

Abstract
Somatic cell reprogramming to pluripotency requires an immediate increase in cell proliferation and reduction in cell size. It is unknown whether proliferation and biomass controls are similarly coordinated with early events during the differentiation of pluripotent stem cells (PSCs). This impasse exists because PSCs grow in tight clusters or colonies, precluding most quantifying approaches. Here, we investigate live cell interferometry as an approach to quantify the biomass and growth of HSF1 human PSC colonies before and during retinoic acid-induced differentiation. We also provide an approach for measuring the rate and coordination of intracolony mass redistribution in HSF1 clusters using live cell interferometry images. We show that HSF1 cells grow at a consistent, exponential rate regardless of colony size and display coordinated intracolony movement that ceases with the onset of differentiation. By contrast, growth and proliferation rates show a decrease of only ∼15% decrease during early differentiation despite global changes in gene expression and previously reported changes in energy metabolism. Overall, these results suggest that cell biomass and proliferation are regulated independent of pluripotency during early differentiation, which is distinct from what occurs with successful reprogramming.
Introduction
Human pluripotent stem cells (hPSCs) generate all embryonic cell types and can be grown for prolonged periods in culture, with their pluripotency/self-renewal program controlled by chromatin structure and a core transcription factor network (1). hPSCs are also small, replicate rapidly, and grow in tight colonies. It remains unknown whether the size and growth rate of hPSCs are regulated by the program that regulates pluripotency/self-renewal, a question that has important implications for our understanding of the relationship between growth rate controls and early differentiation. Interestingly, key features of hPSCs can be reestablished in somatic cells by reprogramming factors (2) and recent, single-cell imaging studies have shown that fibroblasts reprogrammed to hPSCs undergo an increase in proliferation and reduction in area/size within the first cell division (3). This work identified replicative and size barriers that must be rapidly overcome for successful reprogramming. Reprogramming to, and differentiating from, pluripotency are overtly similar but opposite processes (4), and it is unclear whether analogous barriers or checkpoints also exist early during hPSC differentiation. Unfortunately, the growth of hPSCs in tight, multicellular clumps has blocked direct, in situ measurements of cell growth, size, and other fundamental properties, such as intracolony motion during early hPSC differentiation. It therefore remains uncertain whether hPSCs undergo a decrease in proliferation and increase in size with induced differentiation, or if changes in cell proliferation and size (biomass) are coordinated with early hPSC differentiation programs (5).
To date, hPSC size measurements have been imprecise and used to document extreme changes in cross-sectional area by microscopy (3). hPSC growth is typically measured as time to division and not as changes in cell biomass, which is the most direct measure of net cellular growth but requires detection at <5% mass change per hour. Existing optical approaches have also revealed that individual PSC colonies are highly motile, a property that may skew clonogenic counting assays through colony merging (6). Coordinated cell movement within hPSC colonies could provide a basis for colony migration and merging, although this too remains unknown for hPSCs grown in culture (6–8).
In general, cell size has been inferred from the radius or projected area for cells with fixed geometries, such as bacteria, yeasts, and spherical lymphocytes. However, most animal cells, or cells that grow in colonies, have irregular and often changing shapes, resulting in poor estimates of cell biomass based on measurements of projected area (see Fig. S1 in the Supporting Material). Additionally, changes in cell volume or area may result from changes in solute concentrations or flattening against a substrate rather than from changes in biomass. Flow cytometry measures material density gradients and aqueous content to estimate cell size, but cannot quantify the continuous growth of hPSC colonies (9). More promising are micro-electromechanical system microresonators, which can accurately measure the mass of single cells instantaneously and over time (10,11). However, for sufficient sensitivity a resonator must be microns or smaller, making continuous measurements of motile, growing hPSC colonies practically impossible.
To provide an approach for addressing fundamental questions regarding mass and growth control in hPSCs and during early differentiation, we evaluated live cell interferometry (LCI) as a method for quantifying mass distributions within HSF1 hPSC colonies over time. LCI takes advantage of the fact that as light passes through a transparent object, such as a cell, it slows down due to the interaction of light with matter (12,13). This retardation of light from the interaction with cellular biomass causes a phase shift relative to unperturbed light, which is measured using a Michelson interferometer (13). Assuming a cellular average for the relationship between density and changes in refractive index, the measured phase shift at each location inside of the cell is directly proportional to the amount of biomass at that location, resulting in a map of the distribution of mass for all nonaqueous components (dry mass) (12–15). In this study, we assume that the conversion factor between phase shift and mass, the inverse of the specific refractive index, is 5.56 pg/μm3, although we note that this parameter only varies by ∼±10% among the typical contents of a cell (12,13). Assuming the average contents of the cell/colony remain fairly constant over the measurement period, the specific value of this parameter will drop out of any measurements based on relative mass comparisons, such as the specific growth rate. Recent applications of quantitative phase microscopy using other imaging platforms include measurements of cell growth (14), cell death (16), membrane mechanics (17), individual organelles (18), and preliminary imaging of mouse PSCs (19). Prior work with LCI establishes it as a method for repeatable (<2% coefficient of variation) quantification of mass, mass accumulation rate, and mass distributions for large populations of cells on a single cell basis (13,20), but not for cells constrained within hPSC colonies.
