

Abstract
Amyloid fibril accumulation is a pathological hallmark of several devastating disorders, including Alzheimer’s disease, prion diseases, type II diabetes, and others. Although the molecular factors responsible for amyloid pathologies have not been deciphered, interactions of misfolded proteins with cell membranes appear to play important roles in these disorders. Despite increasing evidence for the involvement of membranes in amyloid-mediated cytotoxicity, the pursuit for therapeutic strategies has focused on preventing self-assembly of the proteins comprising the amyloid plaques. Here we present an investigation of the impact of fibrillation modulators upon membrane interactions of β2-microglobulin (β2m) fibrils. The experiments reveal that polyphenols (epigallocatechin gallate, bromophenol blue, and resveratrol) and glycosaminoglycans (heparin and heparin disaccharide) differentially affect membrane interactions of β2m fibrils measured by dye-release experiments, fluorescence anisotropy of labeled lipid, and confocal and cryo-electron microscopies. Interestingly, whereas epigallocatechin gallate and heparin prevent membrane damage as judged by these assays, the other compounds tested had little, or no, effect. The results suggest a new dimension to the biological impact of fibrillation modulators that involves interference with membrane interactions of amyloid species, adding to contemporary strategies for combating amyloid diseases that focus on disruption or remodeling of amyloid aggregates.
Introduction
The transformation of soluble proteins into amyloid fibrils deposited in different organs and tissues is a hallmark of devastating medical disorders, including Alzheimer’s disease, Parkinson’s disease, type II diabetes, and others (1,2). Although the presence of fibrillar aggregates appears to be a universal phenomenon in amyloid diseases, the relationships among amyloid formation, disease progression, and pathogenicity remain unclear.
Amyloid plaques are commonly found extracellularly, often associated with external membrane surfaces (3), although intracellular amyloid deposits are involved in several human disorders (3). A number of recent studies have linked the cytotoxicity of amyloid species with their membrane activity, suggesting that only toxic aggregates bind and disrupt lipid membranes, whereas benign conformers remain inert (4,5). There is an ongoing scientific debate, however, about the nature of pathogenic species. It was initially postulated that large insoluble amyloid plaques are the main culprits of the observed pathological conditions (6). This hypothesis was challenged by findings showing that small oligomeric intermediates, rather than the end-products of the aggregation pathway, represent the primary factors leading to cell damage and death (7,8). This concept was taken further by the suggestion that rapid fibrillation may provide a protective mechanism through formation of inert deposits that reduce the population of transient oligomeric species (9). By contrast with these findings, several recent studies have implicated amyloid fibrils themselves in amyloid diseases. Specifically, fibrils derived from various amyloidogenic proteins have been shown to function as cytotoxic substances that readily bind and permeabilize lipid membranes (10–12), a process that is enhanced by fibril fragmentation (11,13). Preformed amyloid fibrils have also been shown to be internalized by cultured cells and to recruit cytosolic cellular proteins into growing amyloid assemblies (14). In vivo studies demonstrated that mature fibrils induce propagation of amyloidosis and the corresponding pathology in wild-type mouse (15) and human brains (16) through intercellular transmission. Finally, fibrils can be regarded as a source of toxic entities capable of releasing oligomeric species (17), particularly during interaction with lipids (18).
Directly related to the above observations, the mechanistic aspects of amyloid-protein interactions with cellular membranes have been the focus of intense experimental work in recent years (19,20). However, whereas lipid- and membrane-interactions of misfolded proteins appear to be closely related to amyloid cytotoxicity (4,5), development of therapeutic remedies has been directed in a large part toward substances that interfere with the aggregation processes of amyloid precursors into higher-order oligomeric species. Aggregation inhibitor screens have resulted in the discovery of numerous and diverse molecular leads, some of which have been shown to reduce amyloid-mediated cellular toxicity (21–23). Polyphenols, such as resveratrol (found in red grape skins and seeds) (24,25) and epigallocatechin gallate (EGCG, a component of green tea) (26,27) have been among the most widely studied inhibitors of amyloid cytotoxicity and fibril assembly modulators. These molecules have been shown to remodel toxic oligomers into large nontoxic aggregates (28–30) as well as to promote fibril disassembly (29,30). Another group of fibrillation modulators includes glycosaminoglycans (GAGs), anionic polysaccharides widely expressed in different tissue types (31). Heparin, an abundant member of the GAG family (31), has been demonstrated to modulate the fibrillation route and the associated toxicity of various amyloidogenic sequences (32,33). In addition, ionic chelators (21,34), molecular chaperones (35), β-sheet breaking peptides (22), antibodies (23), γ-bodies (36), and polymeric nanoparticles conjugated to functional groups (34,37) have all been used to modulate the course of fibril assembly.
Despite the apparent relationship between membrane interactions of amyloid assemblies and cellular toxicity, the impact of aggregation inhibitors upon membrane activity and lipid-binding properties of amyloid species has been addressed only sparingly (25,38). Here we investigate the relationships among the effects of different polyphenols and the glycosaminoglycans heparin and heparin disaccharide on membrane interactions of amyloid fibrils formed in vitro from β2-microglobulin (β2m). β2m, the noncovalently bound light chain of the MHC-class I complex (39), forms insoluble fibrillar amyloid aggregates that are intimately involved in progression of dialysis-related amyloidosis (11,40,41). Interestingly, recent studies have demonstrated that β2m fibrils, rather than the monomeric protein, are highly membrane-active and putative toxic substances (11). Here, we focus on membrane interactions of short (weight average length <400 nm) β2m fibrils formed by controlled fragmentation of their initially longer counterparts (11,13). In particular, we describe the effects of polyphenols including the widely-studied fibrillation modulators EGCG and resveratrol (42), as well as the synthetic dye bromophenol blue and a second group of compounds consisting of glycosaminoglycans heparin and its building subunit heparin disaccharide (43), upon membrane interactions of β2m fibrils. Furthermore, we examine whether these two distinct classes of molecules exhibit different effects upon membrane interactions of these fibrils.
Materials and Methods
Materials
Chicken egg PC (L-α-phosphatidylcholine), chicken egg PG (L-α-phosphatidylglycerol), and NBD-PE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-n-(7-nitro-2-1,3-benzoxadiazol-4-yl), ammonium salt) were purchased from Avanti Polar Lipids (Alabaster, AL).
TMA-DPH (1-(4-trimethyl ammonium phenyl)-6-phenyl-1,3,5-hexatriene), Laurdan (6-dodecanoyl-2-dimethylaminonaphthalene), and TMR (5-(and-6)-carboxytetramethyl-rhodamine) were purchased from Molecular Probes (Eugene, OR).
Heparin from porcine intestinal mucosa (sodium salt, grade I-A), heparin disaccharide I-A (sodium salt), EGCG ((−)-epigallocatechin gallate, ≥95%), bromophenol blue, and resveratrol (≥99%) were obtained from Sigma-Aldrich (St. Louis, MO). Polymeric chains of full-length heparin supplied by Sigma-Aldrich can range from 18 to 90 monomers (6–30 kDa), whereas the majority of the chains contain 51–57 monomers (17–19 kDa).
