

Abstract
Ischemia causes AKI as a result of ATP depletion, and rapid recovery of ATP on reperfusion is important to minimize tissue damage. ATP recovery is often delayed, however, because ischemia destroys the mitochondrial cristae membranes required for mitochondrial ATP synthesis. The mitochondria-targeted compound SS-31 accelerates ATP recovery after ischemia and reduces AKI, but its mechanism of action remains unclear. Here, we used a polarity-sensitive fluorescent analog of SS-31 to demonstrate that SS-31 binds with high affinity to cardiolipin, an anionic phospholipid expressed on the inner mitochondrial membrane that is required for cristae formation. In addition, the SS-31/cardiolipin complex inhibited cytochrome c peroxidase activity, which catalyzes cardiolipin peroxidation and results in mitochondrial damage during ischemia, by protecting its heme iron. Pretreatment of rats with SS-31 protected cristae membranes during renal ischemia and prevented mitochondrial swelling. Prompt recovery of ATP on reperfusion led to rapid repair of ATP-dependent processes, such as restoration of the actin cytoskeleton and cell polarity. Rapid recovery of ATP also inhibited apoptosis, protected tubular barrier function, and mitigated renal dysfunction. In conclusion, SS-31, which is currently in clinical trials for ischemia-reperfusion injury, protects mitochondrial cristae by interacting with cardiolipin on the inner mitochondrial membrane.
Ischemic AKI occurs in many clinical settings, including shock, sepsis, and cardiovascular surgery, and it leads to increased mortality in critically ill patients.1 Ischemia-reperfusion injury is also a critical issue in organ transplantation, where it can result in delayed graft function, and is a major risk factor for chronic allograft nephropathy.2,3
Tissue injury occurs during ischemia as a result of ATP depletion. The rapid drop in ATP leads to cytoskeletal changes in tubular epithelial cells, because ATP is required for actin polymerization,4 resulting in breakdown of the brush border, loss of cell–cell contact, disruption of barrier function, and cell detachment.5 These cytoskeletal changes are reversible if the duration of ischemia is brief and ATP recovery occurs rapidly on reperfusion. Mitochondrial function is pivotal to the recovery of ATP in proximal tubular cells, because they have minimal glycolytic capacity and must rely on oxidative phosphorylation for ATP synthesis. However, ATP recovery is often delayed on reperfusion, because ischemia results in loss of cristae membranes and mitochondrial swelling.6,7 The recovery of ATP can be further compromised by mitochondrial permeability transition (MPT) during reperfusion, leading to mitochondrial depolarization and cytochrome c (cyt c) release.8
We recently reported on a compound, SS-31 (also known as MTP-131 or Bendavia), that can accelerate ATP recovery after ischemia and reduces ischemic kidney injury.9 Although SS-31 is known to selectively target the inner mitochondrial membrane (IMM) and inhibit MPT,10 its exact mechanism of action in promoting ATP recovery after ischemia remained elusive.11
Because SS-31 carries a 3+ net charge at physiologic pH, we hypothesized that it might bind to cardiolipin (CL), an anionic phospholipid that is uniquely expressed on the IMM.12 Using a polarity-sensitive fluoroprobe, we show that SS-31 selectively binds to CL by both electrostatic and hydrophobic interactions.
CL domains are required for proper cristae formation,12,13 and they are also involved in the organization of respiratory complexes into supercomplexes to facilitate electron transfer and optimize bioenergetic efficiency.14–17 During ischemia, CL peroxidation causes loss of CL and mitochondrial cristae, which would reduce ATP synthesizing capability on reperfusion.18–20 Furthermore, CL peroxidation releases cyt c from the IMM and promotes its release into the cytosol through both MPT-dependent and -independent mechanisms.21–24
It is now recognized that the oxidation of CL by H2O2 is greatly enhanced in the presence of cyt c.25 Approximately 20% of cyt c is tightly bound to CL by hydrophobic interaction, and the insertion of an acyl chain into cyt c exposes the heme iron (Fe) to hydrogen peroxide (H2O2), thereby converting cyt c into a peroxidase.26–28 In this paper, we show that cyt c peroxidase activity is stimulated by high calcium (Ca2+) and H2O2, conditions commonly found in ischemia-reperfusion injury, and this enhanced activity is inhibited by SS-31. Furthermore, we have evidence that the [SS-31/CL] complex inhibits cyt c peroxidase activity by protecting heme ligation in cyt c. These findings shed new light on the target and mechanism of action of SS-31.
Results
SS-31 Selectively Interacts with Hydrophobic Domain of CL
Previous studies have shown that the SS peptides are cell-permeable and selectively concentrate in the IMM.10,29 We hypothesized that SS-31, with its 3+ net charge, interacts electrostatically with anionic phospholipids on the IMM. We used aladan (ald), a polarity-sensitive fluorescent amino acid, to probe whether SS-31 interacts with phospholipids. Ald increases emission intensity as the polarity of its environment decreases, and its emission maximum (λmax) undergoes a blue shift.30 Ald was incorporated into the peptide sequence of SS-31 (D-Arg-Dmt-Lys-Phe-NH2; Dmt=2’,6’-dimethylTyr) by substituting Ald for Phe (D-Arg-Dmt-Lys-Ald-NH2; [ald]SS-31) (Figure 1A). We first examined the ability of [ald]SS-31 to interact with different anionic phospholipids. CL, phosphatidylglycerol (PG), and phosphatidylserine (PS) all increased fluorescence emission of [ald]SS-31 and caused a leftward shift in λmax (Figure 1B). The effect was greatest with CL. Phosphatidylcholine (PC), phosphatidylethanolamine (PE), and cholesterol did not elicit any changes in the emission spectrum of [ald]SS-31 (Figure 1C), suggesting that [ald]SS-31 only interacts with anionic phospholipids. PS is mostly localized to the inner leaflet of the plasma membrane and constitutes only 1% of the IMM.31 Although [ald]SS-31 interacts with PS in vitro, intracellular localization studies with biotinylated SS-31 in cultured renal epithelial cell showed selective perinuclear mitochondrial distribution with no staining of the plasma membrane (Figure 1D). Thus, the selective partitioning of SS-31 to the IMM is most likely caused by its interaction with CL. The binding of CL to 1 μM [ald]SS-31 is saturable (Figure 1E), with KD being 1.87±0.64 μM. To rule out the possibility that interaction of [ald]SS-31 with CL is an artifact caused by the fluoroprobe, we show that ald itself does not interact with CL (Figure 1F). In addition, SS-31 is able to displace nonyl acridine orange, a fluoroprobe known to selectively bind CL32 (Figure 1G). We also show that the addition of CL dose-dependently alters the intrinsic fluorescence of SS-31 (Figure 1H), suggesting that the incorporation of ald did not significantly alter the ability of SS-31 to bind CL.
