

Abstract
Molecular dynamics simulations have been used to characterize the effects of transfer from aqueous solution to a vacuum to inform our understanding of mass spectrometry of membrane-protein-detergent complexes. We compared two membrane protein architectures (an α-helical bundle versus a β-barrel) and two different detergent types (phosphocholines versus an alkyl sugar) with respect to protein stability and detergent packing. The β-barrel membrane protein remained stable as a protein-detergent complex in vacuum. Zwitterionic detergents formed conformationally destabilizing interactions with an α-helical membrane protein after detergent micelle inversion driven by dehydration in vacuum. In contrast, a nonionic alkyl sugar detergent resisted micelle inversion, maintaining the solution-phase conformation of the protein. This helps to explain the relative stability of membrane proteins in the presence of alkyl sugar detergents such as dodecyl maltoside.
Introduction
Membrane proteins play key roles in cell biology, accounting for ∼25% of genes. Advances in structural biology are yielding an increasing number of membrane protein structures (1), with ∼2800 unique structures predicted by 2020 (see, e.g., http://blanco.biomol.uci.edu/mpstruc for a summary). However, relatively few membrane protein structures have been determined in the presence of a lipid bilayer environment. Rather, the majority of biophysical and structural studies are of membrane proteins in the presence of detergents. This in turn has resulted in some discussion about the extent to which the structure of a membrane protein may be altered by changes in its lipid and/or detergent environment (2,3). It is therefore of interest to understand in more detail how the environment presented by detergents may influence membrane protein conformation and stability. In most structural studies, membrane proteins are exposed to bulk aqueous solvent, even when embedded in lipid bilayers or detergent micelles. However, in mass spectrometry (MS), protein complexes are in the gas phase (4), as is also the case for single-particle coherent diffraction imaging using x-ray free-electron lasers (5). In particular, the use of MS is widespread in the characterization of water-soluble proteins, using electrospray ionization (ESI) to transport macromolecular protein complexes into the gas phase (4). It was thought that membrane protein complexes could not be studied using this technique, as their stability requires a membranelike environment (6). However, n-dodecyl-β-D-maltoside (DDM) micelles have been shown to stabilize intact oligomeric membrane protein complexes during the transition to vacuum (7–9). Rapid methodological progress has been made in recent years, such that it is now possible to study not only membrane protein complex composition, but ligand binding and conformational changes of membrane protein complexes (10). It is therefore important to understand the role of the detergent in stabilizing membrane protein complexes in vacuo, given that a vacuum presents an effectively hydrophobic environment. In this context it is important to understand the underlying principles that determine which detergents are able to preserve solution-phase membrane protein interactions while in the gas phase.
Molecular dynamics (MD) simulations provide an in silico approach to explore the interactions of membrane proteins with lipids in bilayers (11), and with detergents both in micelles (12) and in crystals (13). MD simulations have also been employed to explore the behavior of globular proteins in vacuo, revealing the protective effects of detergents, which form an inverted micelle around the (water-soluble) protein (14). To date, only one simulation study of a membrane-protein-detergent complex in vacuo has been published (10). That study was of a monomeric β-barrel membrane protein (OmpA) in an n-dodecylphosphocholine (DPC) micelle. However, most membrane proteins are α-helix bundles rather than β-barrels, and thus are considerably less robust than OmpA to changes in their environment. Furthermore, many membrane proteins are oligomeric. As noted by Friemann et al. (15), further studies are required for a full understanding of the consequences of dehydration on membrane-protein-detergent complexes. It is therefore important to understand the behavior of more typical membrane protein architectures (i.e., α-helical and oligomeric) in an in vacuo environment.
The influenza virus M2 proteins (A/M2 from influenza A and BM2 from influenza B) are homotetramers of transmembrane α-helices that have been studied in some detail via a range of structural and biophysical approaches (16–20). They show some structural sensitivity to their environment (bilayer versus micelle versus crystal (2)), thus providing a test system for membrane protein stability in vacuo.
We have generated models of protein-detergent complexes (PDCs) in vacuo, followed by extended MD simulations to explore the resultant conformational dynamics of the BM2 helix bundle. This enables us to compare the stability of BM2 in complex with the nonionic detergent DDM to its stability in complex with dihexanoyl phosphatidylcholine (DHPC) and with DPC, both zwitterionic detergents widely used for NMR studies of membrane proteins (see, e.g., Wang et al. (18) and Arora et al. (21)). DDM has a maltoside sugar headgroup, whereas DHPC and DPC have phosphocholine headgroups. DDM and DPC have the same length alkyl tail (i.e., 12 carbons) whereas DHPC has two shorter tails (6 carbons). By comparing all three detergents we were able to determine whether differences in stabilization properties are due to differing chain lengths (i.e., the hydrophobic dimension of the micelle) or rather result from the nonionic versus zwitterionic nature of the headgroups. We also used the OmpA-DPC PDC from a previous simulation study (15) to allow comparison of the two membrane protein architectures (i.e., α-helix bundle versus β-barrel). We used a two-stage simulation protocol: 1), steered molecular dynamics (SMD) to transfer the PDC from bulk solution to vacuum; followed by 2), extended simulation in vacuum (i.e., under dehydrating conditions) of the sparingly solvated PDCs generated by the previous stage. These simulations suggest that the structure of the membrane protein is stabilized by DDM, but not by DHPC or DPC.
