

Abstract
Pulmonary hypertension (PH) is a disorder characterized by vascular remodeling and proliferation, a phenotype dependent upon unimpeded growth factor and kinase pathway activation with strong similarities to malignant tumors. This chapter details our novel application of the multikinase inhibitor, sorafenib, in rodent models of PH to improved hemodynamic parameters and attenuates PH structural changes1. Sorafenib is a Raf kinase inhibitor and our biochemical and genomic evidence supported the potential involvement of the MAPK cascade system and TGFB3 in PH development and the response to therapy. Integration of expression genomic analyses coupled with intense bioinformatics identified gene expression and ontology signatures in the development of PH and implicated the role of cytoskeletal protein such as caldesmon or nmMLCK as potentially key participants in PH-induced vascular remodeling and proliferation. Our studies suggest the PKI sorafenib as a potentially novel treatment for severe PH with the MAPK cascade a potential canonical target profoundly effecting vascular cytoskeletal rearrangements and remodeling1.
Keywords: Vascular remodeling, sorafenib, endothelium, cytoskeleton, caldesmon
1 Introduction
Pulmonary hypertension (PH) and cancer pathology share growth factor- and mitogen-activated protein kinase (MAPK) stress-mediated signaling pathways that result in endothelial and smooth muscle cell (SMC) dysfunction and angioproliferative vasculopathy. Protein kinase inhibitors (PKIs) potentially target angiogenic growth factors such as vascular endothelial growth factor receptors (VEGFR-1–3), platelet-derived growth factor receptors (PDGFR-α and -β) as well as key signaling intermediates involved in mediating MAP (mitogen-activated protein) kinase pathway activation such as Raf-1-kinase. We conducted studies with the protein kinase inhibitor (PKI) sorafenib as a potentially novel treatment for severe PH with the MAPK cascade a potential canonical target profoundly effecting vascular cytoskeletal rearrangements and remodeling.1
2 Overview of Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PH) is a lethal syndrome characterized by obstruction of the pulmonary vasculature due to excessive cell proliferation, impaired apoptosis, and vasoconstriction. PH is a disease of small pulmonary arteries (PAs) characterized by intimal hyperplasia, medial hypertrophy, a thickened adventitia, and endothelial proliferative plexiform lesions.2–4 PH occurs in idiopathic and familial forms and is most commonly associated with connective tissue diseases, anorexigen use, HIV, or congenital heart disease. PH typically appears in the third to fifth decade, with increased mortality rates (~50% at 5 years) due to right heart failure as a consequence of increased pulmonary vascular resistance (PVR).2 Elevated PVR reflects reduced cross-sectional area of the vascular bed caused by obstructive vascular remodeling and vasoconstriction. PH is clinically characterized by a progressive increase in pulmonary arterial pressure (PAP) with a mean pressure of greater than 25 mmHg at rest or 30 mmHg during exercise.5 PH can be categorized by the spectrum of its clinical presentation: mild-to-moderate PH, for which PAP is greater than 25 mmHg, or severe PH, for which PAP is greater than 50 mmHg. Severe PH manifests as both an acute and chronic presentation, with acute PH inducing a sudden increase in right ventricle (RV) afterload, with increased end-diastolic volume, and reduced RV ejection fraction.6 Chronic PH leads to progressive RV systolic pressure overload, which leads to dilated RV dysfunction and failure and ultimately death. In 1998, the World Health Organization devised a more clinically useful classification that divides PH into distinct categories, including PH-associated primary pulmonary hypertension (PPH) (familial and sporadic) and PH linked with respiratory system disorders and hypoxemia, among others.5
3 Genetic Studies in Pulmonary Hypertension
The frequency of chronic PH and secondary PH are not known but has been estimated at an annual incidence of 1–2 per million.7 Mutations of the bone morphogenetic protein receptor type 2 gene (BMPR2) have been identified in about 50% of cases of familial PH, with only 20% of individuals with a BMPR2 mutation developing PH. Clearly, other genetic polymorphisms and environmental factors are necessary to initiate the pathological sequence that leads to disease. As external stimuli coupled with undefined genetic susceptibility are likely responsible for the majority of PH cases,5,8–11 this complexity lends itself to the use of high-throughput technologies such as gene microarrays, allowing efficient and accurate simultaneous assessment of the expression of thousands of genes. This technology has been most successfully employed in the investigation of cancer, including the classification of histologically indistinct tumor types with different natural histories.12 Gene microarray strategies permit analysis of the expression profile of lung tissue obtained from patients with PH and the comparison of the gene profile in diseased lungs with that found in the normal lung.9
4 New Concepts in PH Pathophysiology – Neoplastic Vasculopathy