Figure 1.

SS-31 interacts selectively with CL. (A) Chemical structures of SS-31 and [ald]SS-31. (B) Representative fluorescence emission spectra of [ald]SS-31 (1 μM, λex=360 nm) in the presence of different anionic phospholipids (3 μM). A shift in emission maximum (λmax) and increase in fluorescence intensity were observed with the addition of CL, PS, and PG but not with PA. (C) Representative fluorescence emission spectra of [ald]SS-31 in the presence of other lipids (3 μM). Chol, cholesterol. There was no shift in λmax with the zwitterionic phospholipids or cholesterol. (D) Intracellular localization of biotinylated SS-31 in feline kidney cells. Biotin was visualized with streptavidin-AlexaFluor594. (E) The interaction of CL with 1 μM [ald]SS-31 is saturable, with KD=1.87±0.64 μM. (F) Representative fluorescence emission spectra of [ald]SS-31 (1 μM) showing a concentration-dependent increase in fluorescence intensity with the addition of CL. (G) SS-31 displaces nonyl acridine orange (NAO) interaction with CL in a competitive manner. (H) Representative fluorescence emission spectra of 10 μM SS-31 (λex=254 nm) with the addition of CL at different CL:SS-31 ratios. RFU, relative fluorescence units.
CL-Induced Cyt c Peroxidase Activity
Cyt c peroxidase activity has been shown in vitro with the addition of CL in the presence of H2O2.26,27 We measured cyt c peroxidase activity using amplex red, which reacts with H2O2 in the presence of a peroxidase to produce a fluorescent product. Incubation of cyt c (2 μM) in the presence of 10 μM H2O2 alone produced very low fluorescence signal (Figure 2A). However, fluorescence increased 34-fold when 30 μM CL was added to cyt c and 10 μM H2O2 (Figure 2A). Some cyt c peroxidase activity was also detected with PS and PG but not PC or PE. Because no other peroxidase was added, these results confirm that cyt c can act as a peroxidase in the presence of CL and H2O2. SS-31 and [ald]SS-31 both inhibited cyt c peroxidase activity (Figure 2B).
Figure 2.

SS-31 inhibits cyt c peroxidase activity. (A) Effects of various phospholipids on inducing cyt c peroxidase activity in vitro. PC, PE, PG, PS, or CL (30 μM) was added to 2 μM cyt c. Peroxidase activity was measured using amplex red fluorescence in the presence of 10 μM H2O2. (B) SS-31 dose-dependently inhibits cyt c peroxidase activity induced by 30 μM CL (EC50=3.5±0.03 μM). (C) Effects of various phospholipids in inducing peroxidase activity in permeabilized mitochondria. (D) SS-31 dose-dependently inhibits CL-induced peroxidase activity in permeabilized mitochondria (EC50=0.8±0.06 μM). The open square represents inhibition of peroxidase activity produced by 10 μM [ald]SS-31, showing that it has comparable activity with the activity of SS-31. (E) Effects of Ca2+ on CL-induced cyt c peroxidase activity. (F) SS-31 dose-dependently inhibits Ca2+-induced cyt c peroxidase activity (EC50=0.8±0.06 μM). All data are represented as mean ± SEM (n=4–8).
We also used permeabilized mitochondria to determine peroxidase activity of endogenous cyt c in mitochondria. Permeabilized mitochondria showed very low peroxidase activity when incubated in 10 μM H2O2 but increased five- to sixfold on the addition of CL (Figure 2C). Actual mitochondrial H2O2 concentration is not known, but it is clear that CL can serve as a catalyst to induce cyt c peroxidase activity in the presence of any H2O2. This mitochondrial peroxidase activity was also inhibited by SS-31 (effective concentration, 50% [EC50]=0.8±0.06 μM), confirming that SS-31 can potently inhibit endogenous cyt c peroxidase (Figure 2D).
During ischemia, the rapid drop in cellular ATP leads to elevation in cytosolic Ca2+ followed by increase in mitochondrial Ca2+. Calcium induces mitochondrial reactive oxygen species (ROS) and CL peroxidation, and it promotes MPT.22,33 We found that Ca2+ directly stimulates cyt c peroxidase activity in a dose-dependent manner (Figure 2E), and SS-31 inhibited Ca2+-induced cyt c peroxidase activity with EC50=0.8±0.06 μM (Figure 2F).
Mechanism by Which SS Peptides Inhibit Cyt c Peroxidase Activity
SS-31 may interfere with cyt c peroxidase activity by preventing the hydrophobic interaction of CL with cyt c. The addition of CL caused the λmax of [ald]SS-31 to shift from 530 to 500 nm (Figure 3A). When cyt c was added to this [SS-31/CL] complex, the fluorescence signal was dramatically quenched, and another shift was seen in the λmax from 500 to 450 nm. The quenching of the fluorescent signal indicates that this [SS-31/CL] complex must be localized very close to the heme, which is a large resonance acceptor. No quenching was observed in the absence of CL. The additional shift of the λmax from 510 to 450 nm indicates that the complex can penetrate deep into a hydrophobic cavity in cyt c. These findings indicate that SS-31 does not prevent the interaction between CL and cyt c.
Figure 3.

