

Abstract
Primary vesicoureteral reflux (VUR) is the most common congenital anomaly of the kidney and the urinary tract, and it is a major risk factor for pyelonephritic scarring and CKD in children. Although twin studies support the heritability of VUR, specific genetic causes remain elusive. We performed a sequential genome-wide linkage study and whole-exome sequencing in a family with hereditary VUR. We obtained a significant multipoint parametric logarithm of odds score of 3.3 on chromosome 6p, and whole-exome sequencing identified a deleterious heterozygous mutation (T3257I) in the gene encoding tenascin XB (TNXB in 6p21.3). This mutation segregated with disease in the affected family as well as with a pathogenic G1331R change in another family. Fibroblast cell lines carrying the T3257I mutation exhibited a reduction in both cell motility and phosphorylated focal adhesion kinase expression, suggesting a defect in the focal adhesions that link the cell cytoplasm to the extracellular matrix. Immunohistochemical studies revealed that the human uroepithelial lining of the ureterovesical junction expresses TNXB, suggesting that TNXB may be important for generating tensile forces that close the ureterovesical junction during voiding. Taken together, these results suggest that mutations in TNXB can cause hereditary VUR.
Primary vesicoureteral reflux (PVUR) is the most common type of congenital anomaly of the kidney and the urinary tract (CAKUT) and it is characterized by the retrograde flow of urine from the bladder to the ureter and the kidney.1 Its prevalence in children is estimated to be 1%–2%.1 PVUR is the single most important risk factor for pyelonephritis and renal parenchymal scarring and it is a major cause of ESRD in children.2,3 The pathogenesis of PVUR has not been fully elucidated. Developmentally, PVUR and other CAKUT have been incorporated into and explained by the Mackie-Stephens and Wiegert-Meyer hypotheses underlying developmental abnormalities of the genitourinary tract.4,5 Recent murine distal ureter genetic and developmental modeling studies implicated a significant contribution of disordered bladder trigone and common nephric duct interactions in PVUR development.6–13 Although these theories provide explanations for early events during embryogenesis that are likely to give rise to renal dysplasia and VUR, they do not completely explain the pathogenesis of PVUR. VUR is highly heritable, and although many loci have been found, the specific genes encoded by these loci have remained elusive.10,14–17 This is most likely due to a paucity of large pedigrees with adequate statistical power for genome-wide linkage study (GWLS).
We recently identified a 97-member kindred from the United States with at least 16 affected individuals dating back five generations. We performed sequential GWLSs and whole-exome sequencing (WES) on this family and identified mutations in tenascin XB (TNXB) as a cause of VUR. TNXB mutations have been associated with the joint hypermobility variant of Ehlers-Danlos syndrome.18 Studies have revealed that patients with VUR frequently show structural changes within the ureterovesical junction (UVJ) including replacement of the smooth muscle layer by collagenous matrix, suggesting that some patients with VUR may have defects in the extracellular matrix that supports the UVJ.19 Fibroblast cell lines carrying the T3257I mutation identified in the index family demonstrated reduced cell motility and reduced expression of phosphorylated focal adhesion kinase, signifying a defect in focal adhesion disassembly. We also identified a G1331R mutation in the same gene in another kindred with VUR. These findings bring the role of the extracellular matrix and cell adhesion in the development and function of the UVJ into the forefront.
Results
We identified family 6606, a 97-member kindred from the United States dating back at least five generations (Figure 1A). Nine individuals in this family have radiologically confirmed VUR and/or duplex collecting systems. The voiding cystourethrogram (VCUG) and nuclear dimercaptosuccinic acid (DMSA) renal scan from one of the affected individuals is shown in Figure 1B. This individual has bilateral VUR (grade 4 on the left side) and evidence of reflux nephropathy on DMSA scan based on the appearance of the left kidney that shows a diffuse reduction in uptake and scars. In addition, we identified seven family members with recurrent urinary tract infection (UTI) who did not undergo VCUG testing. The clinical characteristics of the affected individuals are summarized in Table 1. We evaluated the statistical power to detect linkage in this family and a power calculation showed that this single pedigree is capable of generating a maximum attainable estimated logarithm of odds score of 4.88 assuming a dominant genetic model.
Figure 1.