The endothelium is dysfunctional in PH and represents one of the key cell types to be studied. An early proapoptotic endothelial insult may promote PH by damaging normal endothelium, thereby selecting apoptosis-resistant clones that ultimately form characteristic plexiform lesions.4 Drawn from drug discovery studies is the observation that severe PH and cancer pathophysiology share common signal transduction pathways leading to abnormal endothelial cell (EC) and SMC interactions and angioproliferative vasculopathy.4 In primary PH, the lung ECs expand in a monoclonal pattern and contain an inactivating mutation of the transforming growth factor receptor II.13 Severe PH can also present with unique tumorlets of ECs that obliterate medium-sized precapillary arteries. The hyper-proliferating ECs often form structures known as plexiform lesions and express angiogenic factors, including vascular endothelial growth factor (VEGF) and its receptor, VEGF receptor 2 (VEGFR-2, KDR).4 Interestingly, the VEGFR-2 inhibitor known as sugen or SU5416 (SU) has been described to augment PH in combination with chronic hypoxia in the rat model and mimic the precapillary arterial EC proliferation, plexiform lesions, and vascular remodeling and hemodynamic effects of severe PH in humans.4 The vascular changes are not reversible on reoxygenation and ultimately evolve into right heart failure and death.4
5 Receptor Tyrosine Kinase Inhibitors as Novel Therapies for Pulmonary Hypertension
Therapeutic options targeted to specific molecular PH mechanisms are sparse but include epoprostenol (Flolan) and iloprost, both prostocyclin (PGI2) analogues, whereas the mainstay of current therapy consists of the use of a combination of agents, including supplemental oxygen, diuretics, anticoagulants, calcium channel blockers, prostanoids, statins, endothelin receptor antagonists, phosphodiesterase 5 inhibitors, or surgical procedures.6,14,15 Despite these advances, PH remains a devastating disease as most approved therapies are expensive, do not reverse the disease remodeling, and consequently offer only limited benefit to exercise capacity. Thus, there is a strong rationale to consider novel therapies related to pathogenic mechanisms such as tyrosine kinase inhibitors.16
Protein phosphorylation is a major posttranslational modification and regulatory (activation, inhibition) mechanism that controls multiple cell functions (transcription, cell growth, proliferation, differentiation, apoptosis, cell cycle) and is catalyzed by a large family of adenosine triphosphate (ATP) phosphotransferases or protein kinases (PKs), which phosphorylate tyrosine (Tyr), serine (Ser), or threonine (Thr) residues. However, PKs potentially undergo abnormal activity by activating cell growth pathways, leading to tumor development. Given their critical role, PKs are now a wide therapeutic target, and several different receptor tyrosine kinase (RTK) inhibitors have been tested in clinical trials, mostly for cancer, and their use has been expanded to rheumatoid arthritis, cardiovascular diseases, diabetes, and more recently PH.1,6,17
5.1 Protein Kinase Inhibitor Effects on Growth Factors and Angiogenesis
RTKs are cell surface receptors that, on binding to several growth factors, activate a cascade of events that ultimately induce cell growth and proliferation. These growth factors include, among many, epidermal growth factor (EGF), insulin growth factor (IGF), and VEGF. On ligation, RTKs form dimers that activate intracellular PK domains, resulting in PK signaling cascades. For example, the RTK/phosphoinositide 3-kinase (PI3K) pathway activates downstream targets such as pyruvate dehydrogenase kinase-isomerase 1 (PDK-1), protein kinase B (AKT), and activation of the transcription factors IκB and nuclear factor κB (NFκB).15 The use of small-molecule PKIs has now expanded as these molecules are competitive receptor antagonists, thereby inhibiting downstream effects. Currently, only eight small-molecule PKIs are approved in the United States, all for cancer treatment: Gleevec (imitinib mesylate), Iressa (gefitinib), Tarceva (erlotinib HCl), Sutent (sunitibin malate), Sprycel (dastinib), Tykerb (lapatinib ditosylate), Nexavar (sorafenib tosylate), and Tasigna (nilotinib HCl monohydrate). Gleevec, dastinib, and nilotinib are inhibitors of the oncogene BCR (Breakpoint cluster region)–ABL (Abelson murine leukemia viral, oncogene homolog 1) fusion PK in chronic myeloid leukemia, whereas Iressa, Tarceva, and Tykerb are EGF receptor (EGFR) family member inhibitors, and Nexavar or sorafenib is a multikinase inhibitor.15 Sorafenib is an oral multikinase inhibitor of the PKs (protein kinases) PKC (protein kinase C)/Ras (Ras sarcoma), Raf, MEK-1,2(mitogen-activated protein kinase/extracellular signal regulated kinase 1,2), extracellular-regulated kinase (ERK) 1 and 2, and Ets-like transcription factor-1 (Elk-1) signaling pathway involved in tumor angiogenesis that has been approved by the Food and Drug Administration (FDA) for intervention of solid tumors in patients with advanced renal cell carcinoma (RCC) and those with unresectable hepatocellular carcinoma (HCC) and currently is undergoing phase II and III clinical evaluation for such other tumors as melanoma and non-small-cell lung cancer.