SS-31 prevents CL from exposing the heme Fe in cyt c to induce peroxidase activity. (A) Change in emission spectrum of [ald]SS-31 on addition of CL and cyt c. Addition of 6 μM cyt c to [ald]SS-02 (1 μM) and CL (3 μM) causes dramatic quenching and additional shift of λmax from 510 to 450 nm, suggesting that the CL–peptide complex resides in a hydrophobic domain in close proximity to the heme. (B) SS-31 prevents the effect of CL on the negative Cotton peak in Soret spectrum of cyt c. CD was carried out with 10 μM cyt c alone (gray) or in the presence of 20 μM CL (black) or CL plus 10 μM SS-31 (red).
Previous studies suggest that the insertion of a CL acyl chain into the heme crevice of cyt c disrupts the Met80-Fe coordinate and exposes the heme Fe.34,35 Disruption of the Met80-Fe ligation can be monitored by circular dichroism (CD).36,37 The addition of CL to cyt c resulted in the loss of the negative Cotton peak at 419 nm and was prevented by the addition of SS-31 in a 1:1 ratio with cyt c (Figure 3B). Thus, SS-31 protects the Met80-Fe bond and prevents CL from exposing the heme Fe to induce peroxidase activity.
SS-31 Protects Proximal Tubular Mitochondrial Cristae during Ischemia
To determine if SS-31 can inhibit CL peroxidation during ischemia-reperfusion, rats were subjected to bilateral renal ischemia for 30 minutes followed by reperfusion. The animals were divided into three groups, with six rats per group. Group I was a sham-operated control group, group II underwent ischemia-reperfusion with saline treatment, and group III received SS-31 during ischemia-reperfusion. SS-31 (2 mg/kg) or saline was injected subcutaneously 30 minutes before ischemia and again at the onset of reperfusion. SS-31 is rapidly absorbed after subcutaneous injection and selectively concentrates in the kidney (Table 1). Electron microscopic examination of sham-operated kidneys revealed elongated mitochondria with densely packed cristae that are organized along the actin cytoskeletal structure (Figure 4A). After 30 minutes of ischemia, mitochondria in proximal tubules of saline-treated kidneys were dramatically swollen, with a threefold increase in mitochondrial area. Matrix density was drastically reduced, and there was almost complete loss of cristae membranes. In contrast, although mitochondria from SS-31–treated kidneys were rounded, they were not swollen and retained mitochondrial density.
Table 1.
Tissue distribution of 125I-SS-31 after subcutaneous administration to rats
| Tissue | 125I-SS-31 (ng/g or ng/ml) | |||
|---|---|---|---|---|
| 0.5 h | 2 h | 6 h | 16 h | |
| Plasma | 5674±894 | 7819±1860 | 407±190 | 145±46 |
| Kidney | 14,060±1428 | 25,326±3497 | 23,539±4603 | 9068±2617 |
| Heart | 833±228 | 1001±144 | 236±37 | 116±18 |
| Liver | 1897±143 | 4606±583 | 4418±653 | 1296±123 |
| Lung | 2600±442 | 2152±812 | 922±134 | 446±163 |
| Fat | 309±52 | 275±61 | 117±23 | 69±9 |
Data are presented as mean ± SD; n=5 for each time point.
Figure 4.

SS-31 protects mitochondrial cristae during ischemia. Representative electron microscopic images of mitochondria in proximal tubular cell from sham-operated controls and saline- or SS-31–treated kidneys (A) immediately after 30 minutes of ischemia and (B) after 5 minutes of reperfusion. Original magnification, ×20,000 in top panel. The boxed area in the top panel is magnified in the corresponding middle panel. Changes in mitochondrial size and matrix density were quantified by image analysis as described in Concise Methods. Data are presented as mean ± SEM. ***P<0.001.
SS-31 Rapidly Restores Normal Mitochondrial Structure after Ischemia
Within 5 minutes of reperfusion, the SS-31–treated mitochondria began to recover their elongated shape, and densely packed cristae are clearly visible (Figure 4B). Mitochondrial area and matrix density were not different from sham. The mitochondria are once again aligned with the actin cytoskeleton. In contrast, mitochondria in saline-treated kidneys remained swollen, with minor recovery of cristae membranes and matrix density, and substantial recovery of mitochondrial structure was not observed, even after 20 minutes reperfusion (data not shown).
SS-31 Promotes Rapid Recovery of the Brush Border after Ischemia
The loss of intracellular ATP interferes with actin polymerization and causes breakdown of the actin cytoskeleton.4,38,39 The regeneration of the brush border is dependent on the restoration of cellular ATP. The microvilli of proximal tubular epithelial cells are long and well organized in the sham-operated kidneys (Figure 5A). After 30 minutes of ischemia, the brush borders of saline-treated proximal tubules are completely disorganized and retracting into the cytosol (Figure 5B). The microvilli remain completely disrupted, with no evidence of actin bundles in the saline-treated kidneys after 5 minutes of reperfusion (Figure 5C). In contrast, long actin bundles are evident in most proximal tubules from SS-31–treated kidneys (Figure 5, D and E). We also followed the recovery of the brush border with phalloidin staining of F-actin. Compared with saline-treated kidneys, F-actin staining was much better preserved at the luminal surface of proximal tubular epithelial cells in SS-31–treated kidneys, and full recovery of brush border was apparent within 5 minutes of the onset of reperfusion (Figure 5F).
Figure 5.

SS-31 promotes restoration of brush border in proximal tubular cells after reperfusion. Representative electron microscopic images of proximal tubules brush border from (A) sham-operated control, (B) saline-treated animal after 30 minutes of ischemia, (C) saline-treated animal after 30 minutes of ischemia and 5 minutes of reperfusion, and (D) SS-31–treated animal after 30 minutes of ischemia and 5 minutes of reperfusion. (E) Actin filaments (shown by arrows) are seen here in higher magnification from the area selected in D. (F) F-actin staining in proximal tubules of (top panel) saline and (bottom panel) SS-31 kidneys (a) at the end of 30 minutes of ischemia and after (b) 5 minutes of reperfusion and (c) 20 minutes of reperfusion. Original magnification, ×600.