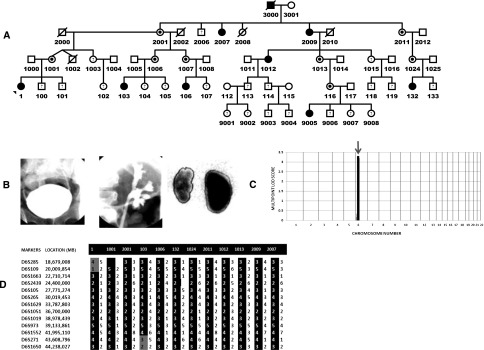
GWLSs in a family with hereditary VUR. (A) Abbreviated pedigree of a 96-member kindred with hereditary VUR. There are at least nine affected individuals with imaging confirmation of VUR/duplex kidney. Squares are male family members and circles are female family members. Obligate heterozygous individuals are shown in partially filled circle/rectangle. (B) Imaging from one of the affected individuals showing grade 4 VUR on the left side. DMSA scan from the same individual showed left renal parenchymal scarring. (C) Genome-wide linkage scan using the Illumina Infinum II HumanLinkage-24 genotyping beadchip assay yields multipoint LOD scores of 3.3 on chromosome 6p in the family. (D) Fine mapping of the chromosome 6p locus and haplotype of individuals defining the chromosome 6P minimal candidate region. Individual 1001 has the ancestral haplotype, the centromeric flanking marker D6S109 is defined by affected individual 1, and telomeric flanking marker D6S271 defined by affected individual 103.
Table 1.
Clinical characteristics of family 6606
| Study Number | Recurrent UTI | VUR/Duplex Collecting System | Reflux Nephropathy |
|---|---|---|---|
| 1 | Yes | VUR | Unknown |
| 103 | Yes | VUR/Duplex | Yes |
| 106 | Yes | VUR | Unknown |
| 132 | Yes | VUR | Unknown |
| 1012 | Yes | Duplex | Unknown |
| 2007 | Yes | Duplex | Unknown |
| 2009 | Yes | Duplex | Unknown |
| 3000 | U | Duplex | Unknown |
| 9005 | Yes | VUR | Unknown |
GWLSs
We obtained a whole-genome multipoint parametric LOD score of 3.3 on chromosome 6p (Figure 1C). This score is in accord for the different penetrance assumptions. This region did not overlap with other loci that have been reported for VUR in the literature (Supplemental Table 1). Haplotype analysis, assuming a dominant model revealed clear evidence that a single disease carrying chromosome segregated with affected individuals in family 6606; the affected haplotype is shown as a black column in Figure 1D.
WES Identifies a Mutation in TNXB
The DNA samples from the proband and affected relative were subjected to WES. We used the following filtering parameters to reduce the number of variants: (1) removal of all variants with minor allele frequency >1%; (2) removal of all variants that are in the dbSNP and 1000 Genome Project; (3) removal of all variants that are found in normal controls; (4) removal of all synonymous variants; and (5) removal of all intronic variants except those with alternate annotations. After applying these parameters genome-wide, 348 potential disease-causing variants were present in at least one individual; however, only 29 of these were present in both individuals. In the linkage region, three variants were present in at least one individual, and all three variants were confirmed by Sanger sequencing. However, only one variant (heterozygous change exon 29. 9770 C>T T3257I in the gene tenascin XB: TNXB) was present in both individuals (Table 2). This variant is present in all affected individuals and all obligate carriers as well as some individuals with recurrent UTI who were classified as unknown for the purpose of linkage analysis. The variant is absent in all unaffected individuals in the family, >800 control chromosomes and also from the publicly available1000 Genome Project dataset (therefore, >2800 control chromosomes). The exons and protein domains of TNXB are shown in Figure 2A. The T3257 amino acid is conserved in evolution (Figure 2, B and C). We directly sequenced all of the coding exons of TNXB and flanking introns in 11 other families with VUR using exon primers (list of primers in Supplemental Table 2). We identified another heterozygous mutation, exon 10. 3991G>A G1331R in a second family, the G1331 amino acid is also conserved in evolution (Figure 2, D and E). We did not find the G1331R mutation in ethnically matched 178 control chromosomes and the change was also absent from the 1000 Genomes Project database (therefore, 2178 control chromosomes). The second kindred is a Caucasian family, and the proband presented with a UTI in the first decade of life and grade 2 reflux on the right side was demonstrated by a VCUG. In addition, her sister also has history of recurrent UTI; however, DNA samples and imaging studies were not available in this individual. The pedigree for the second family and the segregation data are shown in Supplemental Figure 1. Other variants found in the 12 families are listed in Supplemental Table 3.
Table 2.
WES variants in affected individuals in family 6606
| Parameter | 66606/1 | 6606/2009 | Variants Common to Both Individuals |
|---|---|---|---|
| Variants with minor allele frequency <1% | 3996 | 3423 | 630 |
| Variants remaining after filtering | 197 | 151 | 29 |
| Variants in linkage region | 1 | 3 | 1 |
Figure 2.

Missense mutations in TNXB as a cause of VUR. (A) Exons and protein domains of TNXB. (B and C) Missense heterozygous mutation exon 29. 9770 C>T T3257I found in the index family with hereditary VUR; this mutation is conserved in evolution. (D and E) Another kindred with VUR was found to have G1331R mutation in exon 10; this mutation is also conserved in evolution.