Sorafenib, previously known as BAY 43-9006 compound, was originally identified as a Raf kinase inhibitor in vitro18 and subsequently as an inhibitor of ERK phosphorylation but not ERK signaling18–20 in solid tumors. In addition to targeting Raf Ser/Thr kinases, sorafenib also inhibits the Tyr kinase activity of the proangiogenic VEGFR-1, -2, -3 and the platelet-derived growth factor receptor B (PDGFR-B) on human vascular ECs, fibroblasts, and vascular SMCs.21 Thus, sorafenib is now considered a multikinase inhibitor and modifier of tumor growth and progression (Fig. 27.1) with a less-well-elucidated role in apoptosis on tumor endothelium. Downregulation of myeroid cell leukemia-1 protein (Mcl-1) by sorafenib is associated with the release of cytochrome c from mitochondria, caspase activation, and apoptosis, possibly via a MEK/ERK-independent mechanism.19,21 In the clinic, sorafenib demonstrated significant antitumor activity, increasing the medium overall survival,22 and decreased the time to progression in patients with advanced RCC. Overall, sorafenib is well tolerated in patients with solid tumors and even when combined with other chemotherapy.
Fig. 27.1.

Tyrosine kinase (TYK) targeting growth factor and proliferation pathways. Regulation of endothelial cell functions (migration, proliferation, vascular permeability, remodeling) is determined by the activity of protein kinases on the endothelium. Growth factor, angiogenesis/proliferation, and integrin pathways lead to a chain of events that ultimately produce changes in vascular permeability or remodeling. Thus, modulation with therapeutic agents such as statins and sorafenib corresponds to protein tyrosine kinase (PTK), which inhibits the RAS/RAF-1/MAPK pathway, and vascular endothelium growth factor receptor-2 (VEGFR-2) (KDR) blocker prevents vascular remodeling and proliferation. Statins have also been shown to modulate integrin signaling and VEFGR pathways to ultimately affect vascular function
5.2 Sorafenib Effects on Hemodynamic Indices and Vascular Morphology in Rodent PH
Genomic studies for generation of biomarkers still remain to be elucidated for sorafenib in cancer as well as for other sorafenib-responsive entities such as PH. We utilized two preclinical PH rodent models (hypoxia- and hypoxia/SU5416-exposed rats)4 for the study of sorafenib effects on the development of PH. The effect of sorafenib on PA remodeling and RV hemodynamics induced by hypoxia/SU5416 demonstrates significant reductions in PA pressures, RV pressures (Fig. 27.2a, b), and RV hypertrophy (Fig. 27.2c).1 Representative histological observations demonstrated remarkable reductions in medial wall thickening, fibrosis, and luminal obliteration with plexiform-like lesions in hypoxia/SU5416-exposed rat lungs when challenged with sorafenib.1 We further refined the functional roles of sorafenib in lung vascular remodeling by quantifying the levels of both apoptosis (anticleaved caspase 3 antibody) and proliferation (Ki-67 antibody) utilizing tissue microarrays (TMAs) and the Automated Cellular Imaging System (ACIS) apoptosis (Fig. 27.3a). PH-induced apoptosis is mainly localized to the endothelium and the adventitia in the lung vasculature (arrows in Fig. 27.3a). Increased cell proliferation was observed in hypoxia-Sugen-challenged rats compared to normoxia and was evident in all vessel layers with sorafenib-treated rats, demonstrating attenuation of both of these two cellular processes in both PH models1 (Fig. 27.3b).
Fig. 27.2.