Preparation of fibril samples
Fibrils of wild-type human β2m were formed from recombinant protein as previously described in Xue et al. (11). Briefly, lyophilized protein was dissolved in a fibril growth buffer containing 10 mM monosodium phosphate and 50 mM NaCl, pH 2.0, and was syringe-filtered through a 0.2-μm pore size filter. The protein concentration was adjusted to 120 μM and the solution was seeded with 0.1% (w/w) of fragmented β2m fibrils formed under the same conditions, followed by incubation at 25°C under quiescent conditions for 48 h. This procedure was shown to result in formation of long straight β2m fibrils (11). A quantity of 500 μL aliquots of the fibril suspensions was subsequently fragmented by stirring (1000 rpm, 25°C for 48 h) on a custom-made precision stirrer. Fragmented long straight fibrils exhibiting a weight average length of <400 nm (11,13) were used in all experiments. For confocal microscopy, β2m monomers were labeled by TMR as described in the Supporting Material. TMR-labeled fibrils were prepared by mixing unlabeled and labeled monomers such that the final preparation contained 10% of TMR-bound monomer.
Vesicle preparation
Vesicles consisting of egg PC and egg PG (1:1, molar ratio) were prepared in a liposome buffer (50 mM HEPES, 110 mM NaCl, 1 mM EDTA, 0.02% (w/v) NaN3, pH 7.4) at 2-mM total lipid concentration.
Large unilamellar vesicles
Large unilamellar vesicles (LUVs) were prepared by extruding the lipid suspension through a 400-nm pore-size polycarbonate filter as described in the Supporting Material.
Giant vesicles
NBD-PE (0.04%, molar ratio) was added to the lipid mixture for giant vesicle (GV) visualization by confocal microscopy. GVs were prepared using a rapid evaporation method (44). A quantity of 500 μL of aqueous phase containing the liposome buffer supplemented with 0.1 M sucrose was added to 200 μL of lipid-containing solution in chloroform in a round-bottom flask, followed by brief vigorous mixing of the two phases by pipetting. The organic solvent was immediately removed in a rotary evaporator under reduced pressure (40 mbar) for 3 min at room temperature. The resulting vesicle solution exhibited a turbid appearance and was used on the day of preparation.
Vesicle disruption experiments in the presence of small molecules and heparin
Aliquots from the fibril stock solution (120 μM monomer equivalent concentration) were mixed with the vesicles and fibril-membrane interactions were assessed through various spectroscopy and microscopy techniques. In each experiment fibrils were incubated for 3 min with the required amount of the test compound in the liposome buffer before addition to the vesicles using a β2m/test compound ratio of 1:0.4 (w/w) for GAGs (β2m:heparin, heparin disaccharide) or 1:1 (w/w) for polyphenols (β2m: EGCG, bromophenol blue or resveratrol). Stock solutions of the tested small molecules and heparin were prepared in the buffer used for liposome preparations except for resveratrol, which was dissolved in buffer/ethanol 2:1 (v/v). For the control experiments, corresponding amounts of freshly prepared β2m monomer in the fibril-growth buffer, the fibril growth buffer alone, or buffer/ethanol 2:1 mixture were used.
Microscopy imaging
Fibrils preincubated in the liposome buffer alone or with test compounds for 3 min as described above were diluted 10-fold into the vesicle suspension, yielding a 12 μM β2m monomer equivalent concentration and 1.8 mM total lipid concentration at a final pH of 7.4. The images were obtained after 15-min incubation of the fibrils with the vesicles.
Confocal microscopy
Egg PC/PG/NBD-PE (1:1:0.0008 molar ratio) GVs and TMR-labeled β2m fibrils were placed on a glass-bottom Petri dish (MatTek, Ashland, MA) and imaged on an Axiovert 100M confocal laser scanning microscope (Carl Zeiss, Jena, Germany) using a 63×/1.4 N.A. Plan Apochromat DIC oil immersion objective lens (Carl Zeiss). The NBD-PE fluorescent probe was excited with the 488-nm line of an argon laser, while TMR fluorescence was excited with argon-krypton laser at 568 nm. Long-pass (LP) filters LP 505 and LP 580 were employed for acquisition of NBD and TMR fluorescence, respectively.
Cryo-TEM
A drop of a sample solution containing egg PC/PG (1:1) LUVs incubated with fibrils alone or in the presence of the different test compounds was deposited onto a transmission electron microscope (TEM) 300-mesh Cu grid coated with a holey carbon film (Lacey substrate; Ted Pella, Redding, CA). Vitrification was achieved using an electron microscopy (EM) Grid Plunger (Leica Microsystems, Buffalo Grove, IL). The samples were examined at −180°C using a Tecnai 12 G2 TWIN TEM (FEI, Hillsboro, OR) equipped with a model No. 626 cold stage (Gatan, Warrendale, PA), and the images were recorded using a model No. 794 charge-coupled device camera (Gatan) at 120 kV in low-dose mode.
Liposome dye release assay
LUVs were prepared from egg PC/PG (1:1) as described above, except that a buffered carboxyfluorescein (CF) solution (50 mM CF, 50 mM HEPES, 10 mM NaCl, 1 mM EDTA, 0.02% (w/v) NaN3, pH 7.4) instead of liposome buffer was used. After the extrusion, the LUVs were washed three times with liposome buffer by centrifugation at 20,000 g and resuspension to yield a stock solution of 0.5 mM total lipids. A quantity of 2.5 μL aliquots of these LUVs was than diluted into liposome buffer and mixed with fibrils (with or without test compounds as described above) to obtain a total sample volume of 500 μL and a final protein concentration (in terms of β2m monomer equivalent) of 3 μM. The vesicles are saturated by the β2m fibrils under these experimental conditions because further increase of β2m concentration does not affect the extent of LUVs leakage (11). Fluorescence emission of carboxyfluorescein at 517 nm was then recorded for 15 min using an excitation wavelength of 490 nm on a FL920 spectrofluorimeter (Edinburgh Instruments, Edinburgh, Scotland, UK). The percent leakage was calculated as
where I0 is the fluorescence intensity of liposomes alone and I100 is the fluorescence intensity after addition of 10 μL of Triton X-100 (final concentration 0.4% (v/v)), which results in complete vesicle disintegration.
Fluorescence anisotropy
The fluorescence probe TMA-DPH was incorporated into egg PC/PG (1:1) LUVs at final concentration of 0.22% (molar ratio) by mixing the dye dissolved in tetrahydrofuran at 1 mg/mL with the vesicle stock (2 mM) and incubating for 30 min at room temperature. The organic solvent comprised 0.2% (v/v) of the LUV stock solution. Fibrils alone or reacted with different test compounds were combined with 2.5 μL aliquots of egg PC/PG/TMA-DPH LUVs prediluted with liposome buffer to a total sample volume of 500 μL. The final protein concentration was 3 μM (β2m monomer equivalent). TMA-DPH fluorescence anisotropy was measured at 431 nm using an excitation at 360 nm on a FL920 spectrofluorimeter (Edinburgh Instruments). Anisotropy values were automatically calculated by the spectrofluorimeter software. Standard deviation values were obtained from 10 repeats of the anisotropy scans. Changes in anisotropy values (Δ anisotropy) were calculated by subtracting the data for control samples (vesicles with the fibril growth buffer or with the buffer containing the appropriative test compound) from the corresponding fibril-induced anisotropy values.