Materials and Methods
Cell line and growth media
HSF1 human embryonic stem cells (hESCs; 46XY; UC-0001) were grown on feeder-free Matrigel (BD Biosciences, San Jose, CA) in defined cell culture media (StemPro SFM; Invitrogen, Carlsbad, CA) with daily media changes, as described previously in Zhang et al. (21,22). Differentiation with retinoic acid (RA) was induced by replacing bFGF with 10-μM RA (Acros Organics, Fair Lawn, NJ).
Cell counting
Cell counting was performed using a Neubauer hemocytometer after Trypan-blue staining (Invitrogen).
Embryoid body formation assay
Embryoid body (EB) formation was performed as described previously in Zhang et al. (21). Briefly, hESCs were trypsinized to single cells and 106 cells were placed in one well of a AggreWell 400 plate (Stemcell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. The next day, EBs were harvested and grown in Iscove’s Modified Dulbecco’s medium supplemented with 20% fetal bovine serum for 10 days in a Corning (E. I. DuPont de Nemours, Wilmington, DC) CoStar ultra-low attachment six-well plate (Sigma-Aldrich, St. Louis, MO). Media was exchanged every 2–3 days. An inverted light microscope was used to assess and count EBs.
Gene expression profiling
Total RNA was extracted from control or RA-exposed hESCs using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized from total RNA using a Superscript III first-strand cDNA synthesis kit (Invitrogen). Real-time PCR was performed with a SYBR Green PCR kit (Diagenode, Denville, NJ) with denaturation at 94°C for 15 s, annealing at 60°C for 30 s, and extension at 72°C for 45 s over 40 cycles. Primer sequences are available upon request.
Cell cycle analysis
Cells were harvested by trypsinization and stained for DNA content with propidium iodide (Sigma-Aldrich). Single-color flow cytometry was performed on an Accuri C6 flow cytometer (BD Biosciences) and data collected for 30,000–150,000 events. Cell cycle analysis was performed in FlowJo (Tree Star, Ashland, OR) using a Dean-Jett-Fox model fit to the propidium iodide fluorescence intensity histogram.
Live cell interferometry
Live cell interferometry (LCI) imaging was performed on a Contour GT-X8 optical profiler with a through transmissive media attachment (Bruker, Tuscon, AZ). Cell viability was maintained using a custom live cell flow chamber (13,20). A quantity of 20 × 20-mm square silicon substrates were treated with SigmaCote (Sigma-Aldrich) and 1 M NaOH before coating with Matrigel (BD Biosciences) to obtain a surface suitable for hPSC growth (23). HSF1 cells plated as small colonies onto treated silicon were imaged one, five, or seven days after plating (zero, four, or six days after the start of RA exposure). Cells were viable with no visible defects in morphology after >12 h on the interferometer stage under constant illumination. Most colonies were imaged for 2 h at 11× effective magnification. Because of the difficulty in finding colonies which were large, but that did not extend outside of the microscope observation area, most colony growth-rate measurements focus on colonies that spanned at most one or two individual microscope fields of view. A subset of experiments were performed using a large grid of 12 × 15 images with 50-μm overlap between edges for stitching (as seen later in Fig. 2 A). To obtain unaliased estimates of the rate of mass motion, representative smaller colonies were measured at 20× magnification with 30 s between images (seen later in Fig. 6, A–F, and see also Fig. S7, A–F).
Figure 2.
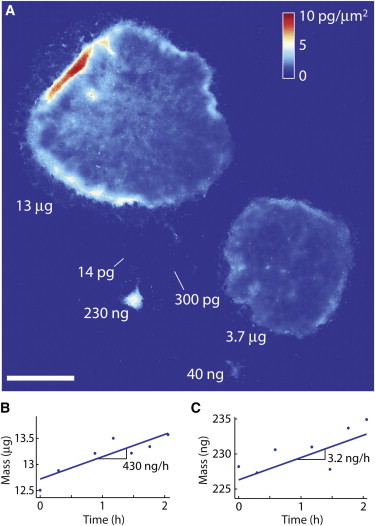
LCI quantifies hPSC colony mass accumulation rates for undifferentiated HSF1 cell clusters spanning five orders of magnitude in mass. (A) Stitched LCI image of HSF1 colonies after seven days in culture showing mass measurements spanning a dynamic range of six orders of magnitude. The largest visible colony has a total mass of 13.5 μg, whereas the smallest visible cellular fragment has a mass of 14 pg. (B and C) Mass versus time plots of two colonies from panel A. Scale bar in panel A is 1 mm.
Figure 6.