Methods
Initial models of BM2/detergent micelles were generated from the NMR structure of the BM2 transmembrane domain (18) (PDB ID 2KIX) via protein/detergent self-assembly simulations (22). In the case of the BM2-DHPC/DPC complexes, 100 ns atomistic (AT) MD simulations with the protein positionally restrained starting from 200 detergent molecules in initial random positions and orientations in the simulation box were used to generate protein-detergent complexes containing ∼70 DHPC and ∼130 DPC detergent molecules. The remaining detergent molecules formed detergent-only clusters, i.e., separate from the main PDC, and thus were discarded before a 100 ns ATMD simulation with the protein free to move. An ∼200 mM concentration of DHPC was chosen to match the experimental conditions used in the NMR structure determination. However, using similar concentrations of DDM and attempting ATMD self-assembly simulations raised issues of incomplete sampling over similar timescales, resulting in failure to form a well-defined PDC. Therefore, an alternative approach was adopted.
The BM2-DDM complex was generated using a two-step process in which a preformed coarse-grained (CG) DDM micelle was built from 150 DDM molecules using Packmol (23) and allowed to interact with BM2, forming a BM2-DDM complex within 1 μs of CGMD simulation time (24,25). CC MD simulations used the MARTINI forcefield (26,27). This was then converted to AT representation using a fragment-based approach (28). We note that previous simulation studies have also adopted a multiscale approach to improve sampling of micelle self-assembly processes (e.g., Brocos et al. (29)). The number of DDM detergent molecules was chosen based on small-angle x-ray scattering data (S.L. Rouse and D. Durand, unpublished) and literature values (30). The use of a preformed micelle may be further justified by consideration of the very low critical micelle concentration (CMC) of DDM (0.15–0.18 mM), which implies that there is a negligible concentration of monomers in solution above the CMC, and that almost all of the detergent present is in micellar form. The resultant PDCs were simulated for 100 ns in aqueous solution before SMD simulation. The protonation states of residues were based on standard solution-state charges. The His27 residues were doubly protonated based on experimental data (7). The OmpA-DPC complex was taken from a previous study (12,22). ATMD simulations were performed using GROMACS (31) (www.gromacs.org) and the OPLS-AA force field (32). The topology and parameters (GAFF) of the detergents DHPC, DDM, and DPC were generated in AMBER format with ANTECHAMBER (33,34) and converted to GROMACS format using the amb2gmx.pl script (35). The detergents were optimized with Gaussian 03 (36) at the high-frequency level with the 6-31G basis set. Restrained electrostatic potential charges (37) were assigned as the atomic partial charges for the detergent by optimizing the charge fitting to quantum-mechanics electrostatic potential maps. A time step of 2 fs was used, and bond lengths were constrained using the LINCS algorithm (38). The BM2-DHPC SMD simulations were also performed using the GROMOS G43A force field, and qualitatively similar behavior was observed (Fig. S1 in the Supporting Material), which is consistent with previous studies that showed agreement between these force fields in vacuo. The SMD and vacuum simulations performed are summarized in Table S1. The full CG and AT DDM parameters are freely available for download and have been deposited in LipidBook (http://lipidbook.bioch.ox.ac.uk) (39).
Transfer of protein-detergent complex from solution to vacuum using SMD
Briefly, this was achieved by 1), extending the simulation box in the z-direction from the endpoint of a simulation of the PDC in solution; 2), coupling a virtual spring to the center of mass of the PDC; and 3), applying a harmonic potential force to the spring such that the spring is pulled in the z-direction away from the bulk solvent. The umbrella sampling option was used such that the force applied to the spring increases with the distance between the spring and the center of mass of the bulk solvent. The pulling rate used was 1 nm/ns and the force was applied using the pull code implemented in GROMACS. The simulation was continued until the PDC and any directly and indirectly interacting water molecules (a molecule is added to the PDC when the distance of any of its atoms from any other atom in the PDC is <3.6 Å) were separate from bulk water (typically 15–20 ns). Pressure coupling was switched off and the integrator chosen was stochastic dynamics with an inverse friction constant of 2 ps. In ESI-MS, the sprayed droplets travel through a collision cell, which is typically at pressures of 10−6–10−7 mbar, so collisions and friction due to the presence of gas particles is expected (40). The use of stochastic dynamics applies a friction term to each atom, mimicking this effect. It also acts as a thermostat, keeping the overall temperature of the system constant. The force constant, pull rate, and pull groups chosen for these simulations were calibrated using the BM2-DHPC system as a test case.
Simulation under dehydrating conditions
After the transfer from bulk solution to vacuum, the main PDC (including any water molecules directly or indirectly in contact using the 3.6 Å criterion previously defined) is extracted and simulated in a vacuum environment, in which 1), there are no periodic boundary conditions; 2), there are no cut offs (interactions between all particles are calculated); and 3), pressure coupling is switched off. In a true vacuum, the temperature coupling would also be switched off. However, the removal of temperature coupling leads to fast evaporation of surface waters, which take a large amount of the internal energy with them. The remaining complex is then at low temperature (in these systems, ∼270 K after a few nanoseconds from a starting temperature of 323 K), and little of the system dynamics or rearrangement can be observed. Therefore, temperature coupling was used to maintain the energy of the system. This allows control of the energy of the complex similar to the control over collision energy in the corresponding ESI experiments. Each component of the system (protein, detergent, and water) was temperature-coupled separately. Center-of-mass translation and rotation was removed every 10 steps. Note that any ions within the initial sparingly solvated PDC were removed before simulation to ensure that each simulation began from an equivalent starting point.
