, whereas the further addition of palmitic acid reduced KC. An interesting result for the HIV membrane mimic and for the HIV-neg membrane mimic as well as for the HIV extracted lipid membrane is that their KC values are quite low (Table 3) compared to many lipid bilayers (37,38) of the same thickness (39). For this study, the most important result is that the peptide variants had little effect on the KC values of the HIV membrane mimic and of the HIV extracted lipid membrane. This supports the conclusion from the form factor results that these peptides do not perturb the HIV membrane mimic.
WAXS
Fig. 4 shows WAXS from four membrane mimics. For three membrane mimics, T-cell, HIV, and HIV-neg, the WAXS is typical of a liquid-ordered phase with intensity centered on the equator at qr ≈ 1.5 Å−1. In contrast, the WAXS data for HIV-chol in Fig. 4 d reveal two separate wide-angle in-plane scattering features: the sharp, intense Bragg rod on the equator at qr ≈ 1.5 Å−1 due to a nontilted gel phase chain-chain correlation and the diffuse arc beginning at qr ≈ 1.4 Å−1 due to a fluid phase chain correlation. Similarly, in the low-angle region at qz ≈ 0.6 Å−1 above the dark beamstop, there are two closely spaced orders indicating two coexisting D-spacings. This gel-fluid phase coexistence prevents determining the bending modulus and the structure as well as the Sxray order parameter. This phase coexistence persisted in all of the HIV-chol samples (even with peptides) at all hydration levels. Interestingly, one role of cholesterol in the HIV membrane may be to prevent a gel-fluid phase separation.
Figure 4.
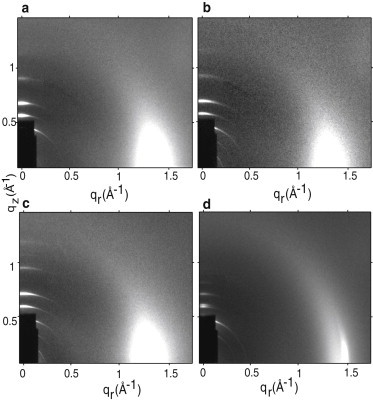
WAXS from lipid membranes at 37°C, a, T-cell, b, HIV, c, HIV-neg, and d, HIV-chol. In d, an intense (white) gel phase peak (qr ≈ 1.5 Å−1) and diffuse Ld (fluid) phase scattering (qr ≈ 1.4 Å−1) indicate gel-fluid phase coexistence.
Sxray
Table 3 also shows the values of the Sxray order parameter obtained by analyzing WAXS data as shown in Fig. 4 for the membrane mimics and in Fig. S4 for all the samples. Sxray is analogous to the NMR SCD order parameter in that it reports molecular orientational order (14,30). Sxray is somewhat smaller for the T-cell membrane mimic than for the HIV and HIV-neg membrane mimics, which is consistent with its lower cholesterol content (30% vs. 45%) (30). The main point of the Sxray results is that, within uncertainty, none of the LLP2 variants significantly changed Sxray of the HIV membrane mimic, consistent with their not entering the HIV membrane mimic. When the CracWTpal peptide was added to the T-cell mimic, the membrane order increased, but it did not increase when just CracWT was added, demonstrating the importance of the palmitoyl group, but not the Crac motif, for increasing membrane order.
CD
CD spectra obtained from hydrated thin films were analyzed for secondary peptide structure. An example of the DichroWEB fit to the CD spectrum from hydrated HIV membrane mimic/CracMX2pal (20:1) is shown in Fig. S7 a. The results of our analysis (Table S1) indicate that the peptides in all three membrane mimics were primarily α-helical, but with significant β-strand and random coil. CD spectra for the 20:1 membrane mimic/peptide samples are shown in Fig. S7, b–d. Interestingly, the helical content of the LLP2 peptides in solution with no lipid was smaller (Table S2); this difference between membrane and solution CD further supports the conclusion drawn from x-ray D-spacing data that LLP2 associates, albeit weakly, with the HIV mimic.