Effect of sorafenib on hemodynamic measurements. (a, b) PA and RV systolic pressures, respectively, obtained in Dahl SS rodents at 3.5-week exposure to normoxia (n = 7), hypoxia/SU5416 (H-SU5416, n = 5), or hypoxia/SU5416/sorafenib treatments (H-SU-Sor, n = 5). (c) RV hypertrophy (RVH) measurements across all of the tested conditions represented by the ratio of RV mass divided by the sum of the mass of left ventricle (LV) plus septum (LV + septum). *p < 0.01, **p < 0.001
Fig. 27.3.
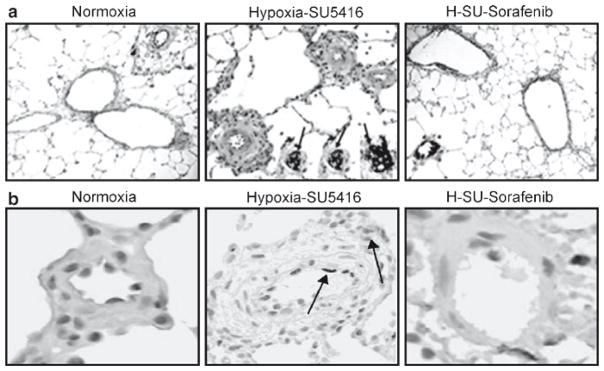
Effect of sorafenib on the development of PH histopathology. Representative images for each group (hematoxylin and eosin staining) with inset (anti-vWF staining) demonstrate that compared to normoxic rats (a), rats exposed to hypoxia/SU5416 showed marked vascular remodeling with medial wall thickening, endothelial cell hyperproliferation, and formation of plexiform lesions with exuberant vWF-positive endothelial cell proliferation (three representative inset arrows). Sorafenib completely prevented the chronic hypoxia-SU5416-induced vascular remodeling. Differences in the level of apoptosis detected by cleaved caspase 3 staining and the level of cell proliferation using Ki67 staining were quantified as brown IOD per 10 μM2 using ACIS. (b) Representative images for apoptosis from each condition. A stepwise increase in staining for apoptosis was demonstrated with hypoxia/SU5416 exposure compared to normoxia. Sorafenib produced dramatic inhibition of apoptosis compared to the hypoxia/SU5416 model, with less attenuation compared to hypoxia. #p < 0.05
6 Role of MAPK Pathway Components in Rodent Pulmonary Hypertension
The MAPK pathway exhibits ubiquitous involvement in cell proliferation and in responses to hemodynamic and environmental stress. Given the putative role of MAPK elements in the development of PH and pharmacodynamics of sorafenib, the posttranslational modification and protein expression levels of phosphorylated and unphosphorylated MEK-1/2 (p-MEK-1/2), ERK, and p38 MAPK (in addition to VEGFR-2) in PH lung homogenates has been studied.1 Levels of p-ERK and p-MEK 1/2 were increased incrementally from hypoxia to hypoxia/SU5416-exposed rats compared to normoxia, while p-p38 showed a similar level of increased posttranslational modification in the two groups. The total amount of MAPK components was not altered compared to normoxia for all three proteins, again consistent with a posttranslational observation. Finally, the reduction in levels of p-VEGFR-2 with hypoxia/SU5416 has been confirmed by the inhibition of VEGFR-2 kinase activity by SU5416. Interestingly, levels of p-VEGFR-2 were increased in hypoxia compared to normoxia but remained downregulated by hypoxia/SU5416/sorafenib exposure, while total VEGFR-2 was not changed significantly throughout the various conditions.
7 Effect of Sorafenib on Lung Gene Expression Profiles in Rodent Pulmonary Hypertension
To study potential mechanisms of sorafenib effects on PH, we integrated bioinformatic analyses of expression profiles that revealed 1,019 transcripts differentially regulated by hypoxia. A comparison between the hypoxia/SU5416 and normoxia groups revealed an additional 465 differentially regulated transcript sets likely involved in the development of the severe PH phenotype. The subsequent comparison of sorafenib treatment in hypoxia/SU5416-exposed rats yielded 38 additional differentially regulated transcripts (Fig. 27.4a) potentially involved in the pathway of sorafenib-mediated PH attenuation. The decrease in the observed list of differentially regulated genes with addition of SU5416 to hypoxia may be explained by a potential drug effect. As a VEGFR inhibitor that disrupts downstream signaling, SU5416 may reduce the expression of numerous genes that are otherwise involved in lung responses to hypoxia and may also explain the potential for an augmented PH phenotype.1
Fig. 27.4.