SS-31 Restores Proximal Tubular Cell Polarity within 5 Minutes of Reperfusion
We also looked at cell–cell contact and attachment of proximal tubular cells to the extracellular matrix during ischemia-reperfusion. E-cadherin is an adhesion molecule that is normally localized to the lateral membrane of tubular cells, whereas β1-integrin is localized to the basal membrane and responsible for cell attachment to the extracellular matrix (Figure 6A). Ischemia led to diffuse cytoplasmic redistribution of E-cadherin from the lateral membrane and β1-integrin from the basal membrane (Figure 6B). Within 5 minutes of reperfusion, both adhesion molecules had returned to their proper locations in the SS-31 group (Figure 6D) but not in the saline group (Figure 6C). Full relocalization of E-cadherin to the lateral membrane was not achieved in the saline group until 20 minutes after reperfusion, whereas β1-integrin remained diffuse even after 60 minutes (data not shown).
Figure 6.

SS-31 promotes rapid recovery of actin cytoskeleton and proximal tubular cell polarity after ischemia. Immunohistochemical staining of E-cadherin and β1-integrin in proximal tubular cells (A) in sham-operated controls, (B) after 30 minutes of ischemia, and after 5 minutes of reperfusion with (C) saline or (D) SS-31 treatment. Original magnification, ×600.
SS-31 Prevents Cell Detachment and Apoptosis after Ischemia-Reperfusion
The rapid reorganization of the cytoskeleton by SS-31 prevented detachment of viable tubular cells that is normally observed 24 hours after ischemia-reperfusion injury (Figure 7). There was also a significant reduction in apoptotic cells in the outer medulla as revealed by terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) stain at 24 hours (Figure 8). As a result, SS-31 treatment completely prevented alterations in renal function caused by 30 minutes of ischemia (Table 2).
Figure 7.
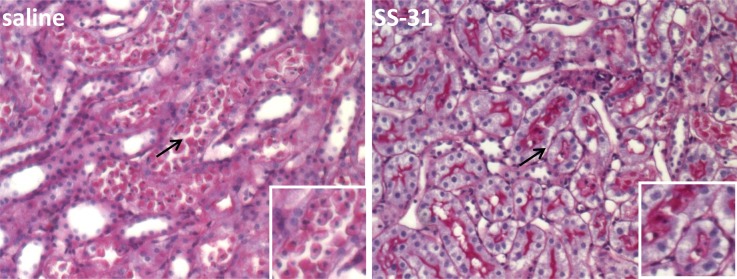
SS-31 prevents tubular epithelial cell detachment caused by 30 minutes of ischemia. Representative periodic acid–Schiff stain showing detachment of viable proximal tubular cells 24 hours after ischemia in saline-treated and SS-31–treated kidneys. Original magnification, ×200. Insets show higher magnifications of areas indicated by the arrows.
Figure 8.

SS-31 prevents tubular cell apoptosis caused by 30 minutes of ischemia. Representative TUNEL staining in the outer medulla 24 hours after ischemia injury from (A) sham, (B) saline-treated, and (C) SS-31–treated kidneys. Original magnification, ×200. TUNEL-positive cells are indicated by arrows. Asterisks show necrotic cells. Apoptotic cells were seen in both proximal (P) and distal (D) tubules. Necrotic cells were seen more often in proximal tubules. SS-31 reduced apoptotic cells in both proximal and distal tubules. Data are represented as mean ± SEM. ***P<0.001.
Table 2.
Renal function 24 hours after ischemia-reperfusion injury
| Biomarker | Sham | Saline | SS-31 |
|---|---|---|---|
| Serum creatinine (mg/dl) | 0.39±0.01 | 1.72±0.55a | 0.62±0.09 |
| BUN (mg/dl) | 13.25±0.59 | 73.17±13.93b | 34.33±7.67 |
| Creatinine clearance (ml/h) | 33.32±2.51 | 10.77±3.39b | 25.08±4.44 |
Data are presented as mean ± SEM (n=6).
P<0.01 compared with sham-operated control.
P<0.001 compared with sham-operated control.
Discussion
Earlier studies have shown that SS-31 is highly effective in minimizing ischemia-reperfusion injury in a number of preclinical models.9,40–44 Although it was known that SS-31 concentrates in the IMM and inhibits MPT,10 its exact mechanism of action remained unclear. Because SS-31 carries a 3+ net charge, we hypothesized that it binds to the anionic phospholipid CL that is uniquely expressed on the IMM. The use of the polarity-sensitive fluoroprobe ald showed that this very polar tetrapeptide can interact with the hydrophobic domain of CL. [ald]SS-31 also interacts in vitro with the other anionic phospholipids, PS and PG, but to a lesser extent compared with CL. There is no interaction with the zwitterionic PE or PC or with cholesterol. This finding suggests that electrostatic interaction plays a role in the partitioning of SS-31 to CL. CL is unique in that it has two phosphate head groups and four acyl chains.12 Figure 9A illustrates how electrostatic interaction between the two basic amino acid residues (Arg and Lys) and the two anionic phosphate head groups on CL aligns the aromatic residues within the hydrophobic acyl chain region. This finding may explain why interaction is stronger for the dimeric CL compared with the monomeric PS and PG. PS is negligible in the IMM, and although PG is an important intermediate for the synthesis of CL, it does not accumulate in mitochondria under normal conditions.12 Thus, we conclude that the selective partitioning of SS-31 to the IMM is primarily caused by its high affinity for CL. Although a number of compounds have been reported to target the mitochondrial matrix in a potential-dependent manner,45 SS-31 is the first compound to selectively target CL on the IMM.
Figure 9.

(A) Proposed model illustrating electrostatic and hydrophobic interactions between (blue) [ald]SS-31 and (red) CL. (B) Proposed model showing how insertion of the acyl chain of CL disrupts the coordination of the heme Fe to Met80. SS peptides form a complex with CL, and the electron-dense aromatic rings help preserve the heme-Met80 coordination.