Discussion
Our most important result is that LLP2 interacts strongly with lipid bilayers that mimic the T-cell membrane and only weakly with bilayers that mimic the HIV membrane or with HIV extracted lipid membranes. This result is most obvious in our LAXS form factor data F(qz) shown in Fig. 2. It is supported by our results in Table 3 that the bending modulus decreases with the addition of LLP2 to the T-cell but not to the HIV membrane mimics or extracted HIV lipid samples, and it is consistent with our results for the effect on the order parameter Sxray obtained from wide angle (WAXS) scattering in Table 3. This dramatic difference in interaction of all of the LLP2 variants with the T-cell mimic compared to the HIV mimic with two different lipid compositions was at first astonishing to us. However, Dick et al. (40) similarly found that HIV-1 Gag protein (and the shorter MA segment) strongly preferred lipids with both acyl chains unsaturated over those with only one chain unsaturated, and the addition of cholesterol increased Gag binding and led to closer packing of phospholipids. Nevertheless, it is important to note that LLP2 does interact, albeit weakly, with the HIV mimic, as indicated by its effect on the lamellar D-spacing and by its having greater helical content than in solution. More detailed discussion regarding this weak binding is elaborated in the last paragraph in the Supporting Material. The cartoon in Fig. 5 summarizes the interaction of LLP2 WT and the mutant MX2 with the two membrane mimics.
Figure 5.
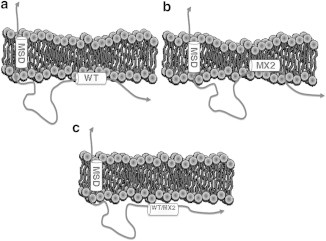
Cartoon showing differences in the interaction of LLP2 with T-cell and HIV membranes. (a) WT LLP2 binds to the outer headgroup region and thins the T-cell membrane. (b) MX2 LLP2 binds more deeply into the hydrocarbon interior and thins the T-cell membrane to a greater extent. (c) WT and MX2 LLP2 only weakly bind to the HIV virion membrane.
This result for the interaction of LLP2 explains, at a biophysical level, why there is a clear difference in the biological effects of the MX2 R->E mutations in LLP2. Although the MX2 mutant did not alter Env incorporation or infectivity, which involve the HIV virion membrane, it did inhibit viral-initiated T-cell death and syncytium formation, which involve only the T-cell membrane (10). In the latter case, there are Env proteins located on the surface of an infected T-cell membrane, but they are not yet localized to a high-cholesterol domain, such as during budding of the new HIV virion. Many of these surface Env proteins get endocytosed before they can be incorporated into virions, but some of them bind to CD4 receptors on other T-cells, causing a large, multinucleated cell that eventually dies. It is remarkable that a peptide in a cytoplasmic location can affect what happens on the exterior of the membrane. Our result implies that interaction with a membrane is important for LLP2 to have a biological effect. This is consistent with the hypothesis that the membrane transduces information embedded in the CTT sequence to the major part of the Env protein that resides on the other side of the membrane. When there is only a weak interaction with the HIV membrane mimic, the MX2 mutant has no effect. When LLP2 interacts with the T-cell membrane mimic our results show that the MX2 mutant embeds differently into the T-cell mimic than the WT. Such a difference is well correlated with the functional inhibition of syncytium formation by the MX2 mutant.