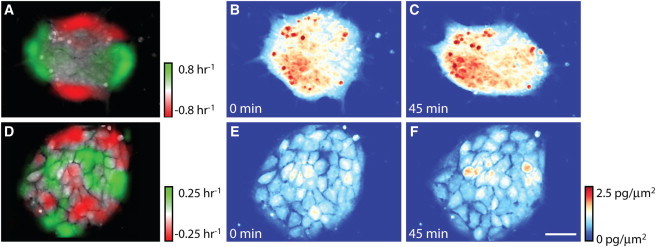
HSF1 hPSCs exhibit larger, faster areas of mass redistribution within colonies, indicating a higher degree of movement coordination, than RA-exposed hPSCs. (A and D) Maps of local specific growth rate (change in mass normalized by average mass) over 45 min for representative (A) undifferentiated and (D) RA-differentiated hPSC colonies (green, mass accumulation; red, mass loss). The pattern of mass redistribution in panel A shows a large change in undifferentiated colony morphology (stretching) along the horizontal axis (B and C) and the generally larger change in mass distribution in undifferentiated versus differentiated colonies. RA-differentiated colonies show a much smaller mass redistribution and little change in overall colony morphology (D–F). Scale bar is 50 μm.
Statistical analysis
Statistical analysis was performed using a two-tailed Student t-test with unequal variances and sample sizes (Welch t-test). Error bars are reported as ± standard error (SE).
Image analysis
All image processing was performed using custom MATLAB (The MathWorks, Natick, MA) scripts, as described below.
Phase unwrapping
Phase errors, integer wavelength errors caused by a one-wavelength ambiguity inherent in quantitative phase imaging (24), were corrected using a random walk-based algorithm that removes integer wavelength jumps and excursions below the background level.
Image stitching
Images were stitched together using an optimization algorithm implemented in the software MATLAB (The MathWorks) that adjusts the offset and planar tilt of each frame to minimize the sum of the differences between the overlapping regions of each adjacent image.
Total colony mass, mass accumulation rate, and area
Colonies were located by local adaptive thresholding based on Otsu’s method (25), and then the mass distributions within these pixels were summed to obtain colony total mass. The slope of a least-squares best-fit line to total mass versus time is the colony mass accumulation rate. Errors in this value are estimated as the standard deviation (SD) of the mean slope (26). Colonies with errors exceeding the estimated growth rate were excluded from the reported average growth rates. Average growth rates including these estimated error rates were determined using a Monte Carlo routine with 10,000 samples, implemented in MATLAB and run twice with different pseudo-random number seeds and compared to check convergence. For each sample in the Monte Carlo simulation, the empirical cumulative density function of the log-transformed specific growth rate data was fit to a Gaussian cumulative density function to estimate the population mean and SD (see Fig. S3 A). The average and SD of these estimated values over all trials is the reported mean growth rate. Additional results from this analysis are presented in the Supporting Material (see Fig. S3 B). Colony area is estimated based on the area of the region determined by Otsu thresholding.
Growth rate versus mass curve fitting
Growth rate versus mass data were fit in MATLAB using the Curve Fit toolbox with a nonlinear least-squares fit and an unconstrained trust region algorithm, weighted by the square of the SE of the growth rate measurements. Confidence intervals on the power-law scaling exponent are based on the Student t-test at a significance level (two-tailed) of p = 0.05.
Local mass accumulation rate
To measure mass accumulation rate distributions within individual colonies, as seen later in Fig. 3, we first remove translational motion by aligning successive colony images based on cross-correlations between frames. We then use Otsu’s method (25) on the location-shifted, time-averaged image to find pixel locations of highest average intensity. This yields a measure of mass versus time m(x,y,t) at each pixel location, x,y. We perform a least-squares fit on the m(t) measurement at each pixel within the colony and estimate the local mass accumulation rate as the slope of this best fit line. Finally, we use a Gaussian low-pass filter with a spatial SD of 10 μm to smooth out high spatial frequency variations while maintaining adequate colony-scale resolution.
Figure 3.
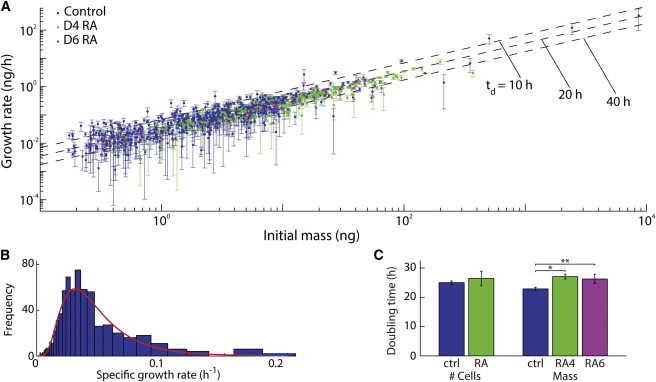
Average mass accumulation rate for untreated and retinoic acid (RA)-exposed hPSC colonies. (A) Growth rate versus initial mass for untreated and RA-exposed hPSC colonies shows an exponential growth rate for mass accumulation across five orders of magnitude in mass. RA-treated colonies show similar mass accumulation rates to untreated colonies. Error bars show the uncertainty in measured growth rate calculated as the SD of the slope of the biomass versus time measurements. Error bars appear uneven due to the logarithmic scale. (B) Histogram of untreated control colony growth rate measurements (normalized by initial mass to provide specific growth rate) with the log-normal curve (red) showing the estimated distribution of specific growth rates that was used to compute doubling time. (C) Doubling time from cell counting is unable to distinguish cell proliferation rates between control and RA-exposed hPSCs during 4–6 days of growth. Doubling time for cell mass shows a decrease of ∼15% with RA treatment (based on measurements of 658 control, 227 RA day-4, and 62 RA day-6 colonies). ctrl = control; RA4 = 4 days of RA treatment, RA6 = 6 days of RA treatment. Error bars show ± SE ∗p < 0.05, ∗∗p < 0.001.
