

Abstract
Improving our understanding of the mechanisms and effects of anesthetics is a critically important part of neuroscience. The currently dominant theory is that anesthetics and similar molecules act by binding to Cys-loop receptors in the postsynaptic terminal of nerve cells and potentiate or inhibit their function. Although structures for some of the most important mammalian channels have still not been determined, a number of important results have been derived from work on homologous cationic channels in bacteria. However, partly due to the lack of a nervous system in bacteria, there are a number of questions about how these results relate to higher organisms. The recent determination of a structure of the eukaryotic chloride channel, GluCl, is an important step toward accurate modeling of mammalian channels, because it is more similar in function to human Cys-loop receptors such as GABAAR or GlyR. One potential issue with using GluCl to model other receptors is the presence of the large ligand ivermectin (IVM) positioned between all five subunits. Here, we have performed a series of microsecond molecular simulations to study how the dynamics and structure of GluCl change in the presence versus absence of IVM. When the ligand is removed, subunits move at least 2 Å closer to each other compared to simulations with IVM bound. In addition, the pore radius shrinks to 1.2 Å, all of which appears to support a model where IVM binding between subunits stabilizes an open state, and that the relaxed nonIVM conformations might be suitable for modeling other channels. Interestingly, the presence of IVM also has an effect on the structure of the important loop C located at the neurotransmitter-binding pocket, which might help shed light on its partial agonist behavior.
Introduction
Pentameric ligand-gated ion channels (pLGICs) constitute an important superfamily of ion channels, both for their paramount role in the central nervous system and because they interact with many allosteric ligands. These properties make them interesting targets for the pharmaceutical industry. Surprisingly, pLGICs appear to be of prokaryotic origin (1), despite the lack of nervous system in these organisms. In eukaryotes, pLGICs are located in the postsynaptic terminal of nerve cells and are responsible for fast neurotransmission. Metazoan pLGICs, unlike their prokaryotic homologs, exhibit a disulfide bridge stabilizing a loop near the extracellular domain (ECD), and are therefore often referred to as Cys-loop receptors. Two early low-resolution structures of torpedo nicotinic acetylcholine receptor were reported by Unwin and co-workers using cryo-electron microscopy (2,1). In addition, there are now x-ray structures of pLGICs for prokaryotic channels; ELIC (closed) (4) and GLIC (open) (5,6). GLIC, in particular, has been an exceptionally good pLGIC testbed for experiments, models, and simulations. All pLGICs subunits share a common topology with four helical segments forming a transmembrane domain (TMD), connected to an extracellular beta sandwich domain (ECD). Five such subunits form a channel, with the second transmembrane helix (M2) in each subunit lining the pore. The neurotransmitter-binding site is located in the ECD, between subunits (7). It is surrounded by loops A-B-C from one (principal) subunit and loops D-E-F from its neighbor (auxiliary) subunit. Loop C is located at the principal side and isolates the neurotransmitter-binding pocket from the surroundings. It is a short loop, including small portions of the β9 and β10 strands. Previous studies have reported conformational changes of loop C upon binding of agonists. When an agonist is bound to the neurotransmitter pocket, loop C closes around the ligand, whereas antagonist binding leads to loop C adopting an uncapped conformation and swinging outward (8–11).
One potential problem for homology modeling of LGICs is that the current prokaryotic structures are all cationic channels, similar to the nicotinic acetylcholine receptor, although many of the most important receptors for anesthetics and alcohol research (in particular GABAAR and GlyR) are anionic channels (11–13). The latter have roughly 30% sequence identity to their prokaryotic homologs, but because they have virtually opposite potentiation and inhibition patterns, it is not trivial to model wide ranges of eukaryotic channels based on the prokaryotic structures. To further complicate the situation, drugs such as alcohol or anesthetics somewhat surprisingly do not bind in the neurotransmitter-binding site in the ECD, but act as allosteric modulators that interact with the TMD. At least three binding pockets have been identified in LGICs; one located between subunits in the ECD, and two others located in the TMD, within and between subunits, respectively (12). Not all channels exhibit all these sites and recent results support the idea of separate potentiating and inhibitory sites (14,15), but also point to important differences between prokaryotic and eukaryotic channel modulation (16).
Until recently, no x-ray structures were available for any eukaryotic LGIC, but this changed with the GluClα structure (17). In theory, this structure should facilitate studies of human channels such as GABAAR and GlyR, because GluClα is both eukaryotic and anionic. However, the GluClα channel in the x-ray structure is stabilized in the open state by the bound ligand ivermectin (IVM). IVM is a widely used drug against parasites and arthropods; it is a semisynthetic macrocyclic lactone, derived from Streptomyces avemitilis. IVM binds to GABAAR and GlyR at high micromolar concentrations (18,19) and to GluCl at nanomolar concentrations (20). As revealed in the crystal structure, IVM appears to act by binding in the intersubunit pocket in the TMD. It locks the channel in a state that facilitates further activation by ligands like glutamate and is therefore considered a partial agonist. However, the large size of IVM also means the molecule is likely to affect the relative positions of subunits and their interactions, which could complicate homology modeling of other channels such as GABAAR or GlyR based on GluCl.
Molecular dynamic studies, in particular, can address this issue and may even be able to generate a stable model without any ligands attached. Therefore, we have used microsecond molecular dynamics simulations to compare the behavior of GluCl in the absence versus presence of IVM. As proposed by Hibbs et al. (17), binding of IVM does indeed increase the intersubunit distance and stabilizes the channel in an open conformation. Furthermore, it appears to induce new conformational changes at the loops located at the neurotransmitter-binding pocket, which might help explain its partial agonist behavior. In the absence of IVM, the allosteric modulator site undergoes a rather striking change with subunits becoming closer together, which in turn decreases the pore radius and tends toward a more closed state.
Methods
The Protein Data Bank (PDB) x-ray structure of GluClα cocrystallized with IVM (PDB ID 3RWH) was used. The Amber99SB-ILDN force field was used for the protein (21). Parameters for IVM were generated and added to the force field by determining atom and bond types with Antechamber (22). Point charges were calculated from quantum chemistry; a semiempirical PM3 Hamiltonian was used to find a local energy minimum conformation for the ligand and a Hartree-Fock HF/6-31G(d) basis set employed to calculate the electrostatic potential in combination with a self-consistent reaction field (23) and an integral equation formalism using 78.39 (water) as the external dielectric constant with a polarizable continuum model. All such calculations were undertaken with GAUSSIAN 03 (24). The point charges were determined from the electrostatic potential with the RESP model (25).
Structures with and without IVM were prepared independently, and inserted into previously relaxed DOPC (1,2-dioleoyl-sn-3-phosphatidylcholine) lipid bilayer systems modeled with the Berger force field parameters (26). After removal of overlapping lipids each system contained 304 DOPC lipids and ∼32,500 waters in a hexagonal box with a side of 11.2 nm and height of 15.2 nm. 59 Na+ anions and 69 Cl− cations were added to neutralize the system and achieve a realistic salt concentration around 100 mM.
The system without IVM was minimized for 10,000 steps excluding protein-protein nonbonded interactions, and then relaxed in a series of three 5 ns simulations with position restraints (1000 kJ/mol/nm2) applied first to all heavy atoms, and then protein backbone, and finally only Cα atoms.
The system with IVM was minimized similarly for 5000 steps, and then another 10,000 steps including all interactions. To better relax protein-IVM interactions, the initial position restraint simulation was extended to 50 ns (all IVM atoms constrained), followed by 10 ns simulations where the protein backbone and IVM heavy atoms were restrained, and finally 10 ns of Cα atoms restraints (IVM unrestrained).
All simulations were run with Gromacs 4.5, using the LINCS algorithm (27) to constrain all bond lengths and 2–2.5 fs time steps. Particle mesh Ewald electrostatics was used with a 10 Å cutoff. For both systems, protein, water and ions, lipids, as well as IVM were coupled separately to temperature baths of 310 K using the Bussi velocity rescaling thermostat (28) and a constant of τT = 0.1ps. Pressure was adjusted with a semiisotropic Berendsen weak barostat to a pressure of 1 bar with τP = 5 ps and compressibility 4.5 × 10−5 bar−1. No constraints were applied to either the protein or IVM in production runs.
The binding pocket volume was analyzed using Mdpocket from the fpocket suite (29). A reference grid was computed from the crystal structure, using only two subunits for both the extracellular and transmembrane binding pockets. This grid was then superimposed on all five subunits. Mdpocket was run with default parameters except that 5,000 Monte Carlo iterations were used instead of 2,500. The pore radius was computed using the HOLE (30) software on frames extracted every nanosecond and the computed radius was averaged over 100 ns windows. Figures illustrating the pore radius were generated with VMD (31) and the remaining with PyMol (32).
Results
Removal of IVM decreases the intersubunit distances at the EMD-TMD interface
To study the distance between subunits, we monitored the distance between Cα atoms of residues G281 in helix M3 and L218 in helix M1′ for each pair of adjacent subunits (prime notion on segments denote the next subunit). In the GluClα crystal structure (with IVM present) this distance amounts to 9.4 Å, although it is substantially shorter (6.4 Å) for the corresponding atoms in GLIC (17). For the GluCl simulation where IVM was kept between all five subunits, this 9.4 Å distance was maintained throughout the simulation, within statistical errors. However, for the simulation where IVM was removed, the M3-M1′’ distance dropped to ∼7.5 Å, and still appeared to be decreasing after 1 μs. (Fig. 1). These residues (M3-G281 in M1′-L218) are located at the extracellular part of the TMD and directly adjacent to the pocket where IVM binds. To study the spacing of the M3 and M1′ segments further down in the TMD, we calculated the distance between residues L292 and V233. Interestingly, this distance is quite stable at 7.2 Å both in the presence and absence of IVM. It is close to the corresponding value in GLIC (7.6 Å), which indicates the subunit motion is more localized to the extracellular region of the TMD. Removal of IVM not only affects the M3-M1′ distance, but also the distance between M2 and M1′. The distance between residues M2-S260 and M1′-Q219 located at the extracellular part of the TMD exhibited at least one large increase in the presence of IVM, whereas the distance between residues M2-T257 and M1′-L227 was similar in both simulations (Fig. 1).
Figure 1.
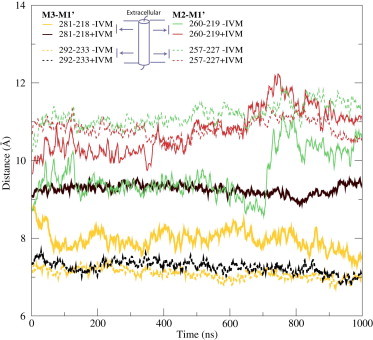
IVM removal brings subunits together. In the absence of IVM (−IVM), the M3 and M1′ helices in adjacent GluCl subunits moved from 9.5 Å to 7.5 Å; a distance close to the TMD-ECD interface (solid thick), compared to a virtually constant distance with IVM (+IVM). The motion was much less pronounced further down in the structure (dashed thick). A similar, but smaller, difference was present between the M2 and M1′ helices (thin lines).
The reduction in intersubunit distances in the absence of IVM was not coupled to corresponding increases in intrasubunit distances; the latter remained stable in both simulations with an exception of the M1-M2 distance close to the TMD-ECD interface (Fig. S1 in the Supporting Material). The secondary structure fluctuations in M2 close to the TMD-ECD interface might have a role in the large M1-M2 distance shifts, both inside and between subunits.
These findings support the conclusion that IVM has a large effect on the stability of the TMD closer to the ECD interface, whereas the rest of the TMD preserves its conformation (Fig. 1, Fig. S1). In addition, the largely constant distances inside subunits suggest that the motion is mainly entire subunits moving closer and inward rather than individual helices changing conformations.
The intersubunit-binding pocket is smaller without IVM
IVM binds to the intersubunit-binding pocket of GluCl, similar to the site where we have previously identified binding sites for anesthetics and alcohols in GlyR (14) and GLIC (15,16). When the IVM ligand is removed before equilibrating the structure, this pocket rapidly shrinks due to occupation by protein side chains and lipid tails. Lipid tails occupy ∼4 out of 5 pockets simultaneously, and 90% of the time, at least one binding pocket is occupied by lipid tails (Fig. S4). The pocket volume in the crystal structure was roughly 700 Å3, after equilibration it dropped to ∼350 Å3, and during the microsecond production simulation it was reduced even further to reach ∼250 Å3 (Fig. 2). The occupation of the binding pocket by the lipids was rather dynamic and no correlation between the volume and the occupancy was observed. The latter volume is close to that estimated for an anesthetic binding site using site-directed mutagenesis (33). This decrease in volume was mainly due to protein backbone motion; in particular, related to the shorter intersubunit distances as described previously. When IVM was kept in the structure, the same binding pocket preserved its initial size throughout the simulation with only minor fluctuations (Fig. 2).
Figure 2.
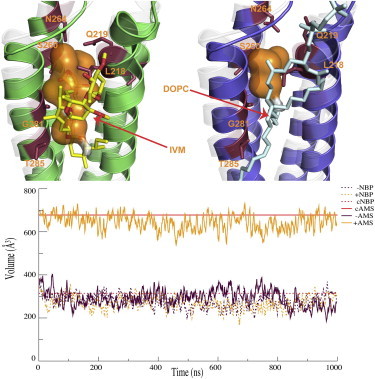
The allosteric site volume drops without IVM. Top: After IVM was removed, side chains and lipid tails occupied the binding pocket, and the helices moved closer together. The upper left panel shows the final structure after 1 μs of simulation with IVM, whereas the right side is the corresponding structure without IVM. The x-ray structure is shown as transparent gray. IVM coordinating residues are shown as sticks. Bottom: The volume of the allosteric modulator site stayed similar to the starting crystal structure (cAMS) with IVM (+AMS) bound, whereas it dropped to less than half its initial size when IVM was removed (−AMS). In contrast, the volume of the neurotransmitter-binding pocket (NBP) was similar to the starting crystal structure (cNBP) with or without IVM present (+NBP,−NBP).
Although there was a clear effect on the allosteric binding pocket, the volume of the neurotransmitter-binding pocket was unaffected and remained around 270 Å3 in simulations both with and without IVM (Fig. 2). Because IVM is a partial agonist, this result could support a mechanism where IVM mainly facilitates (or possibly partially induces) the conformational change in the TMD.
The pore appears to sample more open conformations with IVM
In the GluCl crystal structure, IVM makes 3 H-bonds within each intersubunit cavity, one of which is with S260 (15′ position in the M2 helix). The pore radius in the open GluCl structure is somewhat similar to the presumed-open conformation of GLIC (17). In contrast, the recent locally closed structures of GLIC (34) differ from the open state around the 9′ and 16′ locations in M2, where the pore is in a more closed conformation, similar to the structure of ELIC captured in a closed state (4,6). The presence of IVM in our simulations of GluCl had an effect on the pore conformation starting from 14′ all the way up to the ECD. With IVM present, the channel appeared to explore more open conformations; as open as the GluCl and GLIC crystal structures. On the other hand, in the absence of IVM, the channel rather primarily visits more closed states, but at least in these microsecond-scale simulations the conformations were not quite as closed as the locally closed GLIC structures or ELIC (Fig. S2). Nevertheless, this change clearly supports the assumption that IVM also had an effect on the M2 helices close to the ECD interface by pushing the entire subunits apart and away from the central pore axis to make the channel favor more open conformations.
IVM does not only keep GluCl more open around the 16′ region, but it also causes the channel to be slightly more open at the 9′ region. Both in the simulations with and without IVM, the smallest radius occurred around the 9′ position, where the conformation was as closed as ELIC (Fig. S2). For a small fraction (0.8%) of the simulation with IVM present, the minimum radius shifted to a lower part of the channel (Fig. S5). No such shift was observed without IVM. Partial dehydration or collapse of the pore has been observed in a number of previous simulations of ligand-gated ion channels (35,14,36), but interestingly this behavior too appears to depend on the absence or presence of IVM. In the presence of IVM, the channel still explored more open conformations periodically; after the first 100 ns the minimum radius was ∼2.3 Å. In contrast, when IVM was removed, the minimum radius was only ∼1.7 Å, without any periodic increases. Although the minimum pore radius average was closer in the two simulations (1.4 Å with IVM, 1.2 Å without), their relative difference was found to be statistically significant with a two-tailed Welch t-test. Both the average and tail values of the radius were clearly shifted toward a more open pore due to IVM being bound (Fig. 3).
Figure 3.
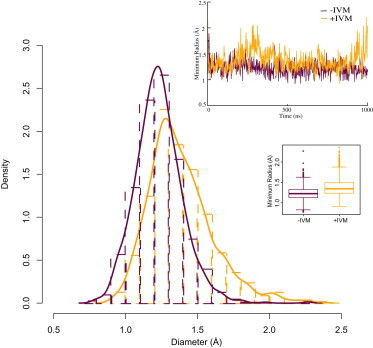
IVM allows the GluCl pore to be more open at 9′. Although the pore is mostly dehydrated after relaxation at the 9′ region, the presence of IVM allowed the channel to sample slightly higher radii, with an average of 1.4 ± 0.2 Å, compared to the simulation without IVM, where the mean was 1.2 ± 0.2 Å. Due to very long correlations, it is not trivial to translate standard deviations to standard errors of means, but using a Welch t-test the difference between the two distributions was found to be statistically significant. The box plot in the inset indicates the first, median, and third quartiles in each box, and whiskers for the smallest/largest values that still are within a factor 1.5 of the box quartiles. Outliers beyond the whiskers are shown as circles.
In the presence of IVM, the channel appeared to fluctuate between an open state and one slightly more closed compared to the open GLIC and GluClα crystal structures. Without IVM bound, the channel moved toward more closed states, although it does not appear to reach as far as the locally closed structures of GLIC at the 16′ region in 1 μs (Fig. 3, Fig. S2).
IVM causes conformational changes in loop C
The partial agonist properties of IVM make it interesting to study its influence on the ECD in more detail. As reported previously, there was no significant effect on the volume of the neurotransmitter-binding pocket volume, but calculation of the root mean-square displacement (RMSD) of Cα coordinates in the ECD indicated higher values at certain time points, e.g., around 250 ns. Average root mean-square fluctuations plotted as a function of residue revealed that the differences in the ECD mainly occurred in the loop regions. These were more flexible in the absence of IVM, and the highest fluctuations were observed for residues from S193 to C202, or loop C.
In both simulations, a small swing-like motion was observed in loop C, where it moved outward from the central channel axis, but there was a difference in the conformations visited in the presence of IVM. In the x-ray structure of GluCl, loop C is relatively well ordered, partly because the antibody used during crystallization binds to this region of the surface. However, without the antibody present, short portions of the β9 and β10 strands that are part of loop C lost their secondary structure and turned into coil. This behavior was first observed in one subunit starting at ∼170 ns (Fig. 4) and this part of the subunit stayed in a disordered state throughout the remainder of the microsecond simulation. Two more subunits started to exhibit gradual fluctuations of the secondary structure in this region, until loop C was finally in a fully disordered state (Fig. 4, top left). In comparison, without IVM present, there were only small motions in loop C (Fig. 4, top right).
Figure 4.
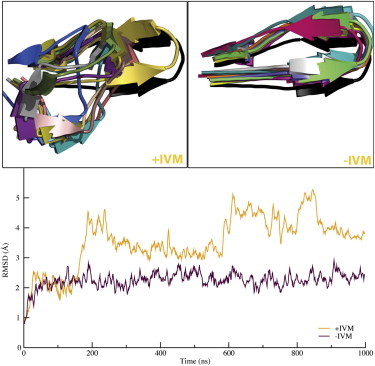
Loop C is more flexible with IVM bound in TMD. Loop C is located next to the neurotransmitter site, almost 40 Å away from the allosteric site. IVM binding still caused loop C to lose beta sheet structure in three subunits (top left), whereas all subunits stayed intact in the absence of IVM (top right). RMSD calculations (bottom) showed that the C loops were mostly rigid throughout the simulations without IVM, but distorted when the ligand was bound. Each color in the two top panels corresponds to a snapshot from each simulation (extracted every 100 ns).
Considering that the original structure was obtained with IVM bound, it is interesting that the removal of this ligand leads to increased rather than reduced ordering in the loop C part of the structure, in particular when this loop surrounds the neurotransmitter-binding pocked. When it loses its ordered structure, loop C exposes a pocket that is more open to the surroundings. This outward motion of Loop C might explain the partial agonist behavior, and why the modulation of GluClα and GluClβ differs (37). That is, the difference in the composition (e.g., hydrophobicity) and length of Loop C in the beta subunit might allow it to be in a disordered state, whereas the alpha subunit would need IVM bound to adopt this state.
IVM disrupts interactions between subunits
The presence of IVM clearly increases the distance between subunits (partly for simple steric reasons), but considering that much smaller molecules, such as ethanol, also act in this region for many channels, it is an interesting question to what extent it affects the interactions between subunits.
The number of H-bonds between subunits increased slightly during the simulation, and there were roughly 1–2 more H-bonds after IVM was removed (Fig. 5). This is partly due to the interactions between the M1′ helix and the adjacent subunit. Side chains of residues L218 to Y221 had fewer H-bonds to the adjacent subunit in the presence of IVM (Fig. S3), and IVM took over some of the disrupted interactions between subunits.
Figure 5.
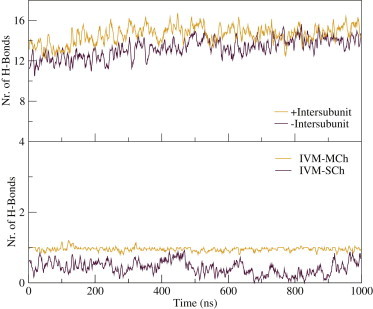
IVM disrupts hydrogen bonds between subunits. Top: The number of hydrogen bonds between each pair of adjacent subunits was 1–2 lower in the presence (black) compared to the absence (red) of IVM. The difference appeared to stabilize at roughly one H-bond after 1 μs of simulation, and was mainly due to interactions with M1 (see Fig. S3). Although the hydrogen bonds between subunits were disrupted, IVM formed new hydrogen bonds with the side chains of residues 219, 260, 264, and 285 (IVM-SCh). The hydrogen bond between the main chain of residue 218 and IVM was conserved throughout the simulation (IVM-MCh).
In the crystal structure, IVM participates in three H-bonds: with the backbone of L218, and the side chains of S260 and T285. The H-bond with the backbone was conserved throughout the simulation, whereas the interactions with the side chains were more dynamic. On average, only one of the H-bonds was fulfilled at any time in the simulation. Residues involved in the H-bonds were located at M1′ (Q219), M2 (S260, N264), and M3 (T285) (Fig. 2). Residues S260 and N264 were competing to interact with IVM. S260 is thought to be important for the activity of the IVM, as GluClβ does not have a serine residue at this position and it is not activated by IVM (37). On the other hand, GlyR and GABAAR have a serine residue at this position and are activated by IVM at micromolar concentrations. GluClβ has an aspargine residue at the same position as residue 264, whereas GlyR and GABAAR have an arginine.
The presence of an amide or acidic residue at position 18′ might be important to facilitate IVM binding, which then would explain the required high concentrations of IVM on GlyR and GABAAAR. The interaction of IVM is weaker with GlyR and GABAAR because there is only one residue for IVM to have an H-bond to (S260), although residue 264 cannot act as an H-bond acceptor when it is an arginine.
No H-bonds were observed to the M2-M3 loop, except short-lived contacts to residue S271, meaning that IVM gets close enough to compete with native protein interactions with the M2-M3 loop. However, the stabilization of the open state might not be due to H-bonds with this particular loop; some other interactions were proposed by Hibbs and Gouaux (17).
Discussion
Although the size of IVM might complicate homology modeling, the strong effect of the ligand makes it a good testbed to understand how the allosteric ligand influences the pore, the binding pocket, the interactions between subunits, and the other structural elements such as loop C. Both the x-ray structure (with IVM) and a conformation where the ligand had been removed were stable over a microsecond of simulation, but there were a number of differences in behavior. In particular, the presence of IVM led to an intersubunit distance of ∼9.4 Å, whereas it was ∼7.5 Å without the ligand bound (Fig. 1). This mechanism is likely to be general, because the IVM binding pocket in the TMD is also one of the sites where anesthetics are thought to bind in GlyR or GABAAR. This intersubunit distance change was limited to the upper part of the TMD, which is the same region exhibiting a significant change between the open and locally closed forms of the GLIC channel (34). A common concern for all models using the GluCl-IVM structure is that the removal of IVM will cause a collapse of the intersubunit pocket and allow M3-M1′ helices to move closer (Fig. 2). This means that homology models would also likely have to go through the same structural change before reaching a more natural conformation for the protein (Fig. 1). Presently, it is difficult to say how severe this would be, e.g., for packing of side chains between subunits in homology models.
The overall RMSD of the TMD did not exhibit any significant differences between the simulations with/without IVM, which supports the hypothesis that the effects are local and easily drown in the fluctuations over five subunits. In contrast, the shrinking intersubunit pocket volume was a direct and clear effect caused by M3-M1′ helices moving closer, lipids entering the pocket, and side-chain reorientation. After 1 μs without IVM, the volume of the pocket was less than half of its initial value and the M3-M1′ helices were 2 Å closer. The distance was still shrinking after 1 μs, and with a longer simulation it might very well reach the same separation as observed in locally closed GLIC (6.5 Å, which requires another 1 Å decrease). Now, on balance, there are also issues with extending simulations to relax the structure without IVM, because this would eventually lead to a completely closed structure rather than an open one (which might not be the target for a homology model).
In this context, it is worth considering that pore dehydration occurs frequently in simulations, which sometimes has been suggested to be channel closure. Several previous studies have reported on a dehydrated pore around the 9′ region, even when open state crystal structures have been used as starting points (14,35,36). One possible explanation could be the use of imperfect protonation states in simulations. Because it is not possible to determine the protonation states from the x-ray structures, protonation states are frequently based simply on pH, which were 7.0 in this study. However, in the case of GLIC there have been quite detailed pKa calculations, and a number of studies with different protonation states have resulted in similar dehydration (35,36,38). This could point to other factors, such as a membrane potential and ionic current through the channel, becoming important to maintain hydration of the open state.
IVM binding has a direct influence on the 9′ region, where it allowed the channel to explore more open conformations both around 9′ and at the extracellular side, above the 15′ region where IVM is bound (Fig. 3 and Fig. S2). Without IVM, the minimum radius of the pore occurred at 9′ with an average of 1.2 ± 0.2 Å (occasionally reaching 1.7 Å), which is even slightly below the ELIC structure. When IVM was present, the minimum radius had an average of 1.4 ± 0.2 Å and periodically exceeded 2 Å. Thus, regardless of hydration or dehydration, the tendency of the channel to visit states that are slightly more open with IVM bound appeared to be important, together with the behavior of the rest of the pore domain.
Somewhat strikingly, the absence or presence of IVM in the allosteric binding site in the TMD appeared to have a significant effect on loop C in the ECD almost 40 Å away, close to the neurotransmitter-binding site. With IVM bound, loop C swung away from the ECD and partly lost its beta sheet secondary structure, which appeared to lead to a more open neurotransmitter-binding pocket. No change was observed in the neurotransmitter binding pocket volume, but a disordered loop C exposed the pocket to the surroundings. This correlates with the findings that loop C has no direct effect on agonist selectivity, but it does affect the efficiency (8–11); after agonist binding, through interactions with the agonist itself, loop C might adopt a more rigid beta hairpin structure and thus help trap the agonist by isolating the cavity from the environment. This agrees with previous models where agonist binding was believed to change loop C from uncapped to capped conformation (10). This type of interaction could potentially also explain how some ligands have double roles as both allosteric modulators and partial agonists (12).
Out of the three H-bonds IVM makes in the x-ray structure, only one (to L218 backbone) was found to be present throughout the simulations. On average, there was always another H-bond formed too, but it alternated between the residues Q219, S260, N264, and T285. This appears to agree with findings of Lynagh et al. (39) who found that GlyR could still be activated by IVM after the mutation S260I. Nevertheless, this result does not rule out the importance of S260, because homomeric GluClβ (where residue 260 is Gln) is not activated by IVM (37). The GluClβ result might also be explained by a valine in position 218, if this side chain affects the backbone flexibility.
In summary, GluClα likely remains one of the best current templates for modeling and simulation of vertebrate ligand-gated ion channels, in particular when the influence of the IVM is taken into account and the structure is allowed to relax without it. In contrast to the prokaryotic GLIC, GluCl is both known to be activated by specific ligands and has a behavior much more similar to GlyR and GABAAR. These attributes should make it possible for future simulations to investigate the entire path from neurotransmitter binding, to ECD influence on the TMD, to allosteric modulator binding, and eventual channel opening in the TMD.
Acknowledgments
This work was supported by grants from the European Research Council (209825), the Swedish Research Council (2010-491,2010-5107), the Foundation for Strategic Research and Swedish e-Science Research Center. Computational resources were provided by the Swedish National Infrastructure for Computing (025/12-32).
Supporting Material
References
- 1.Tasneem A., Iyer L.M., Aravind L. Identification of the prokaryotic ligand-gated ion channels and their implications for the mechanisms and origins of animal Cys-loop ion channels. Genome Biol. 2005;6:R4. doi: 10.1186/gb-2004-6-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unwin N. Nicotinic acetylcholine receptor at 9 A resolution. J. Mol. Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 3.Reference deleted in proof.
- 4.Hilf R.J.C., Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 5.Bocquet N., Nury H., Corringer P.-J. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 6.Hilf R.J., Dutzler R. A prokaryotic perspective on pentameric ligand-gated ion channel structure. Curr. Opin. Struct. Biol. 2009;19:418–424. doi: 10.1016/j.sbi.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Corringer P.-J., Poitevin F., Changeux J.-P. Structure and pharmacology of pentameric receptor channels: from bacteria to brain. Structure. 2012;20:941–956. doi: 10.1016/j.str.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Hansen S.B., Sulzenbacher G., Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celie P.H.N., van Rossum-Fikkert S.E., Sixma T.K. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 10.Velisetty P., Chakrapani S. Desensitization mechanism in prokaryotic ligand-gated ion channel. J. Biol. Chem. 2012;287:18467–18477. doi: 10.1074/jbc.M112.348045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller P.S., Smart T.G. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol. Sci. 2010;31:161–174. doi: 10.1016/j.tips.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Forman S.A., Miller K.W. Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Can. J. Anaesth. 2011;58:191–205. doi: 10.1007/s12630-010-9419-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson A.J., Alqazzaz M., Lummis S.C.R. The pharmacological profile of ELIC, a prokaryotic GABA-gated receptor. Neuropharmacology. 2012;63:761–767. doi: 10.1016/j.neuropharm.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murail S., Wallner B., Lindahl E. Microsecond simulations indicate that ethanol binds between subunits and could stabilize an open-state model of a glycine receptor. Biophys. J. 2011;100:1642–1650. doi: 10.1016/j.bpj.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howard R.J., Murail S., Harris R.A. Structural basis for alcohol modulation of a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. USA. 2011;108:12149–12154. doi: 10.1073/pnas.1104480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murail S., Howard R.J., Lindahl E. Molecular mechanism for the dual alcohol modulation of Cys-loop receptors. PLOS Comput. Biol. 2012;8:e1002710. doi: 10.1371/journal.pcbi.1002710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hibbs R.E., Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shan Q., Haddrill J.L., Lynch J.W. Ivermectin, an unconventional agonist of the glycine receptor chloride channel. J. Biol. Chem. 2001;276:12556–12564. doi: 10.1074/jbc.M011264200. [DOI] [PubMed] [Google Scholar]
- 19.Adelsberger H., Lepier A., Dudel J. Activation of rat recombinant α(1)β(2)γ(2S) GABA(A) receptor by the insecticide ivermectin. Eur. J. Pharmacol. 2000;394:163–170. doi: 10.1016/s0014-2999(00)00164-3. [DOI] [PubMed] [Google Scholar]
- 20.Cully D.F., Vassilatis D.K., Arena J.P. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature. 1994;371:707–711. doi: 10.1038/371707a0. [DOI] [PubMed] [Google Scholar]
- 21.Lindorff-Larsen K., Piana S., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J., Wang W., Case D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Tomasi J., Mennucci B., Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 24.Frisch M.J., Trucks G.W., Pople J.A. Gaussian; Wallingford, CT: 2003. Gaussian 03. [Google Scholar]
- 25.Bayly C.I., Cieplak P., Cornell W. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. (ACS Publications) [Google Scholar]
- 26.Berger O., Edholm O., Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pronk S., Páll S., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 29.Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smart O.S., Neduvelil J.G., Wang X., Wallace B.A., Sansom M.S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996;14:354–360. doi: 10.1016/s0263-7855(97)00009-x. 376. [DOI] [PubMed] [Google Scholar]
- 31.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 32.Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System, Version 1.3r1.
- 33.Jenkins A., Greenblatt E.P., Harrison N.L. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J. Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prevost M.S., Sauguet L., Corringer P.-J. A locally closed conformation of a bacterial pentameric proton-gated ion channel. Nat. Struct. Mol. Biol. 2012;19:642–649. doi: 10.1038/nsmb.2307. [DOI] [PubMed] [Google Scholar]
- 35.LeBard D.N., Hénin J., Brannigan G. General anesthetics predicted to block the GLIC pore with micromolar affinity. PLOS Comput. Biol. 2012;8:e1002532. doi: 10.1371/journal.pcbi.1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng M.H., Coalson R.D. Energetics and ion permeation characteristics in a glutamate-gated chloride (GluCl) receptor channel. J. Phys. Chem. B. 2012;116:13637–13643. doi: 10.1021/jp3074915. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh R., Andersen E.C., Kruglyak L. Natural variation in a chloride channel subunit confers avermectin resistance in C. elegans. Science. 2012;335:574–578. doi: 10.1126/science.1214318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nury H., Poitevin F., Baaden M. One-microsecond molecular dynamics simulation of channel gating in a nicotinic receptor homologue. Proc. Natl. Acad. Sci. USA. 2010;107:6275–6280. doi: 10.1073/pnas.1001832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lynagh T., Webb T.I., Lynch J.W. Molecular determinants of ivermectin sensitivity at the glycine receptor chloride channel. J. Biol. Chem. 2011;286:43913–43924. doi: 10.1074/jbc.M111.262634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.