In Silico Modeling Reveals Deleterious Missense Mutations
In silico modeling with Polyphen 2 software showed that both variants are damaging with deleterious score of 0.999 of a maximum of 1.20 The T3257I mutation is predicted to be in the linker region between the 23rd and 24th fibronectin type III (FnIII) domains of TNXB (Figure 2A).21,22 Modeling the T3257I mutation in the three-dimensional structure of the protein results in some structural changes in FnIII 24 (Supplemental Figure 2A). The G1331 residue is located in the fifth FnIII domain of TNXB. Modeling the G1331R mutation results in a perturbation of the secondary structure of FnIII domain as predicted by I-TASSER software (Supplemental Figure 2B).21,22
Joint Examination for Hypermobility and Skin Biopsy
Although homozygous TNXB deficiency has been associated with a recessive form of Ehlers-Danlos syndrome that includes large joint dislocations, degenerative joint disease, mitral valve prolapse, and rectal and uterine prolapse, about 50% of patients with heterozygous TNXB mutations have been found to have isolated joint hypermobility.23–25 We therefore screened some of the affected and unaffected individuals in the index family for joint hypermobility. A rheumatologist (C.E.R.) blinded to participants’ affectation status and genotype, performed joint hypermobility tests on two affected and one unaffected members of the family using the Beighton hypermobility score.26 The two affected individuals have significant joint hypermobility with scores of 7 of 9 and 5 of 9, respectively, whereas the unaffected individual has normal joint mobility with a score of 0 of 9. There were no symptoms of skin hyperelasticity or easy bruising and no differences in skin biopsy findings between affected and unaffected individuals as determined by light microscopy and electron microscopy. However, fragmentation of elastic tissue was found in both individuals but was subjectively worse in the affected individual (data not shown).
In Vitro Studies Reveal Reduced Cell Motility and Expression of Phosphorylated Focal Adhesion Kinase in Fibroblast Cell Line with the T3257I Mutation
Because TNXB is composed of multiple fibronectin domains, we hypothesized that the T3257I mutation would alter cell adhesion and cell motility. To test this hypothesis, we performed a standard wound healing assay. In Figure 3A, fibroblasts from an affected patient expressing the mutant TNXB (Mut T3257I) migrated significantly less in response to 50 ng/ml of platelet-derived growth factor (PDGF) compared with the wild-type (WT) cell line. The results of these experiments are quantified in Figure 3B, which shows the mean from three different experiments 42.0 (SD 12) versus 60.3 (SD 15) (P=0.04). The size and electrophoretic migration of TNXB from fibroblast cell culture is similar for both the mutant and the WT cell line (Figure 3C). Because TNXB is known to interact with integrins in order to facilitate transduction of mechanosensory information from the extracellular matrix to the intracellular compartment, we postulated that the observed diminished motility observed in fibroblasts expressing the mutant TNXB may be due to impaired transmission of mechanosensory signals necessary for assembly of focal adhesions.27,28 To test this, we evaluated the phosphorylation/activation of focal adhesion kinase (FAK), a ubiquitously expressed nonreceptor tyrosine kinase that is known to play a central role in focal adhesion disassembly and cell motility in response to integrin stimulation.29
Figure 3.

Reduced cell motility and expression of phosphorylated FAK in a fibroblast cell line of an individual with T3257I mutation. (A) Representative images of WT and mutant dermal fibroblast wound healing assays in the presence of PDGF versus vehicle alone. Note the attenuation of motility in TNXB mutant-expressing fibroblasts (Mut T3257I) in response to PDGF relative to the WT cell line. (B) The results of three individual wound healing assays expressed as percent wound healing are quantified. (C) Representative images of immunoblots from WT and mutant fibroblasts with staining for TNXB, FAK, and pY397-FAK. Note that TNXB expression is similar in all groups and that the PDGF-induced phosphorylation of FAK at tyrosine 397 is significantly attenuated in mutant (Mut T3257I) versus the WT fibroblast cell line.
Phosphorylation of FAK at tyrosine 397 (pY397-FAK) is recognized as an activating post-translational modification site that is stimulated by several growth factors including PDGF.29,30 In Figure 3C, immunoblots of unstimulated and PDGF-treated WT and mutant fibroblasts stained for TNXB, pY397-FAK and total FAK are shown. In WT fibroblasts, pY397-FAK staining in response to PDGF is significantly upregulated relative to baseline. Conversely, in fibroblasts expressing the mutant TNXB, pY397-FAK staining is relatively unchanged from baseline in response to PDGF. These findings suggest that the TNXB mutation may attenuate PDGF-induced fibroblast motility via a mechanism involving impaired transmission of extracellular-to-intracellular signals and decreased FAK activation.