Laurdan fluorescence assay
Laurdan probe was dissolved in chloroform and added to the egg PC/PG (1:1) lipid mixture at 0.5% molar ratio before evaporation of the organic solvent. LUVs were then prepared as described above at 2 mM total lipid concentration. A quantity of 2.5 μL aliquots of egg PC/PG/Laurdan LUV stock solution was diluted by liposome buffer (pH 7.4) to a final sample volume of 500 μL, followed by addition of β2m fibrils alone or preincubated with different test compounds at the ratios described above. The final protein concentration was 3 μM (β2m monomer equivalent). Laurdan emission spectra were recorded over a time course of 20 min using excitation at 365 nm on a PTI QuantaMaster spectrofluorimeter (Photon Technology International, Birmingham, NJ). Shift of emission maxima was quantified by general polarization (GP) function (45),
where Iblue and Ired are emission intensities at 435 and 478 nm, respectively. Changes in GP values (Δ GP) were calculated by subtracting the data for control samples (vesicles with fibril growth buffer or with the buffer containing the appropriative test compound) from the corresponding fibril-induced GP values.
Results
Small molecules and heparin modulate fibril-induced membrane permeabilization
The molecules selected for this study belong to two families of well-known fibrillation modulators: polyphenols and glycosaminoglycans (GAGs) (Fig. 1). Specifically, plant-derived polyphenols EGCG and resveratrol were tested for their impact on fibril-membrane interactions, while the synthetic polyphenol bromophenol blue was employed for comparison with these natural compounds. The glycosaminoglycans heparin and heparin disaccharide (a minimal repeat unit of heparin (43) lacking its fibrillation-modulating activities (46)) were also examined. Heparin has been shown to affect amyloid formation of a peptide derived from the human prion protein, wherein aggregation was enhanced at low GAG/protein ratios and inhibited at higher heparin concentrations (46). In addition, heparin, but not its disaccharide, has been shown to stabilize β2m amyloid fibrils (47,48). The physical properties of the molecules used are summarized in Table 1.
Figure 1.
Molecular structures of the compounds studied. Note that both heparin polymer and its disaccharide subunit were used in the studies described.
Table 1.
Physical properties of molecules used in this study (61)
| Compound | pKa | LogD, pH 7 | LogP | Hydrogen bonds |
|
|---|---|---|---|---|---|
| Donors | Acceptors | ||||
| EGCG | 7.75 ± 0.25 | 0.57 | 0.639 ± 0.702 | 8 | 11 |
| Bromophenol blue | 4.12 ± 0.10 | 5.10 | 9.171 ± 1.046 | 2 | 5 |
| Resveratrol | 9.22 ± 0.10 | 3.02 | 3.024 ± 0.267 | 3 | 3 |
| Heparin disaccharide | — | — | — | 2–6 | 12–24 |
LogP is a partition coefficient of nonionized molecule between octanol and water; LogD is octanol/water partition coefficient of ionized and neutral species of a compound formed at a given pH. Total number of hydrogen bonds in a molecule corresponds to the number of hydrogen acceptors. All data are given for 25°C.
Fig. 2 depicts dye release experiments designed to analyze permeation of large unilamellar vesicles (LUVs) composed of PC/PG (1:1) by β2m fibrils, and the effect of the tested compounds upon the membrane disruption processes. The leakage experiments employed vesicle-encapsulated carboxyfluorescein, which initially is weakly fluorescent due to self-quenching at high concentration (49). After vesicle disruption by membrane-active analytes, dye leakage results in increased fluorescence emission. The experiments depicted in Fig. 2 A (long dash) confirm that the β2m fibrils created in vitro interact with lipid membranes and induce membrane defects permeable for the water-soluble fluorescent dye, consistent with previous results (11). The β2m fibrils, however, do not induce complete vesicle disintegration as evident from only partial membrane leakage (Fig. 2 A). This effect can be ascribed to fibril self-association at neutral pH (50), which presumably reduces amount of the fibrils available for membrane binding. An additional factor that may limit dye release by the fibrils includes nonhomogenic distribution of lipid compositions within vesicle population (51). Addition of β2m monomers did not result in vesicle leakage (Fig. 2 A, short dash), underscoring the fact that the β2m monomers do not damage the lipid bilayer, at least as judged at the concentrations and solution/lipid conditions used.
Figure 2.
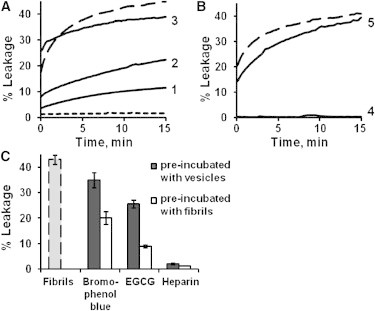
The effect of polyphenols and GAGs on β2m fibril-induced vesicle leakage. Time-dependent increase in fluorescence reflecting leakage of carboxyfluorescein from PC/PG (1:1) LUVs after incubation with β2m. (A) Effects of polyphenols on fibril-induced dye-leakage. (Long dash) β2m fibrils alone (no fibrillation modulators added); (short dash) β2m monomers alone; (1–3) β2m fibrils incubated for 3 min with (1) EGCG, (2) bromophenol blue, and (3) resveratrol. (B) Effects of GAGs on fibril-induced vesicle leakage. (Long dash) β2m fibrils alone; β2m fibrils incubated for 3 min with (4) heparin polymer; and (5) heparin disaccharide. (C) Effect of preincubation of vesicles with different additives on β2m-fibril induced membrane leakage. (Shaded) β2m fibrils alone. (Solid) Fibrillation modulators incubated with vesicles for 30 min before addition of fibrils. (Open) Fibrillation modulators incubated with β2m fibrils for 3 min before addition to the vesicles. Percent leakage corresponds to the end-point of the kinetic curves (see Fig. S3 in the Supporting Material).
Preincubation of the β2m fibrils with the three polyphenols analyzed here (at weight-equivalent concentrations) shows that the effect of EGCG and bromophenol blue on membrane disruption by the fibrils differs significantly from that of resveratrol. Specifically, both bromophenol blue and EGCG inhibit the effect of fibrils on membrane permeability, although not completely (Fig. 2 A, curves 1 and 2). Incubation of the fibrils with either EGCG or bromophenol blue for more prolonged periods did not enhance the inhibitory capacity of the polyphenols (see Fig. S1 in the Supporting Material). Resveratrol, on the other hand, accelerates initial dye release by the fibrils, whereas the long-term extent of the vesicle leakage is slightly reduced (Fig. 2 A, curve 3) as compared with fibrils alone. This enhancement in the initial amplitude of membrane permeability can be ascribed to resveratrol-membrane interactions (52) that may alter lipid bilayer susceptibility to the β2m fibrils. Indeed, binding of resveratrol to LUVs was verified by changes in anisotropy of lipid-incorporated TMA-DPH probe (data not shown).