Heat map and gene ontology (GO) depiction of sorafenib-driven differentially regulated transcripts in the hypoxia-SU5416 pulmonary hypertension rodent model. (a) Microarray analysis of Dahl SS rat lungs (n = 4 chips/group) at 3.5 weeks revealed 38 differentially regulated transcripts between the hypoxia/SU5416 groups with and without sorafenib intervention. These 38 transcripts were assessed for significant directional expression pattern, yielding six genes that appeared across all four conditions. These six genes (Cald1, Pcolce, C1qg, Idi1, Tgfβ3, Ptgis) consistently displayed an increase in expression levels in hypoxia compared to normoxia, with greater expression level in the hypoxia/SU5416 model of PH. In each case, a reduction in gene expression was observed following sorafenib to levels comparable to normoxia. These transcripts were then clustered with a conventional heat map analysis with red blocks representing upregulation and green blocks representing downregulated expression of the relative transcript. (b) The transcript names were manually highlighted to show the significantly enriched GO classes that fell into the six most highly represented general categories and GO superclasses: “Developmental processes,” “muscle development and regulation of muscle contraction,” “defense response/immune system,” “cell proliferation,” “cell differentiation,” and “cellular metabolic processes/metabolism” and apoptosis
To derive the potential biological significance of sorafenib-driven genes, we first identified the significantly overrepresented biological processes in gene ontologies utilizing OntoExpress. While the sorafenib-driven gene profile derived from a comparison between the hypoxia/SU5416 group and hypoxia/SU5416/sorafenib group identified a number of biological process GO terms, a manual examination of these terms revealed six repeatedly represented overarching functional categories: cellular metabolic processes/metabolism (ten genes), developmental processes (seven genes), muscle development and regulation of muscle contraction (six genes), defense response and immune system (five genes), cell differentiation (four genes), and cell proliferation (two genes). These genes are highlighted in the heat map of 38 differentially expressed transcripts between hypoxia/SU5416 and hypoxia/SU5416/sorafenib1 (Fig. 27.4b).
For each of these overarching functional classes observed in the sorafenib-driven genes, we next compared the number of distinct significant biological processes (represented as GO terms) across the two data sets (normoxia vs. hypoxia/SU5416 and hypoxia/SU5416 vs. hypoxia/SU5416/sorafenib). Significance was based on the frequency of these biological process terms representing the differentially expressed genes as compared to those observed for all genes on the microarray chip (adjusted p < 0.05). To compare the relative enrichment of the biological process terms across the interventions, we analyzed the number of GO terms that were identical, related, or unrelated to each other between the different experimental conditions (Fig. 27.4b). The majority of muscle-related and cell proliferation GO terms involved in normoxia versus hypoxia or normoxia versus hypoxia/SU5416 were either related or identical to those affected by the hypoxia/SU5416 versus hypoxia/SU5416/sorafenib set. In addition, for the other four functional GO categories, nearly half of the GO terms could be explained by similar relationships to hypoxia/SU5416 versus hypoxia/SU5416/sorafenib, indicating that sorafenib regulates the majority of biological processes implicated in the hypoxia/SU5416 model of PH. Hierarchical relationships between the significant GO terms, defined as identical or related across all the two group conditions, within the example illustrate how the majority of these terms are related to cell development, immune response, and proliferation of (but not limited to) B lymphocytes, fibroblasts, neuroblasts, or epithelial cells.1
To complement gene ontology (GO) analysis, PubMatrix evaluation (which enumerates associated PubMed citations) was made of a selection of ten microarray-derived sorafenib candidate genes with a pair of genes representative of the most frequently appearing ontologies in our study (Table 27.1). This approach revealed a single previously identified PH candidate gene (Tgfβ3) as well as several potentially novel candidates as defined by the limited or absent number of PubMed citations. Several genes, however, have been reported to be involved in biological pathways that have been linked to PH, as evidenced by the number of citations in search terms such as cell proliferation, cancer, and angiogenesis.
Table 27.1.