CL is important for cristae formation and optimal function of the electron transport chain.12–16 It serves to bind cyt c to the IMM to permit efficient electron transfer between complexes III and IV. This binding of cyt c is lost when CL is peroxidized, and CL peroxidation is recognized as the initial step to opening of the MPT pore.21–23 Ischemia and high Ca2+ stimulate CL peroxidation by increasing mitochondrial ROS.19,33,46 We now show that high Ca2+ can also induce CL peroxidation by directly stimulating cyt c peroxidase activity, and this in vitro activity can be inhibited by SS-31. Furthermore, because SS-31 can concentrate in the IMM by ∼5000-fold,29 it can potently inhibit mitochondrial peroxidase activity at 1–10 μM. Thus, SS-31 may inhibit CL peroxidation by two mechanisms—reducing mitochondrial ROS by scavenging H2O210,47,48 and directly inhibiting cyt c peroxidase activity. Additional experiments are required to show direct inhibition of CL peroxidation in vivo. The ability of SS-31 to inhibit CL peroxidation may be the mechanism behind MPT inhibition.10
In its native state, cyt c is tightly folded, and its peroxidase activity is very low, because all six coordination positions in its heme Fe are occupied. Peroxidase activity increases significantly when the protein is partially unfolded or the Met80-Fe bond is weakened or ruptured by exposure to CL.27,34 Our studies suggest that SS-31 prevents CL from breaking the Met80-Fe coordination in cyt c. A proposed model for this mechanism is illustrated in Figure 9B. Unlike other inhibitors of cyt c peroxidase that have been described,49–52 SS-31 is selectively targeted to CL rather than the mitochondrial matrix and does not alter CL composition or the normal interaction between CL and cyt c.
The IMM is organized in two distinct domains: the inner boundary membrane and the cristae membrane that is enriched with all of the respiratory complexes.20 In tubular cells, like in other cells with high metabolic demand, cristae membrane makes up most of the IMM, with deep invaginations that extend into the matrix. CL is required for proper cristae formation, because its unique conical structure favors formation of curvatures rather than bilayers.12 CL peroxidation during ischemia leads to loss of cristae membranes and inhibition of oxidative phosphorylation.53–55 Mitochondrial ATP depletion compromises osmotic regulation in mitochondria and leads to matrix swelling.56 In the present study, 30 minutes of ischemia dramatically reduced cristae membranes, and mitochondrial size increased by more than threefold. Treatment with SS-31 before ischemia prevented mitochondrial swelling and significantly helped to preserve cristae membranes. Because swelling is caused by failure of energy-driven water efflux mechanisms, these results suggest that renal epithelial cells treated with SS-31 managed to produce sufficient ATP during ischemia to maintain fluid regulation in mitochondria.
Reperfusion is associated with slow recovery of cristae membranes in the saline group, although mitochondria remained very swollen, suggesting that ATP synthesis remains compromised. In contrast, the SS-31–treated mitochondria completely recovered cristae membranes within 5 minutes of reperfusion, and this accelerated ATP recovery led to rapid repair of cytoskeletal structure and preservation of tubular integrity. Furthermore, inhibition of CL peroxidation minimized MPT, inhibited apoptosis, and preserved renal function. Although other mitochondria-targeted antioxidants have been reported to minimize ischemia-reperfusion injury,57,58 none have reported protection of mitochondrial structure and ATP production.
Our results show that SS-31 is very effective in preventing acute ischemia-reperfusion injury and suggest that it may be beneficial for conditions where renal ischemia is predictable, such as sepsis, shock, and cardiovascular surgery. SS-31 may also improve graft survival in transplantation, especially with extended criteria donor kidneys. Although SS-31 was given before ischemia in the present study, it has also been shown to reduce infarct size and prevent no reflow when given after myocardial ischemia40,43,59 and after cerebral ischemia in mice.41 SS-31 can even improve kidney outcome after reperfusion after chronic ischemia in a porcine model of renal artery stenosis.44 The validity of CL as a target for therapeutic development is currently being evaluated in a multinational clinical trial with Bendavia for reperfusion injury in patients with acute coronary events (NCT01572909), and a Phase 2 trial was just initiated to assess the effectiveness of Bendavia on improving renal function after angioplasty for severe renal artery stenosis (NCT01755858).
Concise Methods
Chemicals
SS-31 was supplied by Stealth Peptides Inc., Newton Centre, MA. [ald]SS-31 and biotin-SS-31 were synthesized using standard solid-phase peptide synthesis (Dalton Pharma Services, Toronto, Ontario, Canada); 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine,1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-ethanolamine, and 1,2-dipalmitoyl-sn-glycero-3-phospho-(1'-rac-glycerol) were obtained from Avanti Polar Lipids Inc. (Alabaster, AL). CL from bovine heart (consisting primarily of tetralinoleoyl CL) and all other reagents and assay kits were purchased from Sigma (St. Louis, MO).
Synthesis of Ald
Ald was synthesized from commercially available 6-methoxy-2-acetonaphthone by a modification of known literature procedures. We found 2-iodo-1-[6-(dimethylamino)naphthalen-2-yl]ethanone, an intermediate in the original procedure,30 to be both light and thermally sensitive, making subsequent manipulation difficult. Conversely, 2-bromo-1-[6-(dimethylamino)naphthalen-2-yl]ethanone, an intermediate in another method,60 was much easier to handle and was used as the electrophile in the chiral cinchonidinium bromide-catalyzed enantioselective alkylation of N-(diphenylmethylene)glycine tert-butyl ester. Furthermore, final hydrolysis of the imine and ester functionalities using the reported 6 M HCl suffered from poor yield. Hydrolysis with ethane-1,2-dithiol in trifluoroacetic acid proved to be much more reliable and higher yielding. Addition of the Fmoc (fluorenylmethoxycarbonyl) moiety was accomplished under basic conditions.61 The Fmoc-ald was used for synthesis of [ald]SS-31.
Interaction of SS Peptides with Phospholipids
Interaction of SS-31 with phospholipids was examined by changes in fluorescence spectra of [ald]SS-31, an SS-31 analog that contains the polarity-sensitive fluorophore ald (λex=360 nm; Hitachi F-4500 fluorescence spectrophotometer). All experiments were done in deionized water or 20 mM Hepes (pH 7.4) to optimize electrostatic interactions of peptides with phospholipids. The different solvents used to dissolve the various phospholipids (chloroform, methanol, and ethanol) had negligible effect on the fluorescence spectra. To rule out the possibility that interaction of [ald]SS-31 with CL is an artifact caused by the fluoroprobe, we determined the ability of SS-31 to displace nonyl acridine orange, a fluoroprobe known to selectively bind CL.32 In addition, the interaction of SS-31 with CL was also examined by changes in intrinsic fluorescence of SS-31 caused by its Phe and Tyr residues (λex=257 nm).