Results and Discussion
To mimic the initial desolvation stages of ESI, the four PDCs (Ompa-DPC and BM2-DHPC, BM2-DPC, and BM2-DDM) were pulled from aqueous solution into a vacuum using SMD (Fig. 1 A), generating a sparingly solvated PDC (Fig. 1 B). These simul ations are described in the next section.
Figure 1.
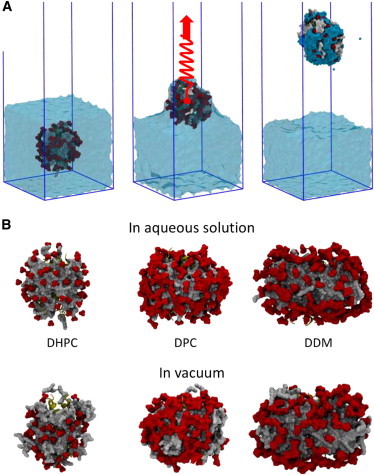
Transfer of PDCs from water to vacuum using SMD simulations. (A) Three stages in the process are shown: 1), a PDC in water, with a water/vacuum interface; 2), application of an external force (red arrow with spring) to the center of mass of the PDC to pull it through the water/vacuum interface; leading to 3), the PDC in the vacuum phase with some water (cyan) remaining bound to the complex and the occasional water molecule (two small cyan spheres) escaping into the vacuum phase. (B) The arrangement of detergent molecules around BM2 in solution and after transfer to vacuum. The detergent tails are shown as a gray surface, the detergent headgroups are red, and the protein is yellow. Water is omitted for clarity.
OmpA is an exceptionally stable β-barrel membrane protein that has been the subject of earlier simulations of PDCs in vacuo (15,41). OmpA shows little conformational sensitivity to its environment in simulations of a lipid bilayer, a detergent micelle, a protein/detergent crystal, or in vacuo (12,13,15). Thus, SMD simulations of the OmpA-DPC PDC provide a suitable control with which to compare BM2. They also allow a direct comparison of our two-stage protocol, described here, with the protocol in a previous simulation study (15,41) that employed manual removal of water molecules.
The PDCs are equilibrated in solution and adopt the normal micelle geometry, in which the hydrophobic tails are sequestered within the core, allowing the headgroups to interact with water. As seen in the previous simulation study (15), as the complex is transferred from solution to vacuum, the DPC detergent molecules reorientate such that headgroup-headgroup interactions are maximized, leading to a disruption of the normal micelle arrangement. The alkyl chains align along the surface of OmpA. As anticipated, there was little change in protein conformation, with a Cα root-mean-square deviation (RMSD) of <1.5 Å for residues in the β-barrel (Fig. S2). Thus, the changes in the OmpA-DPC PDC were largely limited to the detergent, again consistent with previous studies (15). The detergent molecules continued to rearrange during a further >0.3 μs simulation of the sparingly solvated complex (with periodic boundary conditions implemented), such that the headgroups and remaining water molecules localized in the vicinity of the charged side chains of the OmpA loops (Fig. S3). The detergent-detergent and detergent-water interactions dominate, at the expense of protein-detergent and protein-water interactions.
Having established the expected behavior for the OmpA-DPC complex, we now use the above protocol to explore the behavior of the α-helical BM2 protein in each of the three different detergents. The changes in detergent packing during the SMD simulations of the BM2-detergent complexes allow us to dissect the processes that occur during transfer from solution to vacuum. Fig. 1 B shows snapshots of each of the complexes in solution and in vacuum. It is evident that changes in detergent are most marked for DHPC, followed by DPC and then DDM (Fig. 1 B). The driving force behind these changes appears to be maximization of electrostatic interactions. Thus, DHPC and DPC micelles begin to lose their regular orientation, and the headgroups pack closer into the core of the PDC while the detergent tails move toward the surface. This correlates with a change in surface exposure of the protein such that the hydrophobic, membrane-spanning residues tend to become more exposed to vacuum, whereas hydrophilic residues located near the termini of BM2 become less exposed (Figs. S4 and S5). The corresponding trend is observed for the detergent molecules, where the hydrophilic area is reduced as hydrophobic tails become more exposed. In the packing of DDM detergent, these trends were much less pronounced, with little change in exposure of hydrophobic or hydrophilic residues relative to packing trends for the PDC in water. These changes can be quantified as the degree of detergent headgroup clustering in each simulation (Fig. S6). There is a clear decrease in minimum distances between DHPC and DPC headgroups in vacuum compared to solution. In contrast, in the BM2-DDM PDC, there was very little change in detergent headgroup clustering upon the transition to vacuum.
The resultant arrangements of detergent molecules and water molecules in the in vacuo BM2-DHPC, BM2-DPC, and BM2-DDM complexes (Fig. S4) show that the remaining waters are largely located within the headgroup regions, maximizing the number of detergent-water hydrogen bonds on the PDC surface. In contrast to the OmpA-DPC complex in vacuum, the DHPC tails do not appear to align upon the protein surface but splay outward. This orientation allows the glycerol oxygen atoms of each chain to form hydrogen bonds with the remaining water molecules.