PubMatrix evaluation of selected sorafenib-driven genes across PH-related search terms
| Gene name | Gene symbol | Ontology | PH | Proliferation | Hypoxia | Vascular remodeling | Angiogenesis | Cancer |
|---|---|---|---|---|---|---|---|---|
| Actin γ2 | Actg2 | Muscle | 3 | 131 | 3 | 7 | 12 | 180 |
| Tropomyosin 3, γ | Tpm3 | Muscle | 0 | 7 | 1 | 0 | 1 | 33 |
| Complement component 1, q subcomponent, γ polypeptide | C1qg | Defense and immune | 0 | 0 | 0 | 0 | 0 | 1 |
| RT1 class1, CE12 | Rt1-CE12 | Defense and immune | 0 | 0 | 0 | 0 | 0 | 0 |
| Caldesmon 1 | Cald1 | Cell proliferation | 1 | 73 | 7 | 7 | 9 | 212 |
| V-maf musculoaponeurotic fibrosarcoma oncogene homolog | Maf | Cell proliferation | 0 | 95 | 4 | 0 | 8 | 278 |
| TGF-β3 | Tgfb3 | Cell differentiation | 61 | 3,463 | 133 | 129 | 371 | 3,173 |
| Protein tyrosine phosphatase, receptor type R | Ptprr | Cell differentiation | 0 | 13 | 0 | 0 | 2 | 22 |
| 3-Hydroxy-3-methylglutaryl-coenzyme A synthase 1 | Hmgcs1 | Metabolism | 7 | 28 | 8 | 6 | 13 | 32 |
| Isopentenyl-diphosphate δ isomerase | Idi1 | Metabolism | 0 | 3 | 0 | 0 | 0 | 6 |
Given both the ontology and PubMatrix analyses, we next sought to identify a subset of genes that have an increased likelihood of involvement in PH. Each of the 38 transcripts differentially regulated between hypoxia/SU5416 and hypoxia/SU5416/sorafenib groups intersected with those SAM-derived transcripts differentially regulated between normoxia and hypoxia/SU5416 as well as between normoxia and hypoxia. Therefore, the expression of particular genes arising from these three SAM data sets represents a dual screen for PH candidate genes: first in terms of their universal presence in all condition-based comparisons and then as strong statistical significance in expression levels given their SAM-based selection. A directional expression pattern for six genes across all four conditions with the values obtained from the original GCRMA-normalized data for each chip. This pattern is shown in Fig. 27.4a and clearly mirrors both the exacerbating effect of SU5416 on the hypoxic PH phenotype as well as the potent mitigating effect of sorafenib.
8 Comparison of Sorafenib-Modulated Microarray Data Sets with Prior PH Studies
Intersected microarray data with existing previously reported microarray studies of PH evidence the total number of common genes between the two comparison sets derived from our two models of PH and those of four previous published PH studies.9,11,23,24 We observed a total of 57 overlapping hypoxia-driven genes with the Malek et al. report,24 35 common differentially expressed genes with hypoxia/SU5416, and 47 common differentially expressed hypoxia-driven genes in the Gharib et al.9 report, as well as 26 common genes when compared with differentially expressed hypoxia/SU5416-driven rat genes from our study. Comparison with the Geraci et al. human PH report9 revealed five common overlapping genes (Slc25a1, Shmt2, Plcb4, Pts, Fyn) in the hypoxia/SU5416 group. In addition, we also identified two microarray studies11,23 in which we failed to identify any common genes after intersecting the orthologous rat genes and hypoxia/reoxygenation challenge in mice.23 Similarly, we found no overlapping genes with the prior hypoxia versus hypoxia/simvastatin PH rats we previously reported.11 Thus, ortholog approaches are helpful to identify genes that are shared by a significant number of rodent genes differentially expressed in mouse, rat, and human microarray experiments in PH, providing strong validation of the experimental model of our study. Examples of the various comparisons to establish hypoxia and PH candidate genes include bone morphogenetic protein 6, tissue plasminogen activator, and members of the prostaglandin receptor family. Solute carrier family 28 (sodium-coupled nucleoside transporter), member 2 (Slc28a2) was especially unique (identified in three of the five studies), and members of this transporter family were found in all but two total comparisons. Two of the genes highlighted in Fig. 27.4a, a member of the C1q family and isopentenyl-diphosphate delta isomerase (Idi1), are also commonly present in already published multiple comparison sets as well as a gene encoding the cytoskeletal protein caldesmon (Cald1).