A theoretical challenge is to understand how the cytoplasmic CTT influences the extracellular domain of the gp41 Env protein. A potential biophysical mechanism involves the lateral pressure profile of the membrane (41), which would be affected locally by embedded LLP2 and which would then affect other nearby parts of gp41, such as the transmembrane domain (TMD). Perturbation of the TMD of gp41 could affect the conformational changes necessary for fusion as several studies with modified TMDs have demonstrated inhibition of fusion (42–44). With regard to the T-cell membrane, the deeper embedding of MX2 would alter the lateral pressure profile compared to WT and that would affect the TMD of the gp41 Env protein differently, correlating well to the inhibition of cell-cell fusion upon mutation. It is important to emphasize in this theory that it is the difference in the lateral pressure profile between WT and MX2 that modulates biological function, not that MX2 binds more deeply and therefore might affect the membrane more than WT. Finally, with regard to the HIV membrane mimic, the very weak interaction of LLP2, either WT or MX2, predicts that MX2 would have little effect on Env incorporation or infectivity, as observed.
Another potential mechanism might have been the softening of the T-cell membrane mimic by LLP2, which could then have led to highly curved fusion intermediates, as we have hypothesized for the HIV FP23 fusion peptide (39). However, in this work, the bending modulus decreases more with MX2 than with WT, and it increases above that of the T-cell mimic when the Crac motif is added; therefore our data contradict membrane softening as the biophysical mechanism for LLP2’s effect.
It is of interest to correlate the difference in the interaction of LLP2 with the lipid compositions of the membrane mimics. The HIV membrane mimic has less disaturated chain lipid dihydrosphingomyelin (1.7 compared to 2.7 mol %) and less negatively charged phosphoinositol (1.3 mol %) compared to the T-cell (6.3 mol %), decreasing the overall negative charge in the T-cell (0.2e/lipid) to 0.15e/lipid in the HIV membrane mimic. However, the largest difference is 45 mol % cholesterol in the HIV membrane mimic compared to 30 mol % in the T-cell membrane mimic. With such a high concentration of cholesterol it is surprising that the HIV membrane mimic has such a low bending modulus, KC, which indicates more flexibility than the average single component lipid bilayer. This low KC does not depend on negative charge, because HIV-neg had an equally low KC, which is consistent with other studies that have shown that lipid negative charges have a negligible effect on KC (46,47). Because the KC of the HIV membrane mimic is quite low, this softer membrane more easily forms highly curved intermediates during fusion with its host cell (48). Previously, Kol et al. (49) found that the immature HIV virion that had CTT engaged with the p55Gag was twice as stiff as the mature virion that had the CTT disengaged from the p55Gag. Our study did not involve p55Gag, but our result that our HIV membrane mimic and HIV extracted lipids are quite soft when there are no other associated proteins is consistent with Kol et al. (49). Although it may seem counterintuitive that membranes with high cholesterol concentration can be soft, we have shown before that the effect of cholesterol depends on the chain length and saturation of the lipids, not just on the cholesterol concentration (50,51).
It is also of interest to consider the orientation and conformation of LLP2. Our CD results (Fig. S7 and Table 1) show ∼55–70% α-helix for all LLP2 variants in T-cell, HIV, and HIV-neg membrane mimics. A helical structure was observed in WT LLP2 from several clades of HIV-1 gp41 in 10 mM sodium dodecyl sulfate (SDS) or 60% triflouroethanol, although helical content was not quantified in that study (52). The differences in helical content are small, so secondary structure appears unlikely to be the primary cause of differences in the effects of WT and MX2 in the T-cell mimic. All of the peptides are more helical when associated with membrane mimics than in the aqueous phase (see Table S1 and Table S2). LLP2 helices have large hydrophobic moments and would therefore be expected to orient either with the long axis parallel to the surface of membranes as shown in Fig. 5, or as transmembrane bundles consisting of more than one or two peptides. Transmembrane bundles have had prominent in-plane scattering along the qr direction such as occurs for alamethicin (53) and no such scattering was observed for any of our LLP2 samples.
Given our result that LLP2 peptides interact only weakly with the HIV membrane mimic, they would be more likely to oligomerize in solution along their hydrophobic faces, consistent with a study by Lee et al. (54) that has implicated LLP2 in Env multimerization. Other studies have implicated LLP2 interacting with p55Gag during virion formation (55–58). Env incorporation into cholesterol-rich raft-like domains could occur by the interaction of LLP2 with the MA portion of p55Gag, which binds to rafts as other studies have shown (59–63), but our data are silent on these possibilities.