There are some small changes to the BM2 protein during transfer from solution to vacuum (Fig. S2). The overall secondary-structure content remains unchanged in each simulation, but the RMSD from the initial BM2 transmembrane helix bundle solution conformation is slightly higher (∼0.5 Å) for DHPC and DPC than for DDM.
The short (15–20 ns) SMD simulations captured the first changes in detergent reorientation associated with the removal from aqueous solution, yielding partially solvated PDCs in vacuo. However, to evaluate more fully the impact of extensive dehydration on the PDCs, longer (>0.5 μs) simulations in vacuo were performed during which water molecules were free to evaporate (Fig. 2, A and B). Table S2 lists the measured structural properties of the PDCs at the start and end of the dehydration during vacuum simulations. Here, we discuss more fully some of the key results.
Figure 2.
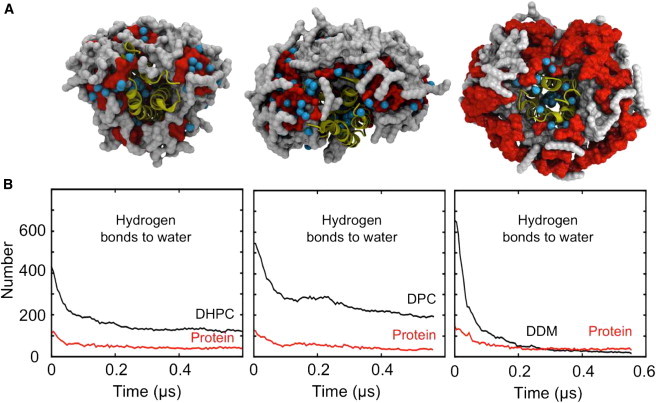
Evaporation of water molecules from PDCs during extended simulations under dehydrating conditions in vacuo. (A) Snapshots from the final frames of each simulation show the protein in yellow cartoon representation and the detergent molecules as a surface with headgroups colored red and tails colored gray. Bound water molecules are shown as cyan spheres. (B) Hydrogen-bonding interactions of water molecules with detergent (black lines) and with protein (red lines) as functions of time.
A large number of water molecules evaporated from the PDCs within the first 100 ns. In each case, the dehydration process had a time constant of ∼100 ns. Although the rate of water loss was similar between the three PDCs, one intriguing difference was observed, namely, that in the case of DDM, the water was preferentially lost from the headgroup region (as opposed to waters interacting with the protein), whereas in the DPC and DHPC complexes, the majority of the remaining water molecules are located in the detergent headgroup region (evident from Fig. 2, A and B). The final numbers of waters that remained bound to the PDC were ∼130, ∼120, and ∼35 for BM2-DHPC, BM2-DPC, and BM2-DDM, respectively (Table S2). The final number of detergent-water H-bonds was ∼75% less in the BM2-DDM simulation than in the BM2-DHPC simulation, whereas the final number of BM2-water H-bonds was similar in each case. Of course, further desolvation may occur during the time course of the MS experiment which is several orders of magnitude longer than the simulations.
Examination of the spatial distributions of detergent headgroup and tail atoms close to the protein surface (Fig. 3) reveals a clear difference between the zwitterionic and nonionic detergents. It is evident that arrangement of DDM molecules adjacent to the protein more closely matches that of the corresponding solution-phase micelle, with the hydrophobic region of BM2 surrounded by detergent tails and the headgroups restricted to the more polar regions at the termini of the transmembrane helices. In contrast, for the DHPC PDC, detergent packing was rearranged such that DHPC headgroups came into close contact with the transmembrane region of BM2, resulting in some of the protein becoming exposed to vacuum (Fig. 4). A similar pattern is seen for BM2 in DPC. Despite the initial sparingly solvated BM2-DPC complex having more of the protein hydrophobic surface covered by detergent (compared to the other two detergents), the DPC molecules rapidly rearrange upon dehydration (Fig. 4). This rearrangement maximizes the electrostatic interactions between detergent headgroups at the expense of protein-detergent interactions. As the DPC micelle tends toward inversion, a large proportion of the protein becomes exposed to vacuum. Thus, the DDM PDC more closely preserves a bilayerlike environment in vacuo.
Figure 3.
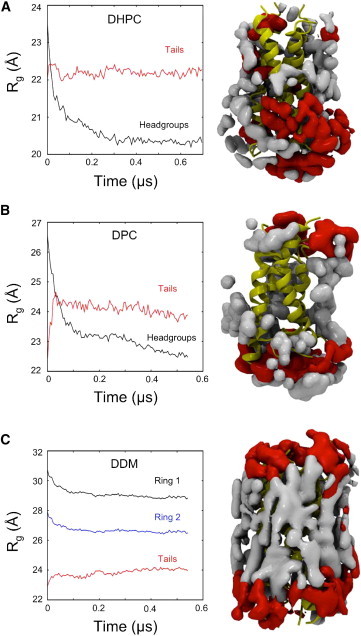
Rearrangement of detergent molecules during simulation under dehydrating conditions. Radii of gyration for detergent headgroups (black trace) and tails (red trace) are shown, highlighting the micelle inversion of DHPC (A) and DPC (B), but not DDM (C). For BM2-DDM, radii are shown for the first (black trace) and second (blue trace) sugar ring of the DDM molecules. Isosurfaces correspond to the mean distribution (over the length of the simulation) of detergent headgroup (red) and tail (gray) atoms within 4 Å of the protein surface for each of the PDCs. In DHPC and DPC, there is partial exposure of the protein to vacuum, whereas in DDM the normal solution packing, in which the hydrophobic portion of the protein is covered by detergent tails, is maintained. The isosurfaces were generated using the VolMap plugin within VMD (51).