TNXB Is Expressed in the UVJs of Normal and Refluxing Ureters
Immunohistochemical staining and immunofluorescence of normal human UVJs obtained from autopsy samples and UVJs obtained from individuals with radiologically demonstrated VUR undergoing re-implantation was carried out using rabbit anti-human TNXB polyclonal antibody. The patients with VUR who consented to these samples did not undergo mutational screening for TNXB, so their status is unknown. TNXB is expressed predominantly in the transitional epithelial cells of the bladder in both refluxing and nonrefluxing units (Figure 4), and appears to be more strongly expressed in the uroepithelial layer of patients with VUR, consistent with another report that showed upregulation of tenascin C (TNC) in the UVJ of patients with VUR.19 On the basis of gene expression data from the GenitoUrinary Development Molecular Anatomy Project (GUDMAP; http://www.gudmap.org), TNXB is expressed in mice during development and in the postnatal period.31,32 Its relative expression in the ureter is more pronounced compared with that of nephrin a key podocyte gene but similar to uroplakin II, a gene that is enriched in developing lower genitourinary tract (Supplemental Figure 3).
Figure 4.

TNXB is expressed in the UVJ of normal and refluxing ureters. Human UVL sections obtained from normal controls and patients undergoing ureteral re-implantation for radiographically demonstrated PVUR. (A) Immunohistochemistry. Left to right: normal (nonrefluxing ureter) negative control, not stained with rabbit anti-TNXB antibody. There is absence of staining in all layers of the urothelial lining. Second panel: Normal control (nonrefluxing) ureter sample, stained with rabbit anti-TNX antibody (1:50). Note positive (brown) and equal (same intensity) staining throughout all of the urothelium. The third panel represents a refluxing vesicoureteral section of ureter, stained with rabbit anti-TNXB antibody (1:50). Note positive staining of all urothelial layers, but with a stronger intensity staining of the basal layer, compared with the normal control sample in the second panel. (B) Immunofluorescence. Left to right: normal (nonrefluxing ureter) negative control, not stained with rabbit anti-TNXB antibody. There is absence of staining in all layers of the urothelial lining, with nonspecific occasional superficial tissue auto fluorescence. Second panel: Normal control (nonrefluxing) ureter sample, stained with rabbit anti-TNXB antibody (1:50). Note positive (red fluorescence) and equal (same intensity) staining throughout all urothelial lining cell layers. The third panel represents a refluxing vesicoureteral section of ureter, stained with rabbit anti-TNX antibody (1:50). Note positive staining of all urothelial layers, which is brighter in the basal layer compared with the normal control sample in the second panel. Scale bar, μm 200.
Discussion
PVUR remains the most common CAKUT seen in children. Despite the fact that many loci have been reported in the last decade, the genes for PVUR remain elusive. We performed sequential GWLS and WES in a large kindred with VUR and identified heterozygous mutations in TNXB as a cause of dominantly inherited VUR and joint hypermobility. We found another mutation in a family with a history of recurrent UTI and VUR. The two mutations are novel and have never been reported in patients with hypermobility syndrome. Greater than 95% of the TNXB gene encodes multiple fibronectin III domains, thus far, all reported missense mutations in the literature as well as the mutations in this study are in fibronectin III domains of TNXB.25
Tenascins (TNXB, TNC, and TNR) are a family of large extracellular matrix proteins.33,34 They possess a similar structure that is characterized by N-terminal assembly domains, EGF-like repeats, multiple fibronectin III domains and a C-terminal fibrinogen-like domain. In contrast to other fibronectins, the fibronectin domains of most tenascins have antiadhesive properties; thus, during development and cell proliferation they appear to control cell adhesion and migration.33,34 TNXB is expressed throughout development and its expression in the mouse fetus is detected at E15,31,32 coinciding with the time when the UVJ is being formed.35
The role of TNXB mutations in the pathogenesis of VUR is unknown. However, autosomal recessive (AR) TNXB mutations have been associated with Ehlers-Danlos syndrome type III, a condition that is characterized by joint hypermobility, hyperelastic skin, and easy bruising.18,24,25 On the other hand, the phenotype associated with heterozygous mutation in the TNXB gene is variable, with one study reporting that only about 50% of individuals with one single mutation will have joint hypermobility and cutaneous stigmata seen in the AR form of the disease is uniformly absent in these individuals.24,25 We are not aware that any of the members of the family have symptomatic joint hypermobility, but examination of two affected individuals in the family revealed asymptomatic joint hypermobility. Therefore, the finding of T3257I mutation that is segregating with VUR in this study will suggest that VUR/duplex kidney may be the predominant phenotype in individuals with heterozygous mutations in TNXB. Searches of the literature revealed that VUR/duplex kidney have not been previously reported to be associated with either classic AR EDS type III or in individuals with haploinsufficiency or heterozygous missense mutation in TNXB.24,25 Interestingly, a recent study showed that 24% of participants with VUR had generalized joint hypermobility