Intracellular Localization of SS-31
Intracellular targeting of SS-31 was determined by incubating feline kidney cells, grown in biotin-free media for 24 hours, with biotinylated SS-31 for 1 hour. Cells were then fixed with 4% paraformaldehyde, permeabilized with 0.1% triton, and treated with Streptavidin-AlexaFluor594 for 30 minutes at room temperature. Fluorescence staining was observed by wide-field fluorescent microscope (Nikon Eclipse TE2000-U).
Cyt c Peroxidase Activity Assay
Assessment of cyt c peroxidase activity was achieved using the Amplex Red assay. Amplex Red is a reagent that reacts with H2O2 in a 1:1 stoichiometry to produce highly fluorescent resorufin (λex/λem=570/585 nm) in the presence of a peroxidase. In our assay, cyt c is added in place of horseradish peroxidase. Cyt c (2 μM) was incubated with different phospholipids (30 μM) for 1 minute in 200 μl Hepes (pH 7.5) before the addition of 10 μM H2O2 and 50 μM Amplex Red reagent, and the reaction was allowed to proceed for an additional 5 minutes. The continuous time course data were obtained using a microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA). Horseradish peroxidase (0.001 U/ml) was used as a control.
CD Spectroscopy
CD spectra were collected with an AVIA 62 DS spectrophotometer equipped with a sample temperature controller. CD spectra of the Soret region (370–450 nm) were recorded with 10-mm path-length cells containing 20 mM Hepes (pH 7.4) and 10 μM cyt c in the presence or absence of 30 μg/ml CL and SS-31. The maximum lipid concentration was kept low to avoid spectral distortions caused by excessive light scattering. All measurements were done at 25°C. All spectra were corrected for background, and the final spectrum shown represents the average of at least three experiments.
Animals
The study was performed using male Sprague–Dawley rats (Charles River Laboratories International, Inc., Wilmington, MA) weighing 250–300 g. Animals were housed in a light-controlled room with a 12:12-hour light–dark cycle and allowed free access to water and standard rat chow. Care of the animals before and during the experimental procedures was conducted in accordance with the policies of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All protocols had received prior approval by the Cornell University Institutional Animal Care and Use Committee.
Mitochondrial Peroxidase Activity Assay
Mitochondria were isolated from the kidneys of male Sprague–Dawley rats according to reported methods.9 Mitochondria were either used fresh immediately after isolation or frozen at −80°C for later use. Permeabilized mitochondria were obtained by freezing freshly isolated kidney mitochondria at −80°C for 24 hours and then thawing them on ice. These once-frozen mitochondria were capable of supporting uncoupled respiration in the presence of either succinate or NADH, suggesting that they were permeable to small molecules. Addition of exogenous cyt c did not enhance respiration, indicating that they were not permeable to large proteins. Permeabilized mitochondria (80 μg in 200 μl Hepes, pH 7.5) were incubated with CL (30 μM) for 1 minute before the addition of 10 μM H2O2 and 50 μM Amplex Red reagent, and the reaction was allowed to proceed for another 10 minutes.
Ischemia-Reperfusion Experimental Protocol
Rats were anesthetized with ketamine/xylazine cocktail (90 mg/kg ketamine and 4 mg/kg xylazine). Bilateral renal ischemia was induced by the application of nontraumatic microvascular clamps around both left and right renal pedicles. Ischemia was confirmed by blanching of the kidneys. After 30 minutes of ischemia, the clamps were removed, and reperfusion was confirmed visually. Sham-operated animals were not subjected to ischemia. After ischemia, the animals were allowed to recover in individual metabolic cages with free access to food and water. Animals were randomly assigned to the following groups: sham-operated, ischemia with saline, or ischemia with SS-31 (2.0 mg/kg). Treatment was administered subcutaneously 30 minutes before onset of ischemia and at the onset of reperfusion. Serum and urine samples were collected at 24 hours and stored at −20°C until analysis by the ALX Laboratory at the Animal Medical Center, New York, NY. Kidneys were harvested at different times after ischemia for histopathology by light and electron microscopy.
Distribution of SS-31 in Rats
A 125I-SS-31 solution of 5.27 mg/ml with specific activity of 4808.63 kBq/mg was prepared, diluted, and administered subcutaneously to five rats at a dose of 3 mg/kg. Plasma and tissue levels were determined by reverse-phase HPLC, and γ-radioactivity was detected by a Packard Radiomatic 500 TR Flow Isotope Detector.
Renal Histopathology
Kidney samples were fixed in 4% paraformaldehyde, embedded in paraffin wax, and sectioned to 3-μm thickness. The sections were stained with periodic acid–Schiff and examined by light microscopy (Nikon Eclipse TE2000-U). F-actin was stained with Rhodamine Phalloidin (Invitrogen, Carlsbad, CA). Apoptotic cells were identified by TUNEL labeling using the In Situ Cell Death Detection Kit (Roche, Indianapolis, IN) according to the manufacturer’s instructions, and slides were developed using the DAB Substrate Kit (Vector, Burlingame, CA). The number of apoptotic cells was counted from 10 different fields for each sample and averaged.
Immunohistochemical Staining for E-Cadherin and β1-Integrin
Kidney sections (4 μm) were deparaffinized and rehydrated by xylene, graded alcohol series, and ddH2O. Briefly, sections were blocked in blocking buffer for 30 minutes at room temperature, incubated with anti–E-cadherin or anti–β1-integrin (Abcam, Cambridge, MA) overnight at 4°C, and then, incubated with secondary biotinylated goat anti-rabbit IgG (Vector, Burlingame, CA) conjugated to Streptavidin-AlexaFluor594 (Invitrogen, Carlsbad, CA) for 30 minutes at room temperature. Images were examined by light microscopy (Nikon Eclipse TE2000-U).