Figure 4.
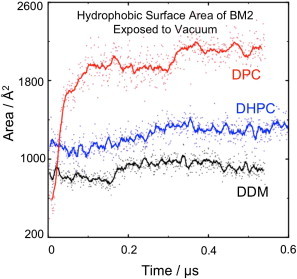
Area of BM2 protein unprotected by detergent molecules during simulations in vacuo. The traces show the hydrophobic surface area of BM2 exposed to vacuum (i.e., not in contact with detergent molecules) during simulation under dehydrating conditions. This surface area is lowest initially for DPC (red trace), but it increases rapidly as water is lost. By the end of the simulations the protein is most exposed in DPC (red trace), followed by DHPC (blue trace) then DDM (black trace).
The marked difference in the extent of detergent rearrangement between DHPC and DDM may be assessed from the radii of gyration of the detergent headgroups and alkyl tails (Fig. 3), demonstrating that the DHPC and DPC micelles tend toward complete inversion (in response to the effectively hydrophobic environment presented by a vacuum), whereas the DDM micelle relaxes initially, without any substantial rearrangement upon further dehydration. For the BM2-DHPC complex, the inversion process and the loss of water-detergent H-bonds occur on the same timescale. For the BM2-DDM PDC, the main consequence of the dehydration is an increase in the number of DDM-DDM H-bonds (Fig. S7), correlating with the loss of water hydrogen-bonding partners. Headgroup-headgroup interactions are maximized for both detergents, but in the case of DDM this may be fulfilled without micelle inversion. In solution phase, the DDM headgroups already form an extended hydrogen-bonding network (42). Upon removal of water molecules, this hydrogen-bonding network can be further extended without the need for drastic rearrangement of the detergent molecules. It is tempting to compare this behavior to that of other sugar-based molecules, such as trehalose, which are known to protect proteins from denaturing during dehydration. The exact mechanism of trehalose protection is not understood, but several factors have been proposed (43,44), such as the sugar fulfilling protein-water hydrogen bonds and/or the DDM-DDM headgroups creating a relatively rigid scaffold that protects the protein upon water loss. Further studies might investigate how trehalose or smaller sugar-based headgroup detergents such as β-octyl glucoside behave upon transfer to vacuum.
The contrasting effects of dehydration on the detergents have correspondingly different effects on the structural integrity of the BM2 helix bundle (Fig. 5). In each simulation the initial rapid water loss results in a slight (∼5%) loss in α-helical content, with a corresponding increase in Cα RMSD to ∼1.5 Å (Fig. 5). However, over the subsequent 0.5 μs, there are major differences between the three systems. In DDM, there is very little further drift in the BM2 Cα RMSD, such that the helix bundle conformation at the end of the in vacuo simulation is essentially the same as at the start (Fig. 4 C). More surprisingly, in the DDM simulation, the protein regains the initially disrupted secondary structure (Table S2 and Fig. S8) within the first 100 ns, and this structure is retained for the remainder of the simulation. For DHPC and DPC, the initial loss in α-helicity marks the beginning of a trend toward gradual reduction in secondary structure. It appears that DDM is able to compensate for the lost hydrogen-bond interactions of the protein with water, again reminiscent of sugar protection during desiccation. In DHPC and DPC, much more pronounced changes in bundle conformation occur (Fig. 5 B), revealed by a continuous increase in Cα RMSD (Fig. 5 A) and associated with loss of secondary structure and a decrease in the number of protein-protein hydrogen bonds. A comparable loss in secondary structure upon (almost) complete dehydration of the PDC was observed in the OmpA-DPC simulation study (15), where 20% secondary structure was lost upon complete dehydration while protein-protein hydrogen bonding increased. Presumably, the increased intramolecular protein hydrogen bonding was largely limited to the loop regions of OmpA, as these displayed the greatest structural deviation upon dehydration.
Figure 5.
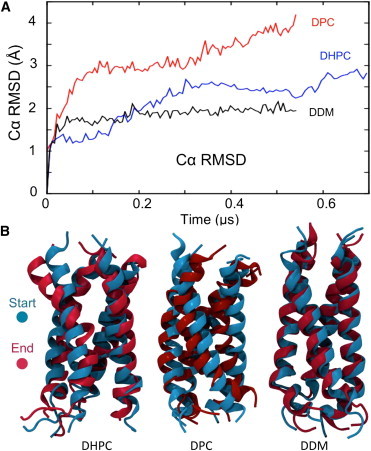
Conformational changes of BM2 protein during simulations in vacuo. (A) Protein Cα atom RMSD as a function of time in DPC (red trace), DHPC (blue trace), and DDM (black trace). (B) Structures of the BM2 helix bundle at the start (cyan) and end (red) of the in vacuo simulations are superimposed. The greatest conformational changes are observed in the zwitterionic detergents DHPC and DPC, whereas the solution-phase conformation is largely maintained in DDM.