compared with 6.7% of normal controls.36 Other investigators have reported a 3-fold increase in the prevalence of VUR in children with isolated hypermobility compared with those without hypermobility.37 In this study, we identified two missense mutations from two families with VUR and we showed that two affected individuals in the index family have asymptomatic joint hypermobility. Our findings, combined with existing epidemiologic data suggest that mutations in TNXB represent a hitherto unrecognized cause of VUR. In this study, we showed through immunohistochemical and confocal microscopy of UVJ from refluxing and nonrefluxing units in humans that TNXB is abundantly expressed at the UVJ. Furthermore, a review of the publicly available database, GUDMAP, showed that TNXB is expressed in the ureter and bladder of mice on days E15, a period that coincides with the time when the UVJ is being formed.31,32,35 Taken together, these data suggest that TNXB is important in the ontology of the UVJ and development of VUR. The mechanisms by which mutations in TNXB are likely to cause VUR remain unknown. It is possible that the TNXB mutations lead to VUR because of dysfunctional fibronectin domains. Fibroblast cell lines isolated from an individual with the T3257I mutation displayed reduced cell motility in response to PDGF and reduced expression of phosphorylated FAK, suggesting persistent and enhanced cell adhesion. On the basis of these findings, we posit that this may be a gain-of-function mutation. In addition, our results suggest that the mutation may alter integrin-mediated adhesion and dysregulate the extracellular matrix and tensile forces needed to close the vesicoureteric junction as part of the antireflux mechanism. Because TNXB appears to regulate collagen synthesis or deposition,23 an alternative mechanism by which mutations in TNXB may cause VUR is by altering the microenvironment in which smooth muscle cells develop or by affecting the tensile strength of the extracellular matrix that they anchor into, thereby affecting the antireflux mechanisms of the UVJ. One study reported that TNC was present in the musculature and connective tissue of the UVJ from individuals with VUR but was absent from individuals without reflux.19 Future detailed studies of the morphology of the UVJ in individuals with TNXB mutations will shed light on the mechanisms by which defects in TNXB cause VUR. In addition, large-scale screening of patients with VUR for TNXB mutations and joint hypermobility will reveal the role of TNXB in the etiology of primary VUR.
In conclusion, we identified mutations in TNXB as a new cause of PVUR in a large kindred using a sequential linkage analysis and WES approach. These findings emphasize the role of tenascins and other extracellular matrix proteins during genitourinary development.
Concise Methods
Clinical Ascertainment
Institutional review board approval was obtained from Duke University Medical Center (Durham, NC). Study participants were classified as affected, unaffected, and unknown. Family members were considered affected if they had VUR on a VCUG. Supportive evidence included history of recurrent UTI and abnormal findings on renal ultrasonography. Individuals were considered unaffected if there was no detectable VUR on screening VCUG performed as part of routine clinical care or if they were unrelated married individuals in the family. Finally, individuals were considered unknown if they were asymptomatic or if they had a history of UTI but with no radiologic investigations.
Power Analyses
Power analysis for the index family 6606 was performed using SIMLINK to determine statistical power using a rare dominant affecteds-only model with disease allele frequency of 0.01 and markers with 2–4 alleles with frequencies of 0.4, 0.3, 0.2, and 0.1.38
GWLSs and Fine Mapping
A GWLS was performed using the 10,000 single nucleotide polymorphism linkage panel obtained from the Illumina Infinum 2.5 million genotyping beadchip assay (Illumina Inc., San Diego, California). We used an autosomal dominant affecteds-only model and a disease-causing allele frequency of 0.01. We ran simulations assuming the parametric linkage analysis for a dominant model under assumptions of both complete and reduced penetrance. Two-point and multipoint LOD scores were calculated for all single nucleotide polymorphisms using the VITESSE statistical program.39 The region of linkage was confirmed by genotyping informative microsatellites in the identified disease locus and haplotype analysis was carried out as previously described.40
WES
WES was performed on the proband and an affected relative (separated from the proband by two generations) using standard protocols provided by the vendor (Agilent Technologies, Santa Clara, CA). We used the Agilent All Exon 50 MB kit using one lane of a HisEquation 2000 sequencer. The average depth of coverage of the exomes for the two individuals sequenced was 90× with 96% of the targeted capture regions having at least 5× coverage. Reads were aligned to the Human Reference Genome (HG 18) using Burrows-Wheeler Alignment software.41 Single nucleotide variants were called using SAMtools.42 The variants were annotated to Ensembl 50_36l using the SequenceVariantAnalyzer43 and were analyzed using ATAV software (http://www.duke.edu/~minhe/atav/).
Sanger Sequencing
All of the potential disease-causing variants and exons of TNXB were sequenced by the Sanger method. Primer sequences are listed in Supplemental Table 2. All sequences were analyzed with the Sequencher software (Gene Codes Corp, Ann Arbor, MI).