Decorrelation timescale
To find the decorrelation timescale, we first estimate the autocorrelation, CXX (27), at every time shift, Δt, by applying a sliding window of width w discrete time steps,
where we use w = 20 (10 min at Δt = 30 s per image) to get adequate resolution of the typical timescales with minimized computational cost. We then average this function CXX across all pixel locations x,y, all starting times t0, and all time shifts, τ, to get an overall averaged autocorrelation CXX(τ) (see Fig. S8 C). Finally, we apply a linear least-squares fit to this function, CXX(τ), and find the slope of this best fit line. This slope is an estimate of the decorrelation rate, or how quickly the mass distribution within the colony is changing over time.
Average cell mass
The mass of individual cells within colonies was measured with LCI, then, while on the microscope stage, colonies were fixed with paraformaldehyde (Sigma), permeabilized in a solution of Triton-X (Sigma), and stained with a fluorescent nuclear stain (YOYO-1; Invitrogen). Cell number was determined by manually counting the number of cells within each colony after processing with a custom MATLAB script (see Fig. S5). The average mass per cell was computed as the total colony mass divided by the number of cells.
Coordination distance
For each stack of colony images, we first identify the colony using Otsu thresholding (25) on a composite mean image of all frames. Then, for each pixel within the colony, we compare the local rate of mass increase at each location to the average colony growth rate, to generate a binary image in which each pixel indicates the sign of mass accumulation relative to the mean colony mass accumulation rate as either positive (seen later as green in Fig. 6, A and D) or negative (seen later as red in Fig. 6, A and D). At each pixel in this binary image, we estimate P(Δx) as
where Δx is the Euclidian displacement of each pixel from the current pixel. Finally, we average the P(Δx) profile estimates from each pixel within each colony in the sample to get the final, empirical P(Δx) profile (such that, for example, P(Δx) for day 0 control represents the average over 7 × 106 pixel-wise estimates of P(Δx)). Here, we approximate P(Δx) as an exponential decay and define the coordination distance as the exponential decay constant.
Results
We quantified the biomass of a total of 728 HSF1 colonies at one-, five-, and seven-day timepoints using LCI (Fig. 1 and see Fig. S1). LCI provides precise and reproducible biomass measurements for both small and large hPSC colonies over a range of six orders of magnitude, from subcellular fragments of ∼10 pg to the largest colonies spanning multiple image frames with masses up to ∼10 μg (Fig. 2 A). The mass of individual colonies was tracked over a period of 2 h at selected timepoints from the first several days in culture, enabling studies of hPSCs and the earliest stages of differentiation (Fig. 2, B and C). LCI quantifies hPSC growth rates within 2 h, which is significantly faster than conventional cell proliferation rate measurements (2–6 days). These measurements are robust to changes in colony morphology and mass distribution observed with LCI (see Fig. S6 and Fig. S8 D).
Figure 1.
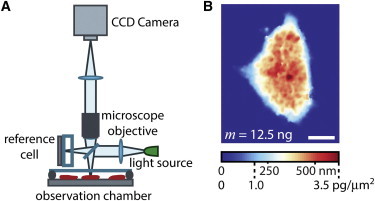
LCI quantifies HSF1 hPSC colony mass. (A) Simplified schematic of the live cell interferometer (LCI) used to obtain dynamic mass distributions of living cells. LCI uses a standard light microscope with a Michelson interferometer between the microscope objective and the temperature and CO2-controlled observation chamber. The interferometer compares lights that pass through cells to light that passes undisturbed through a reference chamber. (B) LCI directly measures the phase shift of light due to its interaction with the material inside a cell. This is converted to cell dry mass using the measured relationship between light phase shift and material density, yielding the mass distribution within each individual colony. Scale bar in panel B is 20 μm.
LCI data show a consistent, exponential rate of hPSC colony mass accumulation under standard self-renewing growth conditions regardless of starting colony mass (Fig. 3 A and see Fig. S2). This result was confirmed by a moderate relationship between colony mass and mass accumulation rate (R2 = 0.87) and a linear power law dependence (power law scaling exponent = 1.05 ± 0.025 at 95% confidence). For large colonies, this relationship is expected independent of the mass trend for individual cells (e.g., linear or exponential increase of mass with time), provided that the mass accumulation rate of each cell is not affected by colony size (see Analysis in the Supporting Material). These data therefore indicate that hPSC colony size does not affect the mass accumulation rate of individual cells within a colony under normal growing conditions either from positive or negative influences.