Examination of the BM2 structure at the end of the BM2-DHPC in vacuo simulation (Fig. 5) reveals a conformational change corresponding to bending of one subunit away from ideal α-helical geometry. In DPC, the BM2 structure after dehydration shows an even greater deviation from the solution-phase structure (Table S2). The trajectories were inspected in detail to determine the likely mechanism of the protein conformational changes. In the case of BM2-DHPC, the decrease in solvation leads to increased electrostatic interactions between the zwitterionic DHPC headgroups and protein residues. Thus, the phosphocholine headgroups may penetrate between helices to interact with water molecules within the central pore of the BM2 helix bundle (Fig. 6), leading to extrusion of the water molecule laterally through the helix bundle. In the BM2-DPC simulation, similar destabilizing interactions between detergent headgroups and the protein occur, in which a single detergent molecule adopts a nonnative orientation and projects into the pore of the BM2 channel, leading to loss of the solution conformation (Fig. 6). Furthermore, as the simulation progresses the protein becomes increasingly expelled from the detergent, leading to greater distortions of the solution conformation.
Figure 6.
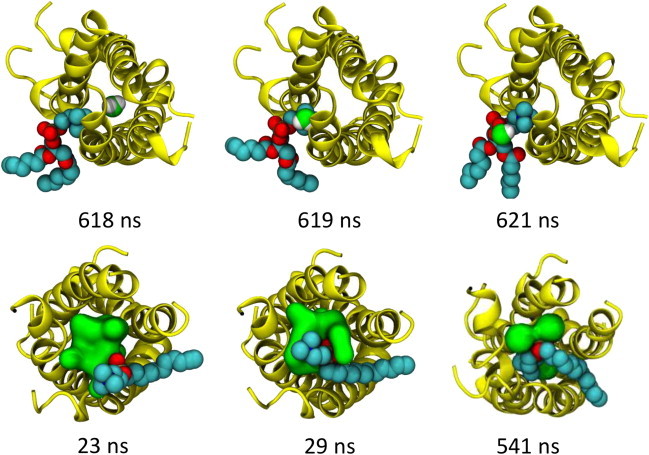
Destabilizing interactions of zwitterionic detergent molecules with BM2 protein during dehydration. (Upper) The change in conformation of the BM2-DDM complex at ∼0.62 μs (see Fig. 5) correlates with extraction of a water molecule (green and white) from the central pore, mediated by interactions with the headgroup of a DHPC molecule. The water and DHPC molecule are shown in spacefilling format, and the protein (yellow) in cartoon format. (Lower) Destabilizing interactions of DPC detergent molecules similar to those depicted above, with the C terminus of BM2 are observed, in which a single detergent molecule gradually becomes lodged between two of the transmembrane helices, interacting with water molecules and remaining within the pore for the duration of the simulation. Water molecules within 4 Å of this His27 residue are shown as a green surface.
The RMS fluctuations of the protein were calculated at varying stages of hydration to evaluate the effects of dehydration on protein dynamics. The BM2 protein displays similar dynamics in the sparingly solvated complex (generated from SMD simulations) compared to the solution phase, as shown by comparison of Cα RMS fluctuations at the start of the dehydration simulation compared to bulk solution simulations (Fig. 7). In all three detergent complexes, the dynamics of the BM2 protein are gradually dampened as the water molecules are lost. This effect is somewhat less pronounced in the case of DDM, where the solution-phase dynamics are lower than in DHPC and DPC to begin with. This perhaps reveals another aspect of the mechanism whereby DDM confers greater stability compared to the other detergents.
Figure 7.
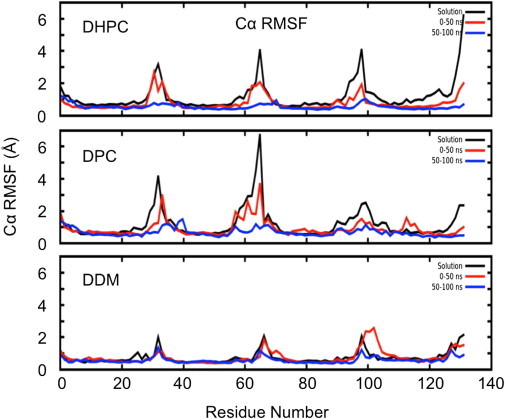
The influence of dehydration on BM2 dynamics. Cα RMS fluctuations are shown at various stages of dehydration. For each PDC, Cα RMS fluctuations in the solution phase are shown in black, those for the first 50 ns in vacuo are in red, and those for 50–100 ns are blue. The loss of water correlates with a decrease in protein dynamics, with some decrease in the first 50 ns, and the dampening effect most pronounced for the 50–100 ns period, during which approximately half of the water molecules in the sparingly solvated complex have evaporated. The dynamics of the protein in DDM vary the least, with minimal difference between the solution phase and the beginning of the simulation under vacuum. The solution-phase protein dynamics are greater in the simulations with DHPC and DPC detergents than in those with DDM.
These simulations reveal why detergents may differ in their ability to protect membrane proteins within an ESI-MS experiment. They reveal a clear distinction between zwitterionic (DHPC and DPC) and alkyl sugar detergents (e.g., DDM). In particular, almost complete dehydration of the DDM PDC did not result in any substantial change in conformation of the BM2 helix bundle, unlike the situation with DHPC and DPC PDCs. This may reflect differences in the numbers of detergents in the micelles with different detergent species. However, it strongly suggests that DDM may be more generally suited to protecting membrane protein conformation and oligomeric state from environmental perturbations. This correlates with the use of DDM in the majority of MS experiments on membrane proteins to date (45), and also with the widespread use of DDM in membrane protein crystallization (46). It should be noted that this protective effect of DDM for membrane proteins is because it retains its micellelike structure, with the hydrophobic tails inward, in vacuo, whereas cationic detergents form inverse micelles (hydrophobic tails outward) in vacuo (14), affording less protection for soluble proteins.
We note that the BM2 α-helix bundle shows a greater sensitivity to the in vacuo conditions in a PDC than does OmpA (15). OmpA is an especially stable β-barrel membrane protein (47), whereas the α-helix bundle of BM2 is more representative of the majority of membrane proteins. A recent comparative study (2) has shown that the closely related A/M2 helix bundle is sensitive to changes in environment between lipid bilayer, detergent micelle, and protein/detergent cocrystal. Despite the relatively high sensitivity of the M2 channel to its environment, the presence of DDM detergent suffices to stabilize the oligomeric structure in vacuo.
Conclusions
Overall, this study reveals the events of dehydration of a PDC in an MS experiment. To model more completely the processes during ESI-MS, it will be necessary to include collisions with neutral gas particles, as well as changes in protonation states of amino acid side chains during dehydration, and to address much longer timescales. Aspects of each of these processes have been studied for simpler systems using MD simulations (see, e.g., (48–50).). However, modeling a combination of these processes will require a multiscale approach. In summary, the results presented here provide a first glimpse of α-helical membrane protein conformational fragility/stability in vacuo, and offer a simulation methodology for predicting the suitability of a given detergent for use in MS and other experiments, including x-ray free-electron laser diffraction experiments. Encouragingly, the results suggest that detergents such as DDM, used in crystallization of membrane proteins, may also help to protect against protein destabilization in vacuo.
Acknowledgments
This work was supported by grants from the Biotechnology and Biological Sciences Research Council, the Engineering and Physical Sciences Research Council (CCPBioSim), the European Union (ScalaLife), and the Wellcome Trust.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.White S.H. Biophysical dissection of membrane proteins. Nature. 2009;459:344–346. doi: 10.1038/nature08142. [DOI] [PubMed] [Google Scholar]
- 2.Cross T.A., Sharma M., Zhou H.X. Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanders C.R., Mittendorf K.F. Tolerance to changes in membrane lipid composition as a selected trait of membrane proteins. Biochemistry. 2011;50:7858–7867. doi: 10.1021/bi2011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benesch J.L.P., Ruotolo B.T. Mass spectrometry: come of age for structural and dynamical biology. Curr. Opin. Struct. Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlichting I., Miao J.W. Emerging opportunities in structural biology with x-ray free-electron lasers. Curr. Opin. Struct. Biol. 2012;22:613–626. doi: 10.1016/j.sbi.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrera N.P., Robinson C.V. Advances in the mass spectrometry of membrane proteins: from individual proteins to intact complexes. Annu. Rev. Biochem. 2011;80:247–271. doi: 10.1146/annurev-biochem-062309-093307. [DOI] [PubMed] [Google Scholar]
- 7.Barrera N.P., Di Bartolo N., Robinson C.V. Micelles protect membrane complexes from solution to vacuum. Science. 2008;321:243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- 8.Zhou M., Morgner N., Robinson C.V. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 2011;334:380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laganowsky A., Reading E., Robinson C.V. Mass spectrometry of intact membrane protein complexes. Nat. Protoc. 2013;8:639–651. doi: 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcoux J., Wang S.C., Robinson C.V. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Natl. Acad. Sci. USA. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stansfeld P.J., Sansom M.S.P. Molecular simulation approaches to membrane proteins. Structure. 2011;19:1562–1572. doi: 10.1016/j.str.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Bond P.J., Sansom M.S.P. Membrane protein dynamics versus environment: simulations of OmpA in a micelle and in a bilayer. J. Mol. Biol. 2003;329:1035–1053. doi: 10.1016/s0022-2836(03)00408-x. [DOI] [PubMed] [Google Scholar]
- 13.Bond P.J., Faraldo-Gómez J.D., Sansom M.S.P. Membrane protein dynamics and detergent interactions within a crystal: a simulation study of OmpA. Proc. Natl. Acad. Sci. USA. 2006;103:9518–9523. doi: 10.1073/pnas.0600398103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y.F., Larsson D.S.D., van der Spoel D. Encapsulation of myoglobin in a cetyl trimethylammonium bromide micelle in vacuo: a simulation study. Biochemistry. 2009;48:1006–1015. doi: 10.1021/bi801952f. [DOI] [PubMed] [Google Scholar]
- 15.Friemann R., Larsson D.S.D., van der Spoel D. Molecular dynamics simulations of a membrane protein-micelle complex in vacuo. J. Am. Chem. Soc. 2009;131:16606–16607. doi: 10.1021/ja902962y. [DOI] [PubMed] [Google Scholar]
- 16.Stouffer A.L., Acharya R., DeGrado W.F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnell J.R., Chou J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J., Pielak R.M., Chou J.J. Solution structure and functional analysis of the influenza B proton channel. Nat. Struct. Mol. Biol. 2009;16:1267–1271. doi: 10.1038/nsmb.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma M., Yi M.G., Cross T.A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pielak R.M., Chou J.J. Influenza M2 proton channels. Biochim. Biophys. Acta. 2011;1808:522–529. doi: 10.1016/j.bbamem.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arora A., Abildgaard F., Tamm L.K. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 22.Bond P.J., Cuthbertson J.M., Sansom M.S.P. MD simulations of spontaneous membrane protein/detergent micelle formation. J. Am. Chem. Soc. 2004;126:15948–15949. doi: 10.1021/ja044819e. [DOI] [PubMed] [Google Scholar]
- 23.Martínez L., Andrade R., Martínez J.M. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009;30:2157–2164. doi: 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- 24.Bond P.J., Sansom M.S.P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 2006;128:2697–2704. doi: 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bond P.J., Holyoake J., Sansom M.S.P. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 2007;157:593–605. doi: 10.1016/j.jsb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Marrink S.J., Risselada H.J., de Vries A.H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 27.Monticelli L., Kandasamy S.K., Marrink S.J. The MARTINI coarse grained force field: extension to proteins. J. Chem. Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 28.Stansfeld P.J., Sansom M.S.P. From coarse-grained to atomistic: a serial multi-scale approach to membrane protein simulations. J. Chem. Theor. Comp. 2011;7:1157–1166. doi: 10.1021/ct100569y. [DOI] [PubMed] [Google Scholar]
- 29.Brocos P., Mendoza-Espinosa P., Pineiro A. Multiscale molecular dynamics simulations of micelles: coarse-grain for self-assembly and atomic resolution for finer details. Soft Matter. 2012;8:9005–9014. [Google Scholar]
- 30.Lipfert J., Columbus L., Doniach S. Size and shape of detergent micelles determined by small-angle x-ray scattering. J. Phys. Chem. B. 2007;111:12427–12438. doi: 10.1021/jp073016l. [DOI] [PubMed] [Google Scholar]
- 31.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 32.Kaminski G.A., Friesner R.A., Jorgensen W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 33.Wang J., Wolf R.M., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 34.Wang J., Wang W., Case D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Mobley D.L., Chodera J.D., Dill K.A. On the use of orientational restraints and symmetry corrections in alchemical free energy calculations. J. Chem. Phys. 2006;125:084902. doi: 10.1063/1.2221683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frisch M.J., Trucks G.W.S., Pople J.A. Gaussian; Wallingford, CT: 2004. Gaussian 03, Revision C.02. [Google Scholar]
- 37.Bayly C.I., Cieplak P., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 38.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 39.Domański J., Stansfeld P.J., Beckstein O. Lipidbook: a public repository for force-field parameters used in membrane simulations. J. Membr. Biol. 2010;236:255–258. doi: 10.1007/s00232-010-9296-8. [DOI] [PubMed] [Google Scholar]
- 40.McCammon M.G., Robinson C.V. Me, my cell, and I: the role of the collision cell in the tandem mass spectrometry of macromolecules. Biotechniques. 2005;39:447–451. doi: 10.2144/05394TE01. 449, 451 passim. [DOI] [PubMed] [Google Scholar]
- 41.van der Spoel D., Marklund E.G., Caleman C. Proteins, lipids, and water in the gas phase. Macromol. Biosci. 2011;11:50–59. doi: 10.1002/mabi.201000291. [DOI] [PubMed] [Google Scholar]
- 42.Abel S., Dupradeau F.Y., Marchi M. Molecular simulations of dodecyl-β-maltoside micelles in water: influence of the headgroup conformation and force field parameters. J. Phys. Chem. B. 2011;115:487–499. doi: 10.1021/jp109545v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Golovina E.A., Golovin A., Faller R. Water replacement hypothesis in atomic details: effect of trehalose on the structure of single dehydrated POPC bilayers. Langmuir. 2010;26:11118–11126. doi: 10.1021/la100891x. [DOI] [PubMed] [Google Scholar]
- 44.Horta B.A.C., Perić-Hassler L., Hünenberger P.H. Interaction of the disaccharides trehalose and gentiobiose with lipid bilayers: a comparative molecular dynamics study. J. Mol. Graph. Model. 2010;29:331–346. doi: 10.1016/j.jmgm.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Barrera N.P., Isaacson S.C., Robinson C.V. Mass spectrometry of membrane transporters reveals subunit stoichiometry and interactions. Nat. Methods. 2009;6:585–587. doi: 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newstead S., Ferrandon S., Iwata S. Rationalizing α-helical membrane protein crystallization. Protein Sci. 2008;17:466–472. doi: 10.1110/ps.073263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arora A., Rinehart D., Tamm L.K. Refolded outer membrane protein A of Escherichia coli forms ion channels with two conductance states in planar lipid bilayers. J. Biol. Chem. 2000;275:1594–1600. doi: 10.1074/jbc.275.3.1594. [DOI] [PubMed] [Google Scholar]
- 48.Ahadi E., Konermann L. Ejection of solvated ions from electrosprayed methanol/water nanodroplets studied by molecular dynamics simulations. J. Am. Chem. Soc. 2011;133:9354–9363. doi: 10.1021/ja111492s. [DOI] [PubMed] [Google Scholar]
- 49.Marklund E.G., Larsson D.S.D., Caleman C. Structural stability of electrosprayed proteins: temperature and hydration effects. Phys. Chem. Chem. Phys. 2009;11:8069–8078. doi: 10.1039/b903846a. [DOI] [PubMed] [Google Scholar]
- 50.Daub C.D., Cann N.M. How are completely desolvated ions produced in electrospray ionization: insights from molecular dynamics simulations. Anal. Chem. 2011;83:8372–8376. doi: 10.1021/ac202103p. [DOI] [PubMed] [Google Scholar]
- 51.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.