In Silico Prediction of Effect of Amino Acid Substitution
The variants identified in the TNXB gene were scored using Polyphen 2 to examine the predicted damaging effect of the amino acid substitution to the function of TNXB. PolyPhen-2 calculates a naïve Bayes posterior probability that any mutation is damaging and this is represented with a score ranging from 0 to 1.20 The effect of amino acid change on secondary structure of the protein was assessed by the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/).
Joint Examination for Hypermobility
Joint hypermobility variant of Ehlers-Danlos syndrome has previously been associated with TNXB mutations; therefore, joint examinations for hypermobility were carried out in two affected and one unaffected family members using the Beighton hypermobility score.18,25,26 The examiner (C.E.R.) was blinded to the renal phenotype and whole-exome findings.
Skin Biopsy and Staining for Elastic Tissue
Skin biopsies were obtained from one individual with the T3257I mutation and from an unaffected age-matched control. Samples were fixed and stained by routine hematoxylin and eosin stain and Fontana-Masson method for elastic tissue staining. The sections were examined with a Zeiss microscope and by transmission electron microscopy.
Scratch Wound Healing Assay
Fibroblast cell lines were established by culturing skin biopsy samples in DMEM. Cells were cultured on collagen 1– coated plates and allowed to grow to confluence before scratch wound creation. Scratch wounds were created and cells were treated with PDGF (Cell Signaling Inc., Beverly, MA). Fibroblast wound healing images were obtained at 0 and 15 hours using an EVOS microscope and wound healing was quantified as the percent wound closure. Immunoblotting was performed using standard methods. Detailed methods are available in the Supplemental Methods.
Immunohistochemical Staining and Immunofluorescence of the UVJ
Human tissue samples with radiographically confirmed PVUR in paraffin were obtained from the pathology department and Pediatric Renal Biobank at the University of Iowa under approval of institutional board review protocol #200901770. Normal controls were obtained from autopsy specimens. Immunohistochemistry and immunofluorescence were performed on slide sections using standard methods. Slides were incubated with rabbit anti-tenascin-XB antibody (Proteintech Inc, Chicago, IL) overnight, rinsed in PBS and incubated with biotinylated anti-rabbit IgG 1:200 (BA-1000; Vector Labs, Burlingame, CA) and rinsed with PBS. Detailed methods are available in the Supplemental Methods.
Disclosures
None.
Supplementary Material
Acknowledgments
We acknowledge Drs. Russell Hall, Heather Yeowell, and James Bristow for their useful suggestions in the study design. We thank Drs. Elizabeth T. Cirulli, David B. Goldstein, and Kevin V. Shianna and the personnel of the Center for Human Genome Variation for assistance with WES. We also thank the following individuals for the contributions of control samples: Dr. James Burke, Dr. Christine Hulette, Dr. Kathleen Welsh-Bohmer, Dr. Francis J. McMahon, Nirmala Akula, Dr. Julie Hoover-Fong, Dr. Nara L. Sobreira, Dr. David Valle, Dr. M. Chiara Manzini, Dr. Annapurna Poduri, Dr. Nicole Calakos, Mr. David H. Murdock and the Murdock Study Community Registry and Biorepository, Dr. Joseph McEvoy, Dr. Anna Need, Mr. Jordan Silver, Ms. Marlyne Silver, Dr. Eli J. Holtzman, Dr. Gianpiero Cavalleri, Dr. Norman Delanty, Dr. Chantal Depondt, Dr. Sanjay Sisodiya, Dr. William B. Gallentine, Dr. Erin L. Heinzen, Dr. Aatif M. Husain, Ms. Kristen N. Linney, Dr. Mohamad A. Mikati, Dr. Rodney A. Radtke, Dr. Saurabh R. Sinha, Ms. Nicole M. Walley, Dr. Deborah Koltai Attix, Ms. Vicki Dixon, Ms. Jill McEvoy, Dr. Vandana Shashi, Dr. Patricia Lugar, Dr. William L. Lowe, Dr. Scott M. Palmer, Dr. Doug Marchuk, Dr. Deborah Levy, Dr. Zvi Farfel, Dr. Doron Lancet, Dr. Elon Pras, Dr. Yong-Hui Jiang, Dr. Qian Zhao, Dr. Joshua Milner, Dr. Demetre Daskalakis, Mr. Arthur Holden, Dr. Elijah Behr, Dr. Robert H. Brown Jr, Dr. Sarah Kerns, and Dr. Harriet Oster. In addition, we thank the personnel of the Center for Human Genetics core facilities, the University of Iowa Pediatric Renal Biobank and its members, and, most importantly, the family members of the Duke VUR project and University of Iowa Renal Biobank.