The observed exponential increase of colony mass is governed by a single parameter, the specific growth rate = (dm/dt)/m (growth rate normalized by mass). We fit the distribution of measured specific growth rate for 658 HSF1 colonies to a log-normal distribution and obtained a mean specific growth rate of 0.03 h−1, with a SD of 0.63 ln(h−1) (Fig. 3 B and see Fig. S3, A and B). This value corresponds to a mass doubling time of 22.8 ± 1.1 h (Fig. 3 C). Cell counting yielded a mean cell number doubling time of 24.9 ± 1.2 h (Fig. 3 C and see Fig. S3 C). This result is not significantly different from our population average mass accumulation rate, although we note that cell counting is intrinsically a bulk measurement and does not reveal the trend of colony growth rate with increasing colony size and typically requires an experiment duration of several doubling times. Using LCI, we were able to make growth rate measurements at multiple distinct timepoints in the seven days total experiment duration, which showed that there was also no significant variation of the mean specific growth rate with time since last passage, despite dramatic differences in colony size (see Fig. S3 D). Additionally, there were no significant differences in mean specific growth rate or the power law scaling exponent of the growth rate versus mass data when only the smallest colonies were considered in the analysis (see Fig. S3, E and F).
We induced trilineage differentiation by HSF1 exposure to 10-μM retinoic acid (RA) starting at 24 h after plating (0 days) to serve as a model of early induced differentiation. Cell cycle analysis showed a substantial increase in the G0/G1 subpopulation (Fig. 4 A and see Fig. S4, B and C) and gene expression profiling showed loss of OCT4 and induction of homeobox family and trilineage differentiation genes MSX2, GATA3, and HAND1 by four days of RA exposure (Fig. 4 B and see Fig. S4 A). Morphological changes were also consistent with RA-induced differentiation (Fig. 5 A). Colonies were imaged with LCI after days 4 and 6 of continuous RA treatment and mass accumulation rates measured, as for untreated colonies (Fig. 3 A and see Fig. S3, B and D). Surprisingly, a decrease of ∼15% in the rate of mass accumulation occurs during RA-induced differentiation (Fig. 3 C), despite the appearance of cells with significantly larger projected areas (Fig. 5 A). The magnitude of this change cannot be explained by the moderate, 3% increase in the fraction of dead cells observed during RA differentiation (see Fig. S4 D). This result contrasts sharply with the almost immediate reduction in cell size and nearly 50% increase in proliferation rate observed at the earliest stages of successful reprogramming to pluripotency (3) and the 100% decrease in proliferation rate typically observed in the differentiation of murine ESCs (5). These results indicate that the net hPSC growth rate before and during early differentiation is maintained independently of programs that regulate self-renewal or early differentiation.
Figure 4.
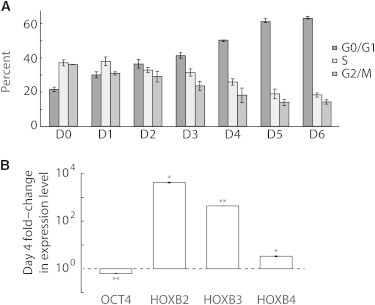
Cell cycle and gene expression profiling of RA-exposed HSF1 hPSC colonies. (A) Cell cycle analysis of RA-treated colonies shows a substantial increase in G0/G1 fraction, and decrease in S and G2/M fractions by four days of RA treatment. (B) Gene expression after four days of RA treatment shows induction of homeobox family genes and loss of pluripotency marker OCT4. Error bars show ± SE. ∗p < 0.05. ∗∗p < 0.01.
Figure 5.
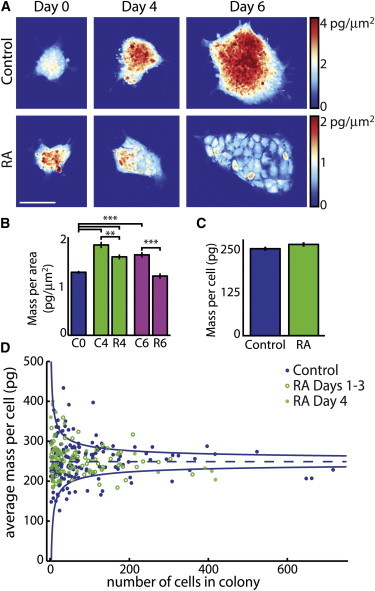
RA-differentiated hPSCs have a lower density, but the same total mass, as pluripotent hPSCs. (A) Colonies showing morphological changes associated with RA-induced differentiation. RA-treated cells visible within colonies have a lower mass per area (note color bar) but much larger area. (B) Comparison of mass per area (density) of hPSC colonies with and without RA treatment. (C) Comparison of average single cell mass shows no significant change in mass per cell during differentiation. (D) Plot of all average single cell masses from untreated and RA-treated colonies. (Solid blue lines) Mean ± SD of the control measurements (μ = 250 pg) assuming σ = σOneCell n−1/2 as expected for the population SD. Scale bar in (A) is 100 μm. Error bars show ± SE. ∗∗p < 0.001, ∗∗∗p < 10−6.