Although natural and experimental variations in Env gp120 and gp41 ectodomain sequences have been studied in detail as determinants of Env structure and functional properties, the current data indicate a need to more rigorously evaluate the role of natural CTT variations as a determinant of Env phenotypes, including antibody neutralization sensitivity, due to its inside-outside signaling ability (64). In addition, the defined role of CTT-membrane mimic interactions as a determinant of overall Env conformation may in part explain the failure of experimental immunizations with HIV Env proteins engineered with deletions of the entire CTT to reduce cytotoxicity and increase the levels of Env protein expression (65). These experimental immunizations with CTT-deleted Env constructs have in general elicited high levels of antibody to the vaccine protein, but relatively low levels of antibody reactivity with the native viral Env protein that fail to neutralize viral infectivity. It is possible that the CTT truncations substantially alter the conformation of the vaccine Env protein compared to the native full length viral Env protein. Thus, these data indicate the importance of designing HIV vaccines to express full-length membrane-associated Env protein to maximize the production of antibody responses that recognize and inactivate the native Env structure on the surface of viral particles and infected cells.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM44976 (PIs J.F.N., S.T.N.) and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI087533(PI RCM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. A.L.B. was partially supported by the Howard Hughes Medical Foundation for undergraduate research. We acknowledge the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation (NSF) and the NIH/NIGMS under NSF award DMR-0936384, and we especially thank Dr. Arthur Woll for facilitating our use of the G1 station. We acknowledge the Center for Molecular Analysis at Carnegie Mellon University for use of the Jasco 715 and for mass spectrometry analysis and the Protein Synthesis Core of the University of Pittsburgh for peptide production. We acknowledge Bradley Treece for help with calculations of Sxray.
Supporting Material
References
- 1.Costin J.M., Rausch J.M., Wimley W.C. Viroporin potential of the lentivirus lytic peptide (LLP) domains of the HIV-1 gp41 protein. Virol. J. 2007;4:1–14. doi: 10.1186/1743-422X-4-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koenig B.W., Ferretti J.A., Gawrisch K. Site-specific deuterium order parameters and membrane-bound behavior of a peptide fragment from the intracellular domain of HIV-1 gp41. Biochemistry. 1999;38:6327–6334. doi: 10.1021/bi982800g. [DOI] [PubMed] [Google Scholar]
- 3.Moreno M.R., Giudici M., Villalaín J. The membranotropic regions of the endo and ecto domains of HIV gp41 envelope glycoprotein. Biochim. Biophys. Acta. 2006;1758:111–123. doi: 10.1016/j.bbamem.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Kliger Y., Shai Y. A leucine zipper-like sequence from the cytoplasmic tail of the HIV-1 envelope glycoprotein binds and perturbs lipid bilayers. Biochemistry. 1997;36:5157–5169. doi: 10.1021/bi962935r. [DOI] [PubMed] [Google Scholar]
- 5.Chen S.S.L., Lee S.F., Wang C.T. Cellular membrane-binding ability of the C-terminal cytoplasmic domain of human immunodeficiency virus type 1 envelope transmembrane protein gp41. J. Virol. 2001;75:9925–9938. doi: 10.1128/JVI.75.20.9925-9938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenberg D., Wesson M. The most highly amphiphilic alpha-helices include two amino acid segments in human immunodeficiency virus glycoprotein 41. Biopolymers. 1990;29:171–177. doi: 10.1002/bip.360290122. [DOI] [PubMed] [Google Scholar]
- 7.Murakami T., Freed E.O. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Natl. Acad. Sci. USA. 2000;97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilk T., Pfeiffer T., Bosch V. Retained in vitro infectivity and cytopathogenicity of HIV-1 despite truncation of the C-terminal tail of the env gene product. Virology. 1992;189:167–177. doi: 10.1016/0042-6822(92)90692-i. [DOI] [PubMed] [Google Scholar]