Negative-stain EM confirmed that the general morphology of β2m fibrils was not affected by incubation with the polyphenols for 5 min (see Fig. S2). EM images, however, could not rule out that subtle structural changes within the fibrils contributed to the observed effects of the molecules tested. The dye-leakage results suggest that bromophenol blue and EGCG disfavor the formation of bilayer lesions by the β2m fibrils, whereas resveratrol appears to have no inhibitory effect on β2m fibril-induced impairment of membrane integrity. Fig. 2 B similarly shows dramatic differences between the effects of full-length heparin (curve 4) and heparin disaccharide (curve 5) upon vesicle leakage induced by β2m fibrils. Specifically, whereas interaction of full-length heparin with β2m fibrils prevents lipid bilayer disruption by these protein aggregates, heparin disaccharide had minor effect on the ability of the fibrils to cause dye release from the vesicles (Fig. 2 B).
Polyphenols are relatively hydrophobic molecules that have been shown to interact with membranes in vitro (53) and in vivo (52). Accordingly, studies conducted on EGCG have shown that it can cross the blood-brain barrier (52) and interact with model membranes without forming pores in the bilayer (53). We also observed membrane activity of EGCG through an increase in anisotropy of the membrane-incorporated fluorescent probe TMA-DPH in the presence of this molecule (data not shown). To determine whether EGCG and bromophenol blue inhibit the membrane activity of β2m fibrils via insertion of these molecules into the lipid bilayer and subsequent stabilization of the membrane, rather than by altering membrane-fibril interactions, the polyphenols were incubated with vesicles before the addition of β2m fibrils. The results of these experiments (Fig. 2 C and see Fig. S3) showed that 30-min preincubation of the polyphenols with LUVs did not enhance their inhibitory activity. On the contrary, the ability of the polyphenols to impair fibril-induced dye-leakage was attenuated compared with preincubation of these molecules with β2m fibrils. Further control experiments confirmed that the polyphenols did not induce any detectable dye-leakage in the absence of fibrils even after the 30-min incubation with vesicles (data not shown). These findings suggest that EGCG and bromophenol blue suppress association of the β2m fibrils with the PC/PG lipid vesicles, presumably by sequestering their exposed hydrophobic regions. By contrast with the action of the polyphenols, full-length heparin showed complete inhibition of membrane permeabilization by the fibrils. This effect occurred whether or not heparin was preincubated with vesicles or with the fibrils (Fig. 2 C), implying rapid binding of this molecule to β2m fibrils.
Fibril-induced lipid bilayer deformation and impact of fibril modulators
The vesicle dye-leakage experiments shown in Fig. 2 report on the permeability of the lipid bilayer after incubation with β2m fibrils. To examine the effects of fibrils on the bilayer integrity, giant vesicles (GVs) composed of PC/PG (1:1) incorporating the fluorescent probe NBD-PE (green) were mixed with β2m fibrils containing rhodamine-labeled monomer (red) (see Materials and Methods). Imaging of the samples using dual-color fluorescence confocal microscopy allows simultaneous analysis of vesicle deformation (such as shape change and bilayer perturbation), as well as the behavior and localization of the β2m fibrils relative to the lipids. Representative images depicting the experiments are shown in Fig. 3, while quantification of the data is summarized in Fig. S4 and Table S1 in the Supporting Material. The images obtained reveal a smooth, round shape of the GVs that is unperturbed after incubation with buffer or with monomeric β2m (Fig. 3, A and B, respectively), consistent with previous results (11,54). Images of the fibrils in the absence of vesicles show evidence for extensive fibril clustering at the pH used (pH 7.4) (Fig. 3 C). β2m fibrils formed at pH 2 tend to bundle through lateral association when transferred to a higher pH (50), presumably due to the reduced positive charge. The fluorescence images shown in Fig. 3 D, (i) and (ii), provide a striking visual depiction of the effects of β2m fibrils that destroy the integrity of the GVs, consistent with previous results (54). Furthermore, the β2m fibril aggregates (displaying the red rhodamine fluorescence) are coated by a thin layer composed of disassembled lipids (exhibiting green fluorescence) that appear to be extracted from the damaged vesicles. The confocal microscopy images in Fig. 3 D thus reveal significant vesicle disruption, consistent with extensive leakage of carboxyfluorescein from LUVs prepared from the same lipid composition (Fig. 2).
Figure 3.
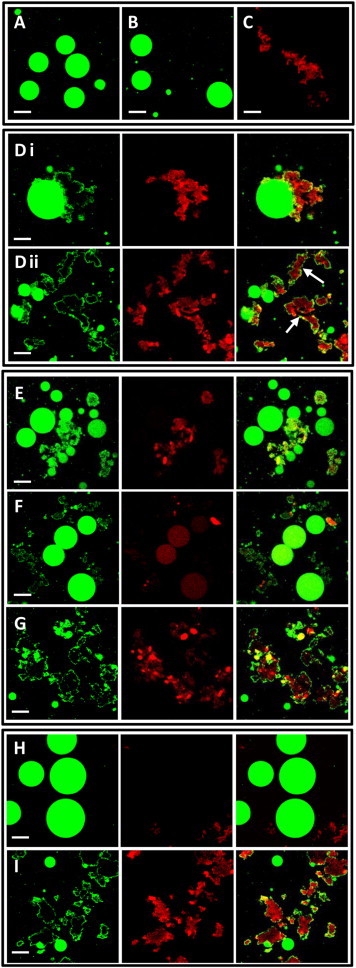
Confocal fluorescence microscopy employing GVs containing NBD-PE (green) and β2m fibrils labeled with TMR (red). (A) Control NBD-PE/PC/PG GVs; (B) GVs incubated with β2m monomers; (C) TMR-β2m fibrils in pH 7.4 buffer. (D-I) (Left images) NBD-PE fluorescence (green); (middle) TMR fluorescence (red). (Right images) (D, i and ii) Superimposition. GVs incubated with TMR-β2m fibrils. D(i) shows an example of a single, large GV, enabling clear visualization of bilayer damage. (Arrows, D ii) Examples of fibrillar aggregates coated by lipids that were presumably derived from disintegrated vesicle(s). (E–I) β2m fibrils preincubated with (E) EGCG, (F) bromophenol blue, (G) resveratrol, (H) heparin, or heparin disaccharide (I) before mixing with GVs. Bars in all images correspond to 20 μm. Note that residual NBD fluorescence is detected in the red channel of the image presented in panel F such that the NBD-labeled GVs appear red.
The confocal microscopy images presented in Fig. 3, E–G, show the effect of preincubating the β2m fibrils with EGCG, bromophenol blue, or resveratrol before their addition to the liposomes. The results show that EGCG impairs β2m-membrane interactions, giving rise to less abundant vesicle destruction compared with GVs incubated with β2m fibrils alone (compare Fig. 3, E and D(ii)). Quantitative analysis assessing ∼100 vesicles in each sample (see Table S1) demonstrated that EGCG reduced the extent of fibril-damaged GVs by approximately five times from 65 to 12% (see Fig. S4). Preincubation of the fibrils with bromophenol blue also resulted in only moderate GV disruption (17% of damaged vesicles, see Fig. S4), with some vesicles remaining intact (Fig. 3 F and see Fig. S4). Note that fluorescence intensity of the TMR probe is significantly quenched in the sample containing β2m fibrils and bromophenol blue (Fig. 3 F), due to fluorescence resonance energy transfer between the emission spectrum of the fluorophore and the absorbance of the polyphenol. To visualize fibrillar aggregates in that sample, gain of the red channel has been increased, resulting in residual NBD signal to become visible as red fluorescence (Fig. 3 F).