HSF1 cells appear small, but their native size in colonies has not been accurately measured (5). An increase in HSF1 cell area is associated with a transition to fibroblast-like appearing cells during induced trilineage differentiation (Fig. 5 A). However, this increase in area is accompanied by a significant decrease in projected mass per area (Fig. 5 B), which is consistent with the near-constant growth rate (biomass accumulation) during this transition. LCI was used in combination with fluorescent nucleus staining to measure the average mass per cell within intact colonies (Fig. 5, C and D, and see Fig. S5, A and B). We also characterized the relationship between average cell mass and the average distance between daughter cells (see Fig. S5 C). As colonies differentiate, the distance between individual cells increases (see Fig. S5 C and Fig. S8 B). We observe no statistically significant relationship between cell mass and distance between adjacent cells, further supporting the conclusion that average mass per cell is approximately constant during early differentiation, across all observed colony morphologies (see Fig. S5, D–G). Despite large changes in the HSF1 gene expression program (Fig. 4 B and see Fig. S4 A) and a previously reported shift from glycolysis to oxidative phosphorylation (21), no significant differences arise between the average mass per cell in pluripotent colonies versus colonies differentiated for four days with RA exposure. Cell doubling time is minimally affected (Fig. 3 C), but the proportion of cells in G0/G1 phase of the cell cycle increases substantially (Fig. 4 A and see Fig. S4 B), preceding changes in proliferation or growth rate (Fig. 3) and cell size (Fig. 5).
LCI quantification reveals where mass is increasing or decreasing within colonies and how quickly the mass distribution changes over time (see Movie S1, Movie S2, Movie S3, and Movie S4 in the Supporting Material). To quantify changing mass distributions over time we imaged selected colonies at a higher data rate and magnification (30 s between each frame, 20× effective magnification, 40 min total observation time). Representative images showing local mass accumulation and loss rates overlaid on colony images show much larger areas of coordinated mass change in HSF1 colonies than in RA-differentiated colonies (Fig. 6, A and D, and see Fig. S7, A—F). The local mass accumulation and loss rates shown in these images represent the sum of local biomass production and destruction, as well as reorganization in the mass of individual cells and the motion of individual cells within colonies. However, due to the relatively short imaging period, in which the total biomass accumulation is only ∼1% of the colony total mass, these images primarily represent the redistribution of mass within colonies, rather than the creation or destruction of biomass. The pattern of mass redistribution shown in Fig. 6 A appears as a stretching of the colony along the horizontal axis with a resulting contraction along the vertical axis, which was not observed in RA-treated colonies (Fig. 6 D and see Fig. S7, D–F).
The autocorrelation function is useful in biological image analysis to quantify the persistence of deviations from the average (28). We measured the decay rate of the autocorrelation of mass versus time for all 2 h colony mass distribution measurements to determine the persistence of fluctuations in intracolony mass distributions. This rate gives an indication of how quickly mass is being reorganized within individual colonies as a result of both mass transport within individual cells and motion of cells relative to one another. The decorrelation rate of undifferentiated and RA-differentiated colonies shows a significantly lower (p < 10−5) value for differentiated colonies, consistent with a much slower reorganization of cellular mass within these colonies as compared to untreated HSF1 colonies (Fig. 7 A).
Figure 7.
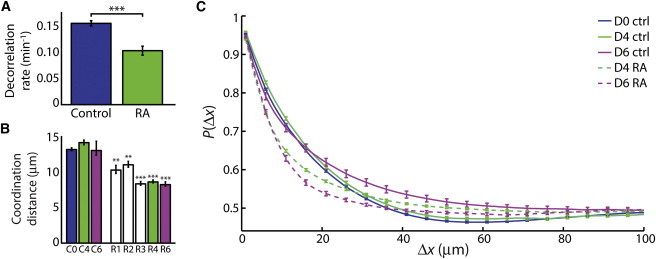
Quantification of mass redistribution rate and coordination within hPSC colonies. (A) Average decorrelation rates for untreated and RA-treated hPSCs reveals that pluripotent colonies redistribute mass significantly faster than RA-differentiated colonies. (B) Coordination distance within a colony is significantly lower in RA-differentiated colonies relative to untreated controls. (C) Probability of agreement in mass increase or decrease as a function of displacement (Δx) as measured in 435 untreated colonies 24 h after plating (day 0), 351 RA-treated, and 131 untreated colonies on day 4, and 75 RA-treated and 88 untreated colonies on day 6 of RA treatment. For clarity, error bars are only shown on every fifth point. Error bars show ± SE. ∗∗p < 0.001, ∗∗∗p < 10−5.