- 9.Postler T.S., Desrosiers R.C. The tale of the long tail: the cytoplasmic domain of HIV-1 gp41. J. Virol. 2013;87:2–15. doi: 10.1128/JVI.02053-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalia V., Sarkar S., Montelaro R.C. Rational site-directed mutations of the LLP-1 and LLP-2 lentivirus lytic peptide domains in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41 indicate common functions in cell-cell fusion but distinct roles in virion envelope incorporation. J. Virol. 2003;77:3634–3646. doi: 10.1128/JVI.77.6.3634-3646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalia V., Sarkar S., Montelaro R.C. Antibody neutralization escape mediated by point mutations in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41. J. Virol. 2005;79:2097–2107. doi: 10.1128/JVI.79.4.2097-2107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y.F., Nagle J.F. Diffuse scattering provides material parameters and electron density profiles of biomembranes. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004;69:040901–040904. doi: 10.1103/PhysRevE.69.040901. (R) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kučerka N., Liu Y.F., Nagle J.F. Structure of fully hydrated fluid phase DMPC and DLPC lipid bilayers using X-ray scattering from oriented multilamellar arrays and from unilamellar vesicles. Biophys. J. 2005;88:2626–2637. doi: 10.1529/biophysj.104.056606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mills T.T., Toombes G.E.S., Nagle J.F. Order parameters and areas in fluid-phase oriented lipid membranes using wide angle X-ray scattering. Biophys. J. 2008;95:669–681. doi: 10.1529/biophysj.107.127845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyatskaya Y., Liu Y.F., Nagle J.F. Method for obtaining structure and interactions from oriented lipid bilayers. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2001;63:011907. doi: 10.1103/PhysRevE.63.011907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brügger B., Glass B., Kräusslich H.G. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. USA. 2006;103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan R., Uchil P.D., Wenk M.R. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J. Virol. 2008;82:11228–11238. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenwood A.I., Pan J.J., Tristram-Nagle S. CRAC motif peptide of the HIV-1 gp41 protein thins SOPC membranes and interacts with cholesterol. Biochim. Biophys. Acta. 2008;1778:1120–1130. doi: 10.1016/j.bbamem.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139:4991–4997. doi: 10.1210/endo.139.12.6390. [DOI] [PubMed] [Google Scholar]
- 20.Epand R.M., Sayer B.G., Epand R.F. Peptide-induced formation of cholesterol-rich domains. Biochemistry. 2003;42:14677–14689. doi: 10.1021/bi035587j. [DOI] [PubMed] [Google Scholar]
- 21.Rousso I., Mixon M.B., Kim P.S. Palmitoylation of the HIV-1 envelope glycoprotein is critical for viral infectivity. Proc. Natl. Acad. Sci. USA. 2000;97:13523–13525. doi: 10.1073/pnas.240459697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C.L., Spies C.P., Compans R.W. The human and simian immunodeficiency virus envelope glycoprotein transmembrane subunits are palmitoylated. Proc. Natl. Acad. Sci. USA. 1995;92:9871–9875. doi: 10.1073/pnas.92.21.9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 24.Steckbeck J.D., Sun C.Q., Montelaro R.C. Topology of the C-terminal tail of HIV-1 gp41: differential exposure of the Kennedy epitope on cell and viral membranes. PLoS ONE. 2010;5:e15261. doi: 10.1371/journal.pone.0015261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tristram-Nagle S.A. Preparation of oriented, fully hydrated lipid samples for structure determination using X-ray scattering. Methods Mol. Biol. 2007;400:63–75. doi: 10.1007/978-1-59745-519-0_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghunathan M., Zubovski Y., Tristram-Nagle S. Structure and elasticity of lipid membranes with genistein and daidzein bioflavinoids using X-ray scattering and MD simulations. J. Phys. Chem. B. 2012;116:3918–3927. doi: 10.1021/jp211904j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kučerka N., Nagle J.F., Katsaras J. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophys. J. 2008;95:2356–2367. doi: 10.1529/biophysj.108.132662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagle J.F., Wiener M.C. Relations for lipid bilayers. Connection of electron density profiles to other structural quantities. Biophys. J. 1989;55:309–313. doi: 10.1016/S0006-3495(89)82806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kučerka N., Kiselev M.A., Balgavý P. Determination of bilayer thickness and lipid surface area in unilamellar dimyristoylphosphatidylcholine vesicles from small-angle neutron scattering curves: a comparison of evaluation methods. Eur. Biophys. J. 2004;33:328–334. doi: 10.1007/s00249-003-0349-0. [DOI] [PubMed] [Google Scholar]
- 30.Mills T.T., Tristram-Nagle S., Feigenson G.W. Liquid-liquid domains in bilayers detected by wide angle X-ray scattering. Biophys. J. 2008;95:682–690. doi: 10.1529/biophysj.107.127910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Y., Huang H.W., Olah G.A. Method of oriented circular dichroism. Biophys. J. 1990;57:797–806. doi: 10.1016/S0006-3495(90)82599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merzlyakov M., Li E., Hristova K. Directed assembly of surface-supported bilayers with transmembrane helices. Langmuir. 2006;22:1247–1253. doi: 10.1021/la051933h. [DOI] [PubMed] [Google Scholar]
- 33.Lobley A., Whitmore L., Wallace B.A. DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics. 2002;18:211–212. doi: 10.1093/bioinformatics/18.1.211. [DOI] [PubMed] [Google Scholar]
- 34.Abdul-Gader A., Miles A.J., Wallace B.A. A reference dataset for the analyses of membrane protein secondary structures and transmembrane residues using circular dichroism spectroscopy. Bioinformatics. 2011;27:1630–1636. doi: 10.1093/bioinformatics/btr234. [DOI] [PubMed] [Google Scholar]
- 35.Mutz M., Helfrich W. Unbinding transition of a biological model membrane. Phys. Rev. Lett. 1989;62:2881–2884. doi: 10.1103/PhysRevLett.62.2881. [DOI] [PubMed] [Google Scholar]
- 36.Helfrich W., Prost J. Intrinsic bending force in anisotropic membranes made of chiral molecules. Phys. Rev. A. 1988;38:3065–3068. doi: 10.1103/physreva.38.3065. [DOI] [PubMed] [Google Scholar]
- 37.Kučerka N., Tristram-Nagle S., Nagle J.F. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2005;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 38.Rawicz W., Olbrich K.C., Evans E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000;79:328–339. doi: 10.1016/S0006-3495(00)76295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tristram-Nagle S., Nagle J.F. HIV-1 fusion peptide decreases bending energy and promotes curved fusion intermediates. Biophys. J. 2007;93:2048–2055. doi: 10.1529/biophysj.107.109181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dick R.A., Goh S.L., Vogt V.M. HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc. Natl. Acad. Sci. USA. 2012;109:18761–18766. doi: 10.1073/pnas.1209408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cantor R.S. Lipid composition and the lateral pressure profile in bilayers. Biophys. J. 1999;76:2625–2639. doi: 10.1016/S0006-3495(99)77415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shang L., Yue L., Hunter E. Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J. Virol. 2008;82:5417–5428. doi: 10.1128/JVI.02666-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.West J.T., Johnston P.B., Hunter E. Mutations within the putative membrane-spanning domain of the simian immunodeficiency virus transmembrane glycoprotein define the minimal requirements for fusion, incorporation, and infectivity. J. Virol. 2001;75:9601–9612. doi: 10.1128/JVI.75.20.9601-9612.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kondo N., Miyauchi K., Matsuda Z. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 2010;285:14681–14688. doi: 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reference deleted in proof.