In contrast with EGCG and bromophenol blue, which appear to suppress β2m/vesicle interactions according to the confocal microscopy data, resveratrol does not show a significant effect on vesicle deformation caused by β2m fibrils (Fig. 3 G and see Fig. S4), consistent with the finding that resveratrol is relatively inefficient in inhibiting β2m-induced LUVs disruption as judged by the carboxyfluorescein dye release experiments (Fig. 2 A). The confocal images recorded after preincubation of the β2m fibrils with heparin (Fig. 3 H) or heparin disaccharide (Fig. 3 I) highlight considerable difference between the impacts of these two compounds on the membrane activity of β2m fibrils, corroborating the dye leakage results presented in Fig. 2 B. Accordingly, preincubation of the fibrils with the heparin polymer completely inhibited liposome disruption with no vesicle damage visible (Fig. 3 H and see Fig. S4). Binding of the full-length heparin to β2m fibrils also resulted in the dispersion of the large fibril aggregates (Fig. 3 H) without alteration of the overall fibrillar appearance (see Fig. S2). Dispersed assemblies of the β2m fibrils exhibit lower protein density and, as such, are not readily visible using fluorescence confocal microscopy. In sharp contrast with these results, heparin disaccharide did not inhibit vesicle damage by β2m fibrils (Fig. 3 I and see Fig. S4), echoing the dye-leakage experiments presented in Fig. 2 B.
Visualizing fibril-vesicle interactions using cryo-TEM
Cryogenic transmission electron microscopy (cryo-TEM) analysis can provide further visual depiction of the interactions of amyloid fibrils with lipid vesicles (54). This technique was used, therefore, to provide further insights into the effects of the polyphenols and GAGs on these interactions. Cryo-TEM images of LUVs created from PC/PG (1:1) are shown in Fig. 4 A. In the absence of fibrils, the lipid vesicles do not adhere readily to an EM grid and hence only few vesicles are found in the control sample, with most of them located in the vicinity of the hydrophobic carbon mesh (Fig. 4 A). Vesicles treated with β2m monomers appear spherical and undamaged, similar to the control sample (Fig. 4 B). Addition of β2m fibrils to the vesicles gave rise to significant changes in liposome morphology and distribution (Fig. 4 C). Accordingly, vesicles visibly accumulated in the fibril-treated samples compared with images obtained of LUVs alone. Moreover, the vesicles appear to associate with the fibrils and to display significant perturbations to their otherwise round shapes, corroborating previous findings (54). Larger vesicles, in general, are more fragile than smaller ones, and therefore GV deformation caused by β2m fibrils is more substantial (Fig. 3 D) than the changes to LUV shapes observed in Fig. 4 C. The cryo-TEM images in Fig. 4, D and E, show the effects of the addition of EGCG and bromophenol blue, respectively, on fibril-membrane interactions. These polyphenols appear to reduce vesicle deformation, consistent with the dye-leakage experiments and confocal microscopy images presented above. Indeed, in the presence of these small molecules, some vesicles remain free of fibrils and mostly retain their round shapes. The images of the heparin-treated fibril samples are even more striking (Fig. 4 F). In these images LUVs accumulation was not apparent and the vesicles appeared generally unperturbed in morphology. Heparin disaccharide, by contrast, had little effect on fibril-vesicle interactions; the image in Fig. 4 G features aggregated and distorted vesicles similar to the effects observed with the liposomes mixed with β2m fibrils in the absence of this GAG.
Figure 4.
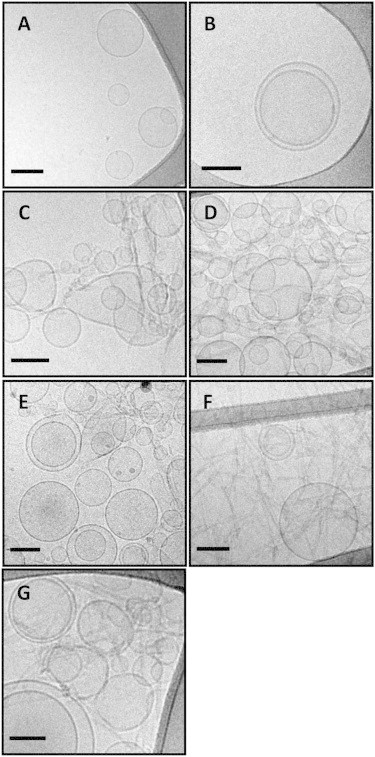
Cryo-TEM images of PGPG LUVs treated with fibrils and different additives. (A) PC/PG (1:1) LUVs (control); (B) vesicles incubated with β2m monomers; (C) vesicles incubated with β2m fibrils; (D–G) preincubation of the β2m fibrils with (D) EGCG; (E) bromophenol blue; (F) full-length heparin; and (G) heparin disaccharide before mixing with the vesicles. Bars in all images correspond to 100 nm.
The effects of fibril binding on lipid dynamics
To investigate further the effect of the β2m amyloid fibrils on membrane bilayer properties and the consequence of preincubation with the fibril modulators, fluorescence anisotropy of PC/PG (1:1) LUVs that incorporate the fluorescence dye TMA-DPH was measured. The fluorescence anisotropy of TMA-DPH fluorophore, which is oriented perpendicular to the lipid bilayer plane (55), constitutes a sensitive probe for bilayer fluidity and dynamics (56). Fig. 5 A depicts the fluorescence anisotropy changes induced by β2m fibrils and β2m fibril/test compound mixtures upon addition to the TMA-DPH/PC/PG vesicles. The results revealed that incubating the vesicles with β2m monomers did not alter the TMA-DPH anisotropy, consistent with the findings that β2m monomers have no effect upon lipid membranes (Figs. 2–4).
Figure 5.
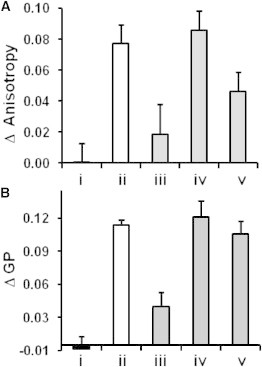
Modulation of bilayer fluidity by β2m amyloid fibrils and different molecules. Changes in (A) fluorescence anisotropy of TMA-DPH and (B) Laurdan emission shift (quantified by GP, Materials and Methods) assayed within PC/PG (1:1) LUVs. The vesicles incubated with (i) β2m monomers, (ii) β2m fibrils, (iii–v) β2m fibrils preincubated with (iii) bromophenol blue, (iv) full-length heparin, and (v) heparin disaccharide before mixing with the vesicles.
By contrast, incubation of β2m fibrils with the TMA-DPH/PC/PG vesicles gave rise to a pronounced increase in anisotropy (Fig. 5 A, ii), indicating reduced bilayer fluidity after binding of the membrane-active fibrils. The effect of bromophenol blue, heparin, and heparin disaccharide upon β2m fibril-induced changes in TMA-DPH anisotropy are also depicted in Fig. 5 A, iii–iv (EGCG and resveratrol gave rise to a significant increase in TMA-DPH anisotropy when incubated with liposomes in the absence of fibrils, ruling out measurements of their effects on β2m-induced changes of lipid dynamics). These experiments showed that preincubation of the fibrils with bromophenol blue substantially lowered β2m fibril-induced increase of lipid bilayer rigidity (Fig. 5 A, iii), consistent with inhibition of fibril-lipids interactions in the presence of this polyphenol.