9 Caldesmon Studies in Rodent Pulmonary Hypertension (Hypoxia/SU5416-Treated Rats)
Our reports indicated that p38 MAPK-mediated caldesmon (CaD) phosphorylation is involved in endothelial cytoskeleton remodeling and motility.25 In our rodent samples with PH, CaD expression was low in normoxia but significantly elevated in hypoxia-SU5416-treated rats and was reduced by sorafenib treatment.1 Figure 27.5 depicts myosin light chain (MLC)-dependent and -independent pathways in endothelium, where disruption of this vascular barrier integrity (i.e., by thrombin, a known vascular barrier-disruptive agent) activates the endothelial cytoskeleton in dynamic modulation of vascular barrier function. Whereas the EC cytoskeleton is composed of three primary components (actin filaments, intermediate filaments, microtubules),26 the actin cytoskeleton exhibits a primary role in EC barrier regulation. Early observations demonstrated that cytochalasin D, which disrupts the actin cytoskeleton, increased EC permeability, while phallacidin, an actin stabilizer, prevented barrier disruption by various agonists.27,28 Actin filaments interact with myosin to generate EC tensile force that drives cell shape changes and barrier regulation. When this cellular contraction occurs along actin stress fibers that span the cell, gaps form between adjacent cells, and increased paracellular permeability ensues. Thrombin, a vascular barrier-disruptive agent, binds to its receptor PAR-1, triggering the Rho-GTP (guanosine triphosphate) activation and Rho kinase activity, which prevents MLC activation and phosphorylation and actin binding. EC activation activates ERK, which promotes CaD activation via phosphorylation. Thus, controlling the functional role of CaD in binding to actin heads, actin polymerization, and stress fiber formation leads to EC barrier regulation and potentially vascular remodeling characteristic of PH. Our studies indicated that actomyosin cross-linking ability of CaD is essential for binding to microfilaments in ECs, and that this cytoskeletal rearrangement is present in rodent PH remodeling of the vascular endothelium, likely involving CaD phosphorylation (Fig. 27.5).
Fig. 27.5.

Endothelial barrier-regulatory mechanisms: role of MLC and caldesmon. Pulmonary endothelial cells (ECs) are characterized by a thin cortical actin ring with associated cell–cell (adherens junctions and cell–matrix or focal adhesions) connections that provide the structural framework for barrier integrity. Thrombin, a known barrier-disruptive agent, results in rapid recruitment of its receptor (PAR-1) into membrane lipid rafts. For activation of PAR-1 receptor, signaling and subsequent activation of the small guanosine triphos-activation is coupled to Gi phatase (GTPase) Rho. Rho stimulates rapid translocation of the actin-binding protein cortactin to the cell periphery, where it interacts with the barrier regulatory enzyme, myosin light chain kinase (MLCK). Phosphorylation of either of these proteins increases their interaction to actin in favor of stress fiber formation. Diacylglycerol (DAG) activates PKC, which activates Raf/MEK/ERK kinase activity. Erk phosphorylation subsequently facilitates the cytoskeletal caldesmon activation and phosphorylation, a key event for actin binding, polymerization, and fiber formation. This sequence of events occurs within minutes of stimulation and results in dramatically increased endothelial dysfunction. These pathways warrant evaluation of a potential therapeutic intervention with PTK such as sorafenib or statins for pulmonary hypertension
10 New Directions and Conclusions
In summary, we described the successful and novel prophylactic application of the multikinase inhibitor sorafenib in rodent models of PH with improved hemodynamic parameters and attenuation of significant structural PH changes1 consistent with other reports that sorafenib improves hemodynamic parameters in other rodent models of PH.17 Our investigation revealed biochemical and genomic evidence supporting the potential involvement of the MAPK cascade system and transforming growth factor β3 (TGFβ3) in PH development and the response to therapy. Integration of expression genomic analyses coupled with intense bioinformatics identified gene expression and ontology signatures in the development of PH and in the attenuation by sorafenib. Studies of sorafenib as a novel therapy in patients suffering from PH is already a well-tolerated, FDA-approved therapy in cancer; the clinical application of this novel therapy for PH can represent an excellent example of bedside-to-bench-to-bedside translation. Moreover, initial studies implicated the role of cytoskeletal protein such as CaD and MLCK (myosin light chain kinase) as key participants in the cytoskeletal rearrangement present during PH-induced remodeling and proliferation in endothelium. These represent potentially novel molecular targets for altering the devastating clinical course of patients with PH.
Acknowledgments
This work was supported by NIH-NHLBI (JGNG P01 HL58064, RO1 HL090860-01) and K22 (LMV 008308-01) awards.