- 46.Song J., Waugh R.E. Bilayer membrane bending stiffness by tether formation from mixed PC-PS lipid vesicles. J. Biomech. Eng. 1990;112:235–240. doi: 10.1115/1.2891178. [DOI] [PubMed] [Google Scholar]
- 47.Rowat A.C., Hansen P.L., Ipsen J.H. Experimental evidence of the electrostatic contribution to membrane rigidity. Europhys. Lett. 2004;67:144–149. [Google Scholar]
- 48.Siegel D.P. The Gaussian curvature elastic energy of intermediates in membrane fusion. Biophys. J. 2008;95:5200–5215. doi: 10.1529/biophysj.108.140152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kol N., Shi Y., Rousso I. A stiffness switch in human immunodeficiency virus. Biophys. J. 2007;92:1777–1783. doi: 10.1529/biophysj.106.093914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pan J.J., Mills T.T., Nagle J.F. Cholesterol perturbs lipid bilayers nonuniversally. Phys. Rev. Lett. 2008;100:198103. doi: 10.1103/PhysRevLett.100.198103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan J.J., Tristram-Nagle S., Nagle J.F. Effect of cholesterol on structural and mechanical properties of membranes depends on lipid chain saturation. Phys. Rev. E. 2009;80:021931. doi: 10.1103/PhysRevE.80.021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steckbeck J.D., Craigo J.K., Montelaro R.C. Highly conserved structural properties of the C-terminal tail of HIV-1 gp41 protein despite substantial sequence variation among diverse clades: implications for functions in viral replication. J. Biol. Chem. 2011;286:27156–27166. doi: 10.1074/jbc.M111.258855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pan J.J., Tristram-Nagle S., Nagle J.F. Alamethicin aggregation in lipid membranes. J. Membr. Biol. 2009;231:11–27. doi: 10.1007/s00232-009-9199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee S.F., Wang C.T., Chen S.S.L. Multimerization potential of the cytoplasmic domain of the human immunodeficiency virus type 1 transmembrane glycoprotein gp41. J. Biol. Chem. 2000;275:15809–15819. doi: 10.1074/jbc.M000601200. [DOI] [PubMed] [Google Scholar]
- 55.Bhatia A.K., Campbell N., Ratner L. Characterization of replication defects induced by mutations in the basic domain and C-terminus of HIV-1 matrix. Virology. 2007;369:47–54. doi: 10.1016/j.virol.2007.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freed E.O., Martin M.A. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J. Virol. 1996;70:341–351. doi: 10.1128/jvi.70.1.341-351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freed E.O., Orenstein J.M., Martin M.A. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 1994;68:5311–5320. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hourioux C., Brand D., Roingeard P. Identification of the glycoprotein 41(TM) cytoplasmic tail domains of human immunodeficiency virus type 1 that interact with Pr55Gag particles. AIDS Res. Hum. Retroviruses. 2000;16:1141–1147. doi: 10.1089/088922200414983. [DOI] [PubMed] [Google Scholar]
- 59.Ono A., Freed E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA. 2001;98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindwasser O.W., Resh M.D. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 2001;75:7913–7924. doi: 10.1128/JVI.75.17.7913-7924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ono A., Waheed A.A., Freed E.O. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: use of a novel Gag multimerization assay. J. Virol. 2005;79:14131–14140. doi: 10.1128/JVI.79.22.14131-14140.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patil A., Gautam A., Bhattacharya J. Evidence that Gag facilitates HIV-1 envelope association both in GPI-enriched plasma membrane and detergent resistant membranes and facilitates envelope incorporation onto virions in primary CD4+ T cells. Virol. J. 2010;7:1–5. doi: 10.1186/1743-422X-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saad J.S., Miller J., Summers M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wyss S., Dimitrov A.S., Hoxie J.A. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J. Virol. 2005;79:12231–12241. doi: 10.1128/JVI.79.19.12231-12241.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Checkley M.A., Luttge B.G., Freed E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011;410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.