Surprisingly, preincubating β2m fibrils with full-length heparin did not attenuate the large increase in anisotropy observed when the fibrils were incubated with liposomes in the absence of any additives (Fig. 5 A, iv), despite the substantial evidence that heparin is able to protect LUVs and GVs from fibril-induced disruption. Thus, the anisotropy experiments suggest that heparin does not prevent the binding of the β2m fibrils to the lipid bilayer, but instead interferes with the ability of the fibrils to cause bilayer disruption. Indeed, the cryo-TEM experiments depicted above indicate that association of heparin-coated β2m fibrils with lipid vesicles appears to be attenuated (Fig. 4 F) relative to the binding of the untreated fibrils (Fig. 4 C). Accordingly, the image of the heparin/fibril mixture incubated with LUVs shows depletion of lipid vesicles (Fig. 4 F), consistent with impaired liposome-fibril interactions.
Addition of heparin disaccharide reduced the impact of the β2m fibrils upon bilayer fluidity, as judged by TMA-DPH anisotropy, but to a lesser extent than was observed with bromophenol blue. The small heparin oligomer presumably interferes to some degree with membrane interactions of β2m, but is not able to prevent bilayer disruption.
Changes in lipid bilayer fluidity after interactions with β2m fibrils were also assessed using a different, complementary approach utilizing membrane-embedded Laurdan as a probe of lipid dynamics (Fig. 5 B). The fluorescence of Laurdan is sensitive to the polarity of the surrounding medium and thus is blue-shifted in more rigid lipid environments due to exclusion of water molecules from the probe proximity (45). The spectral shift is quantified using the general polarization (GP) function (45), which is proportional to the blue/red fluorescence ratio (Materials and Methods). The results in Fig. 5 B corroborate the TMA-DPH anisotropy data by demonstrating that β2m fibrils induce an increase in GP values of Laurdan/PC/PG vesicles. This change in GP remained largely unaltered after preincubation of the β2m fibrils with full-length heparin, reflecting a comparable reduction in lipid mobility in both cases (Fig. 5 B and see Fig. S5). Bromophenol blue, by contrast, largely blocked fibril-induced reduction of membrane fluidity, whereas heparin disaccharide exhibited marginal effect on fibril-lipid interactions. The β2m monomer did not affect lipid bilayer dynamics, confirming that the monomeric protein is not membrane-active under the conditions employed here, consistent with the TMA-DPH anisotropy data.
Discussion
This study sheds light on an important question in the search for therapeutic solutions to amyloid diseases, namely the relationship between fibrillation modulators and the interactions of amyloid fibrils with membranes in the presence of these agents. Although the impact of inhibitors of amyloid formation on the aggregation pathways of amyloidogenic proteins has been studied extensively (27,29,57), the possibility that the same compounds may disrupt fibril-membrane interactions has not been investigated in depth before, to our knowledge. Here we focus on the interaction of in vitro-formed β2m amyloid fibrils with PC/PG (1:1) lipid vesicles. We specifically chose β2m fibrils for this study because these assemblies have been shown previously to be cytotoxic and to be capable of permeabilizing lipid membranes (11). Previous results have demonstrated that electrostatic interactions are important determinants that mediate membrane disruption by β2m fibrils because increasing the fraction of negatively charged lipids within model membranes significantly enhances lipid bilayer permeabilization by these amyloid aggregates (11).
A recent study has revealed that interactions of fragmented β2m fibrils with model membranes give rise to breakage or blebbing of the outer lipid leaflet, accompanied by appearance of small vesicles associated with the fibrils (54). These findings shed light on a possible mechanism by which β2m fibrils elicit membrane permeabilization and disruption. Small lipid structures (presumably vesicles or micelles) have also been detected within other amyloid protein systems during the fibrillation process in the presence of LUVs (58). Furthermore, previous results have shown that the formation of β2m fibrils is not affected by the small molecules examined here (59), whereas heparin (but not heparin disaccharide) stabilizes fibrils against depolymerization at physiological pH (47,48). Moreover, the molecules tested in this study have all been shown to have no detectable effect on fibril appearance (see Fig. S2). Accordingly, for these fibril samples, at least, modification of membrane interactions can be assessed without interference from the effects of the small molecules on fibril assembly.
The results presented demonstrate that β2m fibrils display distinct abilities to interact with, and disrupt, membranes when incubated with the different compounds assessed in this study. Particularly intriguing is the observation that incubation with small molecules belonging to similar structural and functional classes results in different membrane interactions with β2m fibrils. Thus, although resveratrol did not inhibit membrane interactions of β2m fibrillar aggregates, EGCG and bromophenol blue hampered membrane disruption, presumably by binding to the fibrillar aggregates and impeding their association with lipid bilayer, rather than by membrane stabilization mediated by the polyphenol molecules themselves.
The potency of the three polyphenols tested here to prevent lipid bilayer disruption is distributed in the following order:
These differences can be attributed to the distinct structural properties of the assessed compounds. EGCG, the most efficient inhibitor among the three polyphenols, has a pKa value of 7.75 (Table 1). At the pH used in this study (pH 7.4), a significant fraction of EGCG molecules is negatively charged, which presumably mediates favorable electrostatic interactions with β2m fibrils. Resveratrol, which did not alter lipid interactions of the fibrils, has a higher pKa of 9.15 (Table 1), remaining nonionized under the same conditions. Further examination of the structures reveals that EGCG can form the largest number of hydrogen bonds of the three polyphenol compounds studied (11 bonds, Table 1), whereas resveratrol is able to make only three such bonds. Bromophenol blue, which demonstrated moderate inhibitory activity on membrane interactions of β2m fibrils, is fully charged at pH 7.4 (pKa 3.5, Table 1); however, this molecule can form an intermediate amount of hydrogen bonds (five bonds, Table 1) compared with the other polyphenols studied here. EGCG is also the most hydrophilic polyphenol examined, as judged by its low partition coefficient between octanol and water (LogD, Table 1). Together, these results suggest that electrostatic interactions and hydrogen bonding, rather than hydrophobic forces per se, are important determinants that govern the association of the polyphenols with β2m fibrils and, thereby, attenuate membrane disruption by these fibrillar aggregates. When comparing EGCG and bromophenol blue with a GAG of similar molecular weight (heparin disaccharide), it is evident that the latter failed to inhibit membrane activity of β2m fibrils despite having a substantial number of negatively charged substituents and potentially more hydrogen-bond donors and acceptors than the polyphenols studied here (Table 1). Our findings imply that a combination of hydrophobic/aromatic interactions with electrostatic and hydrogen bonds is required for sequestering β2m fibrillar aggregates by these small molecules. Neither of these factors alone is sufficient to rationalize the effect of polyphenols and heparin disaccharide on β2m fibrils-membrane interactions.