Electron Microscopy
Pieces of renal cortical and medullary tissue were fixed in 4% paraformaldehyde, postfixed in 1% osmium tetroxide, dehydrated in graded alcohols, and embedded in Epon. Ultrathin sections (200–400 Å) were cut on nickel grids, stained with uranyl acetate and lead citrate, and examined using a digital electron microscope (JEOL USA JEM-1400). Mitochondrial area and matrix density were quantified using National Institutes of Health Image J. Five sections representing proximal epithelial cells were obtained from each group, and five representative mitochondria from each section were selected for analysis. Each mitochondrion was traced and analyzed for area and pixel density, and the results were averaged.
Statistical Analyses
All results are expressed as mean ± SEM unless otherwise indicated. Statistical analyses were carried out using Prism software (GraphPad Software, Inc., San Diego, CA). Multiple group comparisons were performed using ANOVA followed by a Tukey posthoc test.
Disclosures
The SS peptides described in this article are licensed for commercial research and development to Stealth Peptides Inc., a clinical stage biopharmaceutical company, in which H.H.S. and the Cornell Research Foundation have financial interests. The Research Program in Mitochondrial Therapeutics was established with a gift from Stealth Peptides Inc.
Acknowledgments
We thank Esther Breslow for many helpful discussions. Special thanks to David Eliezer for technical guidance with circular dichroism and Lee Cohen-Gould at the Electron Microscopy and Histology Core Facility at Weill Cornell Medical Center. Finally, we thank all the students who helped with the experiments, including Evan Chang, Yinge Zhao, Christopher Pardee, Maggie Ren, and Felix Rozenberg.
This research was supported, in part, by National Institutes of Health Grants P01-AG001751 and R01-HL101186 and the Research Program in Mitochondrial Therapeutics at Weill Cornell Medical College.
Some of results presented in this manuscript were presented at the American Society of Nephrology meeting, October 30–November 4, 2012, in San Diego, CA.
Footnotes
A.V.B. and S.L. contributed equally to this work.
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Maintaining Mitochondrial Morphology in AKI: Looks Matter,” on pages 1185–1187.
References
- 1.Bagshaw SM, Laupland KB, Doig CJ, Mortis G, Fick GH, Mucenski M, Godinez-Luna T, Svenson LW, Rosenal T: Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: A population-based study. Crit Care 9: R700–R709, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bon D, Chatauret N, Giraud S, Thuillier R, Favreau F, Hauet T: New strategies to optimize kidney recovery and preservation in transplantation. Nat Rev Nephrol 8: 339–347, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Chapman JR, O’Connell PJ, Nankivell BJ: Chronic renal allograft dysfunction. J Am Soc Nephrol 16: 3015–3026, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Atkinson SJ, Hosford MA, Molitoris BA: Mechanism of actin polymerization in cellular ATP depletion. J Biol Chem 279: 5194–5199, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Sharfuddin AA, Molitoris BA: Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 7: 189–200, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Puri PS, Varley KG, Kim SW, Barwinsky J, Cohen M, Dhalla NS: Alterations in energy metabolism and ultrastructure upon reperfusion of the ischemic myocardium after coronary occlusion. Am J Cardiol 36: 234–243, 1975 [PubMed] [Google Scholar]
- 7.Schmiedl A, Schnabel PA, Mall G, Gebhard MM, Hunneman DH, Richter J, Bretschneider HJ: The surface to volume ratio of mitochondria, a suitable parameter for evaluating mitochondrial swelling. Correlations during the course of myocardial global ischaemia. Virchows Arch A Pathol Anat Histopathol 416: 305–315, 1990 [DOI] [PubMed] [Google Scholar]
- 8.Halestrap AP: A pore way to die: The role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans 38: 841–860, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV: Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol 22: 1041–1052, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH: Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279: 34682–34690, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Hall AM: Pores for thought: New strategies to re-energize stressed mitochondria in acute kidney injury. J Am Soc Nephrol 22: 986–989, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Osman C, Voelker DR, Langer T: Making heads or tails of phospholipids in mitochondria. J Cell Biol 192: 7–16, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frey TG, Mannella CA: The internal structure of mitochondria. Trends Biochem Sci 25: 319–324, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Mileykovskaya E, Dowhan W: Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277: 43553–43556, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schägger H: Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278: 52873–52880, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Kiebish MA, Yang K, Sims HF, Jenkins CM, Liu X, Mancuso DJ, Zhao Z, Guan S, Abendschein DR, Han X, Gross RW: Myocardial regulation of lipidomic flux by cardiolipin synthase: Setting the beat for bioenergetic efficiency. J Biol Chem 287: 25086–25097, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claypool SM: Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim Biophys Acta 1788: 2059–2068, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, Turkaly PJ, Hoppel CL: Ischemic injury to mitochondrial electron transport in the aging heart: Damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys 385: 117–128, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Lesnefsky EJ, Minkler P, Hoppel CL: Enhanced modification of cardiolipin during ischemia in the aged heart. J Mol Cell Cardiol 46: 1008–1015, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Vogel F, Bornhövd C, Neupert W, Reichert AS: Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol 175: 237–247, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott M, Zhivotovsky B, Orrenius S: Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ 14: 1243–1247, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Petrosillo G, Ruggiero FM, Pistolese M, Paradies G: Ca2+-induced reactive oxygen species production promotes cytochrome c release from rat liver mitochondria via mitochondrial permeability transition (MPT)-dependent and MPT-independent mechanisms: Role of cardiolipin. J Biol Chem 279: 53103–53108, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G: Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochrome c release. FEBS Lett 580: 6311–6316, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW: Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: Mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem 286: 26334–26343, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiswedel I, Gardemann A, Storch A, Peter D, Schild L: Degradation of phospholipids by oxidative stress—exceptional significance of cardiolipin. Free Radic Res 44: 135–145, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova II, Pacheco AA, Winnica DE, Peterson J, Bayir H, Waldeck DH, Kagan