The regions of local mass increase or decrease in pluripotent colonies are substantially larger than the regions of mass increase or decrease in RA-differentiated colonies (Fig. 6, A and D, and see Fig. S7, A–F). To quantify this appearance, we analyzed the distributions of local mass increase and decrease by measuring the frequency with which two locations within a colony were either both increasing or both decreasing in mass as a function of displacement within the colony, defined here as P(Δx). This quantity, P(Δx), should be equal to 1.0 at very short distances (meaning adjacent locations are highly likely to increase or decrease in mass together), and should fall to 0.5 at large distances (because when two locations are very far from one another, by random chance they will both happen to be rising or falling together ∼50% of the time). The rate of decay of this probability from 1.0 to 0.5 with displacement indicates the degree of coordination of mass motion within colonies, with more coordinated colonies showing a slower rate of decay. Pluripotent colonies measured up to six days in culture show a significantly slower decrease in P(Δx) and a significantly larger coordination distance than RA-differentiated colonies—characteristics that are lost during the first 1–2 days of RA exposure (1080 total colonies; Fig. 7, B and C, and see Fig. S8 A). The distance over which coordination decays in RA-treated colonies is approximately the distance between neighboring cells, whereas untreated colonies show coordination up to approximately two cells away (Fig. 7 B and see Fig. S8 B). These data reveal that coordinated cell movements, detected as shifts in colony mass distributions, occur in hPSCs and stop early during RA-induced differentiation.
Discussion
Cells appear to get larger and grow more slowly early during hPSC differentiation (5). However, our results using a HSF1 model of early induced differentiation show that transitions in gene expression, colony morphology, and cell-cycle phase durations during early differentiation (29) occur with a nearly constant cell mass and rate of mass accumulation (Fig. 3 C and Fig. 5 C). Additionally, this flat or slightly negative growth rate change occurs in early HSF1 differentiation during the transition from glycolytic to oxidative energy metabolism (21). Although the more glycolytic metabolism of hPSCs, reminiscent of the Warburg effect in cancer, may support a slightly higher growth rate (30), its impact on cell mass appears minimal. This is particularly striking in light of cell volume measurements (5), which have shown an increase in cell size during differentiation. This suggests that during early differentiation, cell mass is perhaps regulated independently of cell volume, giving rise to a change in cellular density, as has been observed in recent measurements of the mass and volume of budding yeast (31). This should be investigated further in future work.
hPSCs in vitro show coordinated mass redistribution/intracolony cell movement, which is reminiscent of, but not identical to, cell patterning in early embryonic development (7,8). The loss of motion coordination after RA treatment suggests that these programs are silenced during early hPSC differentiation, in contrast to the program that regulates cell size/biomass, which is maintained. Cytoskeletal components, including the ρ-associated kinase, and adhesion molecules, such as E-cadherin, regulate asymmetric hPSC colony growth, intercolony movement, and colony fusion events (6) and may play a role in this observed coordination. The label-free and quantitative nature of the LCI technique means it can be used to track the motion of all biomass, rather than specific, labeled features or edges, in a consistent way from single cells to entire colonies. This method may therefore have utility in studying the impact of cell mass redistribution in other models of hPSC differentiation, beginning with the earliest specifying events.
Spatial autocorrelations of quantitative phase images have previously been used to assess the mechanical properties of red blood cells (RBCs) undergoing morphological changes (17). In the RBC case, the relatively simple cell composition and single cell dispersion allows for derivation of a direct relationship between quantitative phase data and cellular mechanical properties. In particular, at low frequencies, Park et al. (17) note a moderate anticorrelation in the spatial phase thickness autocorrelation, which is suppressed with increasing RBC stiffness. We note the presence of a similar anticorrelation in the LCI P(Δx) parameter for control colony measurements on days 0 and 4.
Overall, our results show that large changes in HSF1 colony morphology and motion occur during early RA-induced differentiation, preceding any potentially large shifts in cell size and growth rate during later stages of lineage-specific differentiation. Revealing these features using, what is to our knowledge, a novel LCI approach improves our understanding of early hPSC differentiation, which may have practical importance for using differentiated hPSCs in regenerative therapies.
Acknowledgments
We thank K. Niazi and S. Rabizadeh (NantWorks and California NanoSystems Institute, University of California at Los Angeles) for helpful discussions.
This study is supported by a University of California Discovery Biotechnology Award (No. Bio07-10663), a California Institute for Regenerative Medicine Basic Biology 1 Award (No. CIRM RB1-01397), the Broad Stem Cell Research Center at UCLA Innovator Award, the National Institutes of Health Roadmap for Medical Research Nanomedicine Initiative (No. PN2EY018228), the National Institutes of Health/National Institute of General Medical Sciences (grants No. T32CA009120, No. T32CA009056, No. P01GM081621, No. K25CA157940, and No. R01GM094388), and is partially supported by a Translational Acceleration Grant from the Caltech-UCLA Joint Center for Translational Medicine (www.jctm.caltech.edu).
Footnotes
Jin Zhang’s present address is Harvard Stem Cell Institute, Harvard University, Cambridge, MA.
Contributor Information
Jason Reed, Email: jcreed@vcu.edu.
Michael A. Teitell, Email: mteitell@ucla.edu.