This study was supported by grants from the National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (K08DK082495-03 to R.G.) and the American Recovery and Reinvestment Act (ARRA) (RC4DK090937 to P.D.B. and D.H.) and grants from Nephcure foundation (to R.G.). R.G. is the recipient of a Doris Duke Clinical Scientist Development Award. This work was also supported by grants from the Bayden Collins Pediatric Kidney Disease Research Fund, Duke University Medical Center, Duke University School of Medicine core voucher program, and the University of Iowa Pediatric Renal Biobank and sequencing core. In addition, this study was supported in part by the Intramural Research Program of the Center for Research on Genomics and Global Health (CRGGH). The CRGGH is supported by funds from the Office of the Director, the NIDDK, and the National Human Genome Research Institute (Z01HG200362). The sequenced controls used for this study were funded in part by the ARRA (1RC2NS070342-01), the Bryan Alzheimer's Disease Research Centers of the National Institute on Aging (P30AG028377), the National Institutes of Mental Health (RC2MH089915), and the National Institute of Allergy and Infectious Diseases (NIAID) (1R56AI098588-01A1). This research was supported in part by funding from the Division of Intramural Research of the NIAID.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2012121148/-/DCSupplemental.
References
- 1.Arant BS, Jr: Vesicoureteric reflux and renal injury. Am J Kidney Dis 17: 491–511, 1991 [DOI] [PubMed] [Google Scholar]
- 2.Smellie JM, Normand IC: Bacteriuria, reflux, and renal scarring. Arch Dis Child 50: 581–585, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.North American Pediatric Renal Trials and Collaborative Studies: NAPRTCS 2010 Annual Transplant Report, Boston, MA, NAPRTCS, 2010
- 4.Kume T, Deng K, Hogan BL: Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development 127: 1387–1395, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Ichikawa I, Kuwayama F, Pope JC, 4th, Stephens FD, Miyazaki Y: Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int 61: 889–898, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Batourina E, Gim S, Bello N, Shy M, Clagett-Dame M, Srinivas S, Costantini F, Mendelsohn C: Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat Genet 27: 74–78, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Batourina E, Choi C, Paragas N, Bello N, Hensle T, Costantini FD, Schuchardt A, Bacallao RL, Mendelsohn CL: Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat Genet 32: 109–115, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Batourina E, Tsai S, Lambert S, Sprenkle P, Viana R, Dutta S, Hensle T, Wang F, Niederreither K, McMahon AP, Carroll TJ, Mendelsohn CL: Apoptosis induced by vitamin A signaling is crucial for connecting the ureters to the bladder. Nat Genet 37: 1082–1089, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Viana R, Batourina E, Huang H, Dressler GR, Kobayashi A, Behringer RR, Shapiro E, Hensle T, Lambert S, Mendelsohn C: The development of the bladder trigone, the center of the anti-reflux mechanism. Development 134: 3763–3769, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Lu W, van Eerde AM, Fan X, Quintero-Rivera F, Kulkarni S, Ferguson H, Kim HG, Fan Y, Xi Q, Li QG, Sanlaville D, Andrews W, Sundaresan V, Bi W, Yan J, Giltay JC, Wijmenga C, de Jong TP, Feather SA, Woolf AS, Rao Y, Lupski JR, Eccles MR, Quade BJ, Gusella JF, Morton CC, Maas RL: Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet 80: 616–632, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murawski IJ, Watt CL, Gupta IR: Vesico-ureteric reflux: Using mouse models to understand a common congenital urinary tract defect. Pediatr Nephrol 26: 1513–1522, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Hains DS, Sims-Lucas S, Carpenter A, Saha M, Murawski I, Kish K, Gupta I, McHugh K, Bates CM: High incidence of vesicoureteral reflux in mice with Fgfr2 deletion in kidney mesenchyma. J Urol 183: 2077–2084, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper CS, Birusingh KK, Austin JC, Knudson MJ, Brophy PD: Distal ureteral diameter measurement objectively predicts vesicoureteral reflux outcome. J Pediatr Urol 9: 99–103, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Kelly H, Molony CM, Darlow JM, Pirker ME, Yoneda A, Green AJ, Puri P, Barton DE: A genome-wide scan for genes involved in primary vesicoureteric reflux. J Med Genet 44: 710–717, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gimelli S, Caridi G, Beri S, McCracken K, Bocciardi R, Zordan P, Dagnino M, Fiorio P, Murer L, Benetti E, Zuffardi O, Giorda R, Wells JM, Gimelli G, Ghiggeri GM: Mutations in SOX17 are associated with congenital anomalies of the kidney and the urinary tract. Hum Mutat 31: 1352–1359, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feather SA, Malcolm S, Woolf AS, Wright V, Blaydon D, Reid CJ, Flinter FA, Proesmans W, Devriendt K, Carter J, Warwicker P, Goodship TH, Goodship JA: Primary, nonsyndromic vesicoureteric reflux and its nephropathy is genetically heterogeneous, with a locus on chromosome 1. Am J Hum Genet 66: 1420–1425, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weng PL, Sanna-Cherchi S, Hensle T, Shapiro E, Werzberger