References
- 1.Moreno-Vinasco L, Gomberg-Maitland M, Maitland ML, et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics. 2008;33:278–291. doi: 10.1152/physiolgenomics.00169.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higenbottam T, Cremona G. Acute and chronic hypoxic pulmonary hypertension. Eur Respir J. 1993;6:1207–1212. [PubMed] [Google Scholar]
- 3.Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol. 2005;98:390–403. doi: 10.1152/japplphysiol.00733.2004. [DOI] [PubMed] [Google Scholar]
- 4.Taraseviciene-Stewart L, Kasahara Y, Alger L, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 5.Raiesdana A, Loscalzo J. Pulmonary arterial hypertension. Ann Med. 2006;38:95–110. doi: 10.1080/07853890600622143. [DOI] [PubMed] [Google Scholar]
- 6.Rhodes CJ, Davidson A, Gibbs JS, Wharton J, Wilkins MR. Therapeutic targets in pulmonary arterial hypertension. Pharmacol Ther. 2009;121:69–88. doi: 10.1016/j.pharmthera.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Adatia I. Recent advances in pulmonary vascular disease. Curr Opin Pediatr. 2002;14:292–297. doi: 10.1097/00008480-200206000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Bull TM, Coldren CD, Nana-Sinkam P, et al. Microarray analysis of peripheral blood cells in pulmonary arterial hypertension, surrogate to biopsy. Chest. 2005;128:584S. doi: 10.1378/chest.128.6_suppl.584S. [DOI] [PubMed] [Google Scholar]
- 9.Geraci MW, Gao B, Hoshikawa Y, Yeager ME, Tuder RM, Voelkel NF. Genomic approaches to research in pulmonary hypertension. Respir Res. 2001;2:210–215. doi: 10.1186/rr59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girgis RE, Li D, Zhan X, et al. Attenuation of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Heart Circ Physiol. 2003;285:H938–H945. doi: 10.1152/ajpheart.01097.2002. [DOI] [PubMed] [Google Scholar]
- 11.Girgis RE, Ma SF, Ye S, et al. Differential gene expression in chronic hypoxic pulmonary hypertension: effect of simvastatin treatment. Chest. 2005;128:579S. doi: 10.1378/chest.128.6_suppl.579S. [DOI] [PubMed] [Google Scholar]
- 12.Bull TM, Coldren CD, Moore M, et al. Gene microarray analysis of peripheral blood cells in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170:911–919. doi: 10.1164/rccm.200312-1686OC. [DOI] [PubMed] [Google Scholar]
- 13.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 14.Archer SL, Michelakis ED. An evidence-based approach to the management of pulmonary arterial hypertension. Curr Opin Cardiol. 2006;21:385–392. doi: 10.1097/01.hco.0000231410.07426.9b. [DOI] [PubMed] [Google Scholar]
- 15.Grant SK. Therapeutic protein kinase inhibitors. Cell Mol Life Sci. 2009;66:1163–1177. doi: 10.1007/s00018-008-8539-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431. doi: 10.1161/CIRCULATIONAHA.104.503540. [DOI] [PubMed] [Google Scholar]
- 17.Klein M, Schermuly RT, Ellinghaus P, et al. Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation. 2008;118:2081–2090. doi: 10.1161/CIRCULATIONAHA.108.779751. [DOI] [PubMed] [Google Scholar]
- 18.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral anti-tumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 19.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280:35217–35227. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 20.Rahmani M, Davis EM, Crabtree TR, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27:5499–5513. doi: 10.1128/MCB.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahmani M, Nguyen TK, Dent P, Grant S. The multikinase inhibitor sorafenib induces apoptosis in highly imatinib mesylate-resistant bcr/abl+ human leukemia cells in association with signal transducer and activator of transcription 5 inhibition and myeloid cell leukemia-1 down-regulation. Mol Pharmacol. 2007;72:788–795. doi: 10.1124/mol.106.033308. [DOI] [PubMed] [Google Scholar]
- 22.Grant S. Cotargeting survival signaling pathways in cancer. J Clin Invest. 2008;118:3003–3006. doi: 10.1172/JCI36898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gharib SA, Luchtel DL, Madtes DK, Glenny RW. Global gene annotation analysis and transcriptional profiling identify key biological modules in hypoxic pulmonary hypertension. Physiol Genomics. 2005;22:14–23. doi: 10.1152/physiolgenomics.00265.2004. [DOI] [PubMed] [Google Scholar]
- 24.Malek RL, Wang HY, Kwitek AE, et al. Physiogenomic resources for rat models of heart, lung and blood disorders. Nat Genet. 2006;38:234–239. doi: 10.1038/ng1693. [DOI] [PubMed] [Google Scholar]
- 25.Mirzapoiazova T, Kolosova IA, Romer L, Garcia JG, Verin AD. The role of caldesmon in the regulation of endothelial cytoskeleton and migration. J Cell Physiol. 2005;203:520–528. doi: 10.1002/jcp.20244. [DOI] [PubMed] [Google Scholar]
- 26.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 27.Phillips PG. Thrombin-induced alterations in endothelial cell cytoarchitectural and functional properties