Supporting Material
References
- 1.Kim J., Chu J., Orkin S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Smith Z.D., Nachman I., Meissner A. Dynamic single-cell imaging of direct reprogramming reveals an early specifying event. Nat. Biotechnol. 2010;28:521–526. doi: 10.1038/nbt.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loh K.M., Lim B. Recreating pluripotency? Cell Stem Cell. 2010;7:137–139. doi: 10.1016/j.stem.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Singh A.M., Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L., Wang B.H., Wang L. Individual cell movement, asymmetric colony expansion, ρ-associated kinase, and E-cadherin impact the clonogenicity of human embryonic stem cells. Biophys. J. 2010;98:2442–2451. doi: 10.1016/j.bpj.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller P.J., Schmidt A.D., Stelzer E.H. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- 8.Lawson K.A., Meneses J.J., Pedersen R.A. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development. 1991;113:891–911. doi: 10.1242/dev.113.3.891. [DOI] [PubMed] [Google Scholar]
- 9.Tzur A., Moore J.K., Kirschner M.W. Optimizing optical flow cytometry for cell volume-based sorting and analysis. PLoS ONE. 2011;6:e16053. doi: 10.1371/journal.pone.0016053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godin M., Delgado F.F., Manalis S.R. Using buoyant mass to measure the growth of single cells. Nat. Methods. 2010;7:387–390. doi: 10.1038/nmeth.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park K., Millet L.J., Bashir R. Measurement of adherent cell mass and growth. Proc. Natl. Acad. Sci. USA. 2010;107:20691–20696. doi: 10.1073/pnas.1011365107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross K.F.A. St. Martin’s Press; New York: 1967. Phase Contrast and Interference Microscopy for Cell Biologists. [Google Scholar]
- 13.Reed J., Chun J., Teitell M.A. Rapid, massively parallel single-cell drug response measurements via live cell interferometry. Biophys. J. 2011;101:1025–1031. doi: 10.1016/j.bpj.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mir M., Wang Z., Popescu G. Optical measurement of cycle-dependent cell growth. Proc. Natl. Acad. Sci. USA. 2011;108:13124–13129. doi: 10.1073/pnas.1100506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchison J.M. The growth of single cells. I. Schizosaccharomyces pombe. Exp. Cell Res. 1957;13:244–262. doi: 10.1016/0014-4827(57)90005-8. [DOI] [PubMed] [Google Scholar]
- 16.Pavillon N., Kühn J., Marquet P. Early cell death detection with digital holographic microscopy. PLoS ONE. 2012;7:e30912. doi: 10.1371/journal.pone.0030912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park Y., Best C.A., Popescu G. Measurement of red blood cell mechanics during morphological changes. Proc. Natl. Acad. Sci. USA. 2010;107:6731–6736. doi: 10.1073/pnas.0909533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bon P., Savatier J., Monneret S. Optical detection and measurement of living cell morphometric features with single-shot quantitative phase microscopy. J. Biomed. Opt. 2012;17:076004. doi: 10.1117/1.JBO.17.7.076004. [DOI] [PubMed] [Google Scholar]
- 19.Anand A., Chhaniwal V.K., Javidi B. Imaging embryonic stem cell dynamics using quantitative 3-D digital holographic microscopy. IEEE Photon. J. 2011;3:546–554. [Google Scholar]
- 20.Reed J., Troke J.J., Gimzewski J.K. Live cell interferometry reveals cellular dynamism during force propagation. ACS Nano. 2008;2:841–846. doi: 10.1021/nn700303f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J., Khvorostov I., Teitell M.A. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30:4860–4873. doi: 10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J., Nuebel E., Teitell M.A. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012;7:1068–1085. doi: 10.1038/nprot.2012.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohen N.T., Little L.E., Healy K.E. Characterization of Matrigel interfaces during defined human embryonic stem cell culture. Biointerphases. 2009;4:69–79. doi: 10.1116/1.3274061. [DOI] [PubMed] [Google Scholar]
- 24.Ghiglia D.C., Pritt M.D. John Wiley; New York: 1998. Two-Dimensional Phase Unwrapping: Theory, Algorithms, and Software. [Google Scholar]
- 25.Otsu N. A threshold selection method from gray-level histograms. IEEE T. Syst. Man. Cyb. 1979;9:62–66. [Google Scholar]
- 26.Zar J.H. Pearson Prentice Hall; Upper Saddle River, NJ: 2010. Biostatistical Analysis. [Google Scholar]
- 27.Bendat J.S., Piersol A.G. John Wiley; New York: 2000. Random Data: Analysis and Measurement Procedures. [Google Scholar]
- 28.Sund S.E., Axelrod D. Actin dynamics at the living cell submembrane imaged by total internal reflection fluorescence photobleaching. Biophys. J. 2000;79:1655–1669. doi: 10.1016/S0006-3495(00)76415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowry W.E., Richter L., Plath K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou W., Choi M., Ruohola-Baker H. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012;31:2103–2116. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryan A.K., Goranov A., Manalis S.R. Measurement of mass, density, and volume during the cell cycle of yeast. Proc. Natl. Acad. Sci. USA. 2010;107:999–1004. doi: 10.1073/pnas.0901851107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.