A, Caridi G, Izzi C, Konka A, Reese AC, Cheng R, Werzberger S, Schlussel RN, Burk RD, Lee JH, Ravazzolo R, Scolari F, Ghiggeri GM, Glassberg K, Gharavi AG: A recessive gene for primary vesicoureteral reflux maps to chromosome 12p11-q13. J Am Soc Nephrol 20: 1633–1640, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J: Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet 17: 104–108, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Schwentner C, Oswald J, Lunacek A, Pelzer AE, Fritsch H, Schlenck B, Karatzas A, Bartsch G, Radmayr C: Extracellular microenvironment and cytokine profile of the ureterovesical junction in children with vesicoureteral reflux. J Urol 180: 694–700, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P: Prediction of deleterious human alleles. Hum Mol Genet 10: 591–597, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Leahy DJ, Hendrickson WA, Aukhil I, Erickson HP: Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science 258: 987–991, 1992 [DOI] [PubMed] [Google Scholar]
- 22.Roy A, Kucukural A, Zhang Y: I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, Rubin EM, Bristow J: Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat Genet 30: 421–425, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J: Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet 73: 214–217, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zweers MC, Dean WB, van Kuppevelt TH, Bristow J, Schalkwijk J: Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin Genet 67: 330–334, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Smits-Engelsman B, Klerks M, Kirby A: Beighton score: A valid measure for generalized hypermobility in children. J Pediatr 158: 119–123, 123, e1–e4, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Midwood KS, Schwarzbauer JE: Tenascin-C modulates matrix contraction via focal adhesion kinase- and Rho-mediated signaling pathways. Mol Biol Cell 13: 3601–3613, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiquet M, Gelman L, Lutz R, Maier S: From mechanotransduction to extracellular matrix gene expression in fibroblasts. Biochim Biophys Acta 1793: 911–920, 2009 [DOI] [PubMed] [Google Scholar]
- 29.Schaller MD, Parsons JT: Focal adhesion kinase and associated proteins. Curr Opin Cell Biol 6: 705–710, 1994 [DOI] [PubMed] [Google Scholar]
- 30.Chen HC, Appeddu PA, Isoda H, Guan JL: Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem 271: 26329–26334, 1996 [DOI] [PubMed] [Google Scholar]
- 31.McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW, Grimmond S, Lessard JL, Little MH, Potter SS, Wilder EL, Zhang P: GUDMAP: The genitourinary developmental molecular anatomy project. J Am Soc Nephrol 19: 667–671, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Harding SD, Armit C, Armstrong J, Brennan J, Cheng Y, Haggarty B, Houghton D, Lloyd-MacGilp S, Pi X, Roochun Y, Sharghi M, Tindal C, McMahon AP, Gottesman B, Little MH, Georgas K, Aronow BJ, Potter SS, Brunskill EW, Southard-Smith EM, Mendelsohn C, Baldock RA, Davies JA, Davidson D: The GUDMAP database – an online resource for genitourinary research. Development 138: 2845–2853, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bristow J, Carey W, Egging D, Schalkwijk J: Tenascin-X, collagen, elastin, and the Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet 139C: 24–30, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Chiquet-Ehrismann R, Tucker RP: Tenascins and the importance of adhesion modulation. Cold Spring Harb Perspect Biol 3: 1–19, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendelsohn C: Using mouse models to understand normal and abnormal urogenital tract development. Organogenesis 5: 306–314, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Eerde AM, Verhoeven VJ, de Jong TP, van de Putte EM, Giltay JC, Engelbert RH: Is joint hypermobility associated with vesico-ureteral reflux? An assessment of 50 patients. BJU Int 109: 1243–1248, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Adib N, Davies K, Grahame R, Woo P, Murray KJ: Joint hypermobility syndrome in childhood. A not so benign multisystem disorder? Rheumatology (Oxford) 44: 744–750, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Boehnke M, Ploughman LM: SIMLINK [computer program]. A program for estimating the power of a proposed linkage study by computer simulation. Version 4.12. Ann Arbor, MI: University of Michigan Center for Statistical Genetics; 1997
- 39.O’Connell JR, Weeks DE: The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet 11: 402–408, 1995 [DOI] [PubMed] [Google Scholar]
- 40.Pericak-Vance MA: Analysis of genetic linkage data for mendelian traits. In: Current Protocols in Human Genetics, edited by Dracopoli NC, Haines JL, Koprk BR, Moir DT, Morton CC, Seidman CE, Seidman JG, Smith DR, New York, John Wiley and Sons, 1997, pp 1.4.1–1.4.30 [Google Scholar]
- 41.Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup : The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ge D, Ruzzo EK, Shianna KV, He M, Pelak K, Heinzen EL, Need AC, Cirulli ET, Maia JM, Dickson SP, Zhu M, Singh A, Allen AS, Goldstein DB: SVA: Software for annotating and visualizing sequenced human genomes. Bioinformatics 27: 1998–2000, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.