Remarkable experimental outcomes were also found for fibrils incubated with heparin and its building unit, heparin disaccharide. Full-length heparin was found to be the most powerful inhibitor of β2m fibril-induced damage of model membranes among all of the compounds tested. Unlike the small molecules, heparin abolished membrane disruption by β2m fibrils and was able to disperse the large fibrillar aggregates observed at neutral pH. The inhibitory activity of heparin can be ascribed to efficient binding of its multiple negatively-charged sulfated and carboxylic units to β2m fibrils that presumably impede their electrostatic interactions with negatively charged lipids. The remarkable difference in inhibitory potency of heparin and heparin disaccharide highlights the crucial role of the higher local concentration of functional groups in promoting interactions between the compound of interest and the β2m amyloid fibrils. Thus, water-soluble polymers decorated by species possessing the ability to suppress membrane damage by amyloid aggregates may provide a promising strategy in the quest to design potent inhibitors of cell membrane disruption by amyloid fibrils. Interestingly in this regard, application of polymeric compounds conjugated to functional components such as fluorine or metal-chelating groups has been shown to impair the amyloidogenesis and cytotoxicity mediated by Aβ peptide (34,37). Finally, and importantly, comparison of the results of fluorescence spectroscopy assays reporting upon lipid dynamics with those of membrane damage, visualized by dye release, fluorescence microscopy, and cryo-TEM, suggests that heparin modulates, rather than eliminates, β2m fibril-membrane association.
In conclusion, the spectroscopic and microscopic data presented underscore the significant and divergent effects of the different fibril modulators tested upon membrane interactions of β2m fibrils. Additional studies are required to assess whether our findings have a generic nature and are pertinent to other amyloidogenic proteins. In light of the emerging realization concerning the significance of membrane interactions upon the pathological profiles in protein misfolding diseases (3,19,60), the results suggest that an important facet of any study to develop inhibitors of amyloid diseases is the inclusion of analysis of the effect of potential inhibitors on amyloid-lipid interactions.
Acknowledgments
We thank Dr. Yael Kalissman (Ilse Katz Institute for Nano-Scale Science and Technology) for excellent technical assistance with cryo-EM experiments, Dr. Paul Beales (University of Leeds), and members of our laboratories for many helpful discussions.
T.S. was supported by the Marie Curie Intra-European Fellowship (No. 276621). We also acknowledge the Wellcome Trust (grants No. 075675 and No. 080707/z/06/z), the Biotechnology and Biological Sciences Research Council (grant No. BB/526502/1), and the British Council (BIRAX award) for funding this project.
Footnotes
Wei-Feng Xue’s current address is School of Biosciences, University of Kent, Canterbury, Kent CT2 7NZ, UK.
Contributor Information
Sheena E. Radford, Email: s.e.radford@leeds.ac.uk.
Raz Jelinek, Email: razj@bgu.ac.il.
Supporting Material
References
- 1.Huang Y., Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meissner W.G., Frasier M., Bezard E. Priorities in Parkinson’s disease research. Nat. Rev. Drug Discov. 2011;10:377–393. doi: 10.1038/nrd3430. [DOI] [PubMed] [Google Scholar]
- 3.Stefani M. Generic cell dysfunction in neurodegenerative disorders: role of surfaces in early protein misfolding, aggregation, and aggregate cytotoxicity. Neuroscientist. 2007;13:519–531. doi: 10.1177/1073858407303428. [DOI] [PubMed] [Google Scholar]
- 4.Campioni S., Mannini B., Chiti F. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010;6:140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 5.Ladiwala A.R., Litt J., Tessier P.M. Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J. Biol. Chem. 2012;287:24765–24773. doi: 10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caughey B., Lansbury P.T. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 7.Winner B., Jappelli R., Riek R. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 9.Cheng I.H., Scearce-Levie K., Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 10.Pieri L., Madiona K., Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012;102:2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue W.F., Hellewell A.L., Radford S.E. Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 2009;284:34272–34282. doi: 10.1074/jbc.M109.049809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada T., Ikeda K., Matsuzaki K. Formation of toxic Aβ (1-40) fibrils on GM1 ganglioside-containing membranes mimicking lipid rafts: polymorphisms in Aβ(1-40) fibrils. J. Mol. Biol. 2008;382:1066–1074. doi: 10.1016/j.jmb.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 13.Xue W.F., Hellewell A.L., Radford S.E. Fibril fragmentation in amyloid assembly and cytotoxicity: when size matters. Prion. 2010;4:20–25. doi: 10.4161/pri.4.1.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren P.H., Lauckner J.E., Kopito R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luk K.C., Kehm V., Lee V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J.Y., Englund E., Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 17.Cremades N., Cohen S.I., Klenerman D. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martins I.C., Kuperstein I., Rousseau F. Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008;27:224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auluck P.K., Caraveo G., Lindquist S. α-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 20.Jelinek R. Wiley-VCH; Weinheim, Germany: 2011. Lipids and Cellular Membranes in Amyloid Diseases. [Google Scholar]
- 21.Pithadia A.S., Kochi A., Lim M.H. Reactivity of diphenylpropynone derivatives toward metal-associated amyloid-β species. Inorg. Chem. 2012;51:12959–12967. doi: 10.1021/ic302084g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng P.N., Liu C., Nowick J.S. Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 2012;4:927–933. doi: 10.1038/nchem.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Härd T., Lendel C. Inhibition of amyloid formation. J. Mol. Biol. 2012;421:441–465. doi: 10.1016/j.jmb.2011.12.062. [DOI] [PubMed] [Google Scholar]
- 24.Han Y.S., Zheng W.H., Quirion R. Neuroprotective effects of resveratrol against β-amyloid-induced neurotoxicity in rat hippocampal neurons: involvement of protein kinase C. Br. J. Pharmacol. 2004;141:997–1005. doi: 10.1038/sj.bjp.0705688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evers F., Jeworrek C., Winter R. Elucidating the mechanism of lipid membrane-induced IAPP fibrillogenesis and its inhibition by the red wine compound resveratrol: a synchrotron x-ray reflectivity study. J. Am. Chem. Soc. 2009;131:9516–9521. doi: 10.1021/ja8097417. [DOI] [PubMed] [Google Scholar]
- 26.Rezai-Zadeh K., Arendash G.W., Tan J. Green tea epigallocatechin-3-gallate (EGCG) reduces β-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008;1214:177–187. doi: 10.1016/j.brainres.2008.02.107. [DOI] [PubMed] [Google Scholar]
- 27.Ehrnhoefer D.E., Duennwald M., Wanker E.E. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006;15:2743–2751. doi: 10.1093/hmg/ddl210. [DOI] [PubMed] [Google Scholar]
- 28.Ehrnhoefer D.E., Bieschke J., Wanker E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 29.Ladiwala A.R., Lin J.C., Tessier P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 2010;285:24228–24237. doi: 10.1074/jbc.M110.133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng F., Abedini A., Raleigh D.P. The flavanol (−)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry. 2010;49:8127–8133. doi: 10.1021/bi100939a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lever R., Page C.P. Novel drug development opportunities for heparin. Nat. Rev. Drug Discov. 2002;1:140–148. doi: 10.1038/nrd724. [DOI] [PubMed] [Google Scholar]
- 32.Bravo R., Arimon M., Fernàndez-Busquets X. Sulfated polysaccharides promote the assembly of amyloid β (1-42) peptide into stable fibrils of reduced cytotoxicity. J. Biol. Chem. 2008;283:32471–32483. doi: 10.1074/jbc.M709870200. [DOI] [PubMed] [Google Scholar]
- 33.Pérez M., Wandosell F., Avila J. Sulphated glycosaminoglycans prevent the neurotoxicity of a human prion protein fragment. Biochem. J. 1998;335:369–374. doi: 10.1042/bj3350369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu G., Men P., Smith M.A. Nanoparticle-chelator conjugates as inhibitors of amyloid-β aggregation and neurotoxicity: a novel therapeutic approach for Alzheimer disease. Neurosci. Lett. 2009;455:187–190. doi: 10.1016/j.neulet.2009.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mannini B., Cascella R., Chiti F. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA. 2012;109:12479–12484. doi: 10.1073/pnas.1117799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ladiwala A.R., Bhattacharya M., Tessier P.M. Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc. Natl. Acad. Sci. USA. 2012;109:19965–19970. doi: 10.1073/pnas.1208797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saraiva A.M., Cardoso I., Brezesinski G. Controlling amyloid-β peptide(1-42) oligomerization and toxicity by fluorinated nanoparticles. ChemBioChem. 2010;11:1905–1913. doi: 10.1002/cbic.201000237. [DOI] [PubMed] [Google Scholar]
- 38.Högen T., Levin J., Giese A. Two different binding modes of α-synuclein to lipid vesicles depending on its aggregation state. Biophys. J. 2012;102:1646–1655. doi: 10.1016/j.bpj.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burmeister W.P., Gastinel L.N., Bjorkman P.J. Crystal structure at 2.2 Å resolution of the MHC-related neonatal Fc receptor. Nature. 1994;372:336–343. doi: 10.1038/372336a0. [DOI] [PubMed] [Google Scholar]
- 40.Bellotti V., Stoppini M., Ferri G. β2-microglobulin can be refolded into a native state from ex vivo amyloid fibrils. Eur. J. Biochem. 1998;258:61–67. doi: 10.1046/j.1432-1327.1998.2580061.x. [DOI] [PubMed] [Google Scholar]
- 41.Porter M.Y., Routledge K.E., Hewitt E.W. Characterization of the response of primary cells relevant to dialysis-related amyloidosis to β2-microglobulin monomer and fibrils. PLoS ONE. 2011;6:e27353. doi: 10.1371/journal.pone.0027353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevenson D.E., Hurst R.D. Polyphenolic phytochemicals—just antioxidants or much more? Cell. Mol. Life Sci. 2007;64:2900–2916. doi: 10.1007/s00018-007-7237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones C.J., Beni S., Larive C.K. Heparin characterization: challenges and solutions. Annu. Rev. Anal. Chem. 2011;4:439–465. doi: 10.1146/annurev-anchem-061010-113911. [DOI] [PubMed] [Google Scholar]
- 44.Moscho A., Orwar O., Zare R.N. Rapid preparation of giant unilamellar vesicles. Proc. Natl. Acad. Sci. USA. 1996;93:11443–11447. doi: 10.1073/pnas.93.21.11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parisio G., Marini A., Mennucci B. Polarity-sensitive fluorescent probes in lipid bilayers: bridging spectroscopic behavior and microenvironment properties. J. Phys. Chem. B. 2011;115:9980–9989. doi: 10.1021/jp205163w. [DOI] [PubMed] [Google Scholar]
- 46.Bazar E., Jelinek R. Divergent heparin-induced fibrillation pathways of a prion amyloidogenic determinant. ChemBioChem. 2010;11:1997–2002. doi: 10.1002/cbic.201000207. [DOI] [PubMed] [Google Scholar]
- 47.Myers S.L., Jones S., Radford S.E. A systematic study of the effect of physiological factors on β2-microglobulin amyloid formation at neutral pH. Biochemistry. 2006;45:2311–2321. doi: 10.1021/bi052434i. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto S., Yamaguchi I., Naiki H. Glycosaminoglycans enhance the trifluoroethanol-induced extension of β 2-microglobulin-related amyloid fibrils at a neutral pH. J. Am. Soc. Nephrol. 2004;15:126–133. doi: 10.1097/01.asn.0000103228.81623.c7. [DOI] [PubMed] [Google Scholar]
- 49.Chen R.F., Knutson J.R. Mechanism of fluorescence concentration quenching of carboxyfluorescein in liposomes: energy transfer to nonfluorescent dimers. Anal. Biochem. 1988;172:61–77. doi: 10.1016/0003-2697(88)90412-5. [DOI] [PubMed] [Google Scholar]
- 50.White H.E., Hodgkinson J.L., Saibil H.R. Globular tetramers of β (2)-microglobulin assemble into elaborate amyloid fibrils. J. Mol. Biol. 2009;389:48–57. doi: 10.1016/j.jmb.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larsen J., Hatzakis N.S., Stamou D. Observation of inhomogeneity in the lipid composition of individual nanoscale liposomes. J. Am. Chem. Soc. 2011;133:10685–10687. doi: 10.1021/ja203984j. [DOI] [PubMed] [Google Scholar]
- 52.Han Y.S., Bastianetto S., Quirion R. Specific plasma membrane binding sites for polyphenols, including resveratrol, in the rat brain. J. Pharmacol. Exp. Ther. 2006;318:238–245. doi: 10.1124/jpet.106.102319. [DOI] [PubMed] [Google Scholar]
- 53.Sun Y., Hung W.C., Huang H.W. Interaction of tea catechin (−)-epigallocatechin gallate with lipid bilayers. Biophys. J. 2009;96:1026–1035. doi: 10.1016/j.bpj.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Milanesi L., Sheynis T., Saibil H.R. Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2012;109:20455–20460. doi: 10.1073/pnas.1206325109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pebay-Peyroula E., Dufourc E.J., Szabo A.G. Location of diphenyl-hexatriene and trimethylammonium-diphenyl-hexatriene in dipalmitoylphosphatidylcholine bilayers by neutron diffraction. Biophys. Chem. 1994;53:45–56. doi: 10.1016/0301-4622(94)00075-1. [DOI] [PubMed] [Google Scholar]
- 56.Rodrigues C., Gameiro P., de Castro B. Interaction of rifampicin and isoniazid with large unilamellar liposomes: spectroscopic location studies. Biochim. Biophys. Acta. 2003;1620:151–159. doi: 10.1016/s0304-4165(02)00528-7. [DOI] [PubMed] [Google Scholar]
- 57.Vilasi S., Sarcina R., Sirangelo I. Heparin induces harmless fibril formation in amyloidogenic W7FW14F apomyoglobin and amyloid aggregation in wild-type protein in vitro. PLoS ONE. 2011;6:e22076. doi: 10.1371/journal.pone.0022076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sciacca M.F., Kotler S.A., Ramamoorthy A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woods L.A., Platt G.W., Radford S.E. Ligand binding to distinct states diverts aggregation of an amyloid-forming protein. Nat. Chem. Biol. 2011;7:730–739. doi: 10.1038/nchembio.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams T.L., Serpell L.C. Membrane and surface interactions of Alzheimer’s Aβ peptide—insights into the mechanism of cytotoxicity. FEBS J. 2011;278:3905–3917. doi: 10.1111/j.1742-4658.2011.08228.x. [DOI] [PubMed] [Google Scholar]
- 61.SciFinder. 2013. Physical properties of molecules used in the study http://www.scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf Accessed January 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.