VE: Cardiolipin switch in mitochondria: Shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry 46: 3423–3434, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, Basova LV, Peterson J, Kurnikov IV, Kagan VE: Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 45: 4998–5009, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG: Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1: 223–232, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Zhao K, Luo G, Giannelli S, Szeto HH: Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol 70: 1796–1806, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Cohen BE, McAnaney TB, Park ES, Jan YN, Boxer SG, Jan LY: Probing protein electrostatics with a synthetic fluorescent amino acid. Science 296: 1700–1703, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Leventis PA, Grinstein S: The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 39: 407–427, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Petit JM, Maftah A, Ratinaud MH, Julien R: 10N-nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur J Biochem 209: 267–273, 1992 [DOI] [PubMed] [Google Scholar]
- 33.Paradies G, Petrosillo G, Paradies V, Ruggiero FM: Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 45: 643–650, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Vladimirov YA, Proskurnina EV, Izmailov DY, Novikov AA, Brusnichkin AV, Osipov AN, Kagan VE: Mechanism of activation of cytochrome C peroxidase activity by cardiolipin. Biochemistry (Mosc) 71: 989–997, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Sinibaldi F, Howes BD, Piro MC, Polticelli F, Bombelli C, Ferri T, Coletta M, Smulevich G, Santucci R: Extended cardiolipin anchorage to cytochrome c: A model for protein-mitochondrial membrane binding. J Biol Inorg Chem 15: 689–700, 2010 [DOI] [PubMed] [Google Scholar]
- 36.Sinibaldi F, Fiorucci L, Patriarca A, Lauceri R, Ferri T, Coletta M, Santucci R: Insights into cytochrome c-cardiolipin interaction. Role played by ionic strength. Biochemistry 47: 6928–6935, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Santucci R, Ascoli F: The Soret circular dichroism spectrum as a probe for the heme Fe(III)-Met(80) axial bond in horse cytochrome c. J Inorg Biochem 68: 211–214, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Zuk A, Bonventre JV, Brown D, Matlin KS: Polarity, integrin, and extracellular matrix dynamics in the postischemic rat kidney. Am J Physiol 275: C711–C731, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Schwartz N, Hosford M, Sandoval RM, Wagner MC, Atkinson SJ, Bamburg J, Molitoris BA: Ischemia activates actin depolymerizing factor: Role in proximal tubule microvillar actin alterations. Am J Physiol 276: F544–F551, 1999 [DOI] [PubMed] [Google Scholar]
- 40.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK: Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis 18: 215–220, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT: A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem 282: 4634–4642, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Thomas DA, Stauffer C, Zhao K, Yang H, Sharma VK, Szeto HH, Suthanthiran M: Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J Am Soc Nephrol 18: 213–222, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, Gorman JH, 3rd, Sloan RC, Frasier CR, Watson CA, Bostian PA, Kypson AP, Brown DA: Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J Am Heart Assoc 1: e001644, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eirin A, Li Z, Zhang X, Krier JD, Woollard JR, Zhu XY, Tang H, Herrmann SM, Lerman A, Textor SC, Lerman LO: A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 60: 1242–1249, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Smith RA, Hartley RC, Cochemé HM, Murphy MP: Mitochondrial pharmacology. Trends Pharmacol Sci 33: 341–352, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ: Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294: C460–C466, 2008 [DOI] [PubMed] [Google Scholar]
- 47.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD: Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119: 573–581, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ: Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med 39: 1749–1759, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borisenko GG, Kapralov AA, Tyurin VA, Maeda A, Stoyanovsky DA, Kagan VE: Molecular design of new inhibitors of peroxidase activity of cytochrome c/cardiolipin complexes: Fluorescent oxadiazole-derivatized cardiolipin. Biochemistry 47: 13699–13710, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kagan VE, Bayir A, Bayir H, Stoyanovsky D, Borisenko GG, Tyurina YY, Wipf P, Atkinson J, Greenberger JS, Chapkin RS, Belikova NA: Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: A new strategy in anti-apoptotic drug discovery. Mol Nutr Food Res 53: 104–114, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atkinson J, Kapralov AA, Yanamala N, Tyurina YY, Amoscato AA, Pearce L, Peterson J, Huang Z, Jiang J, Samhan-Arias AK, Maeda A, Feng W, Wasserloos K, Belikova NA, Tyurin VA, Wang H, Fletcher J, Wang Y, Vlasova II, Klein-Seetharaman J, Stoyanovsky DA, Bayîr H, Pitt BR, Epperly MW, Greenberger JS, Kagan VE: A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat Commun 2: 497, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Okonkwo DO, Cheng JP, Alexander H, Clark RS, Kochanek PM, Wipf P, Kagan VE, Bayır H: Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci 15: 1407–1413, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL: Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 280: H2770–H2778, 2001 [DOI] [PubMed] [Google Scholar]
- 54.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM: Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic Biol Med 27: 42–50, 1999 [DOI] [PubMed] [Google Scholar]
- 55.Lesnefsky EJ, Chen Q, Slabe TJ, Stoll MS, Minkler PE, Hassan MO, Tandler B, Hoppel CL: Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: Role of cardiolipin. Am J Physiol Heart Circ Physiol 287: H258–H267, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Kaasik A, Safiulina D, Zharkovsky A, Veksler V: Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol 292: C157–C163, 2007 [DOI] [PubMed] [Google Scholar]
- 57.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA: Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 19: 1088–1095, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Plotnikov EY, Chupyrkina AA, Jankauskas SS, Pevzner IB, Silachev DN, Skulachev VP, Zorov DB: Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim Biophys Acta 1812: 77–86, 2011 [DOI] [PubMed] [Google Scholar]
- 59.Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, Brown DA: Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol 52: 1009–1018, 2012 [DOI] [PubMed] [Google Scholar]
- 60.Nitz M, Mezo AR, Ali MH, Imperiali B: Enantioselective synthesis and application of the highly fluorescent and environment-sensitive amino acid 6-(2-dimethylaminonaphthoyl) alanine (DANA). Chem Commun (Camb) 17: 1912–1913, 2002 [DOI] [PubMed] [Google Scholar]
- 61.Chen H-R, Guo X-K, Zhong X-B: Syntheses of aladan and [ald6]ioloatin C and study on their fluorescent properties. Chin J Chem 24: 1411–1417, 2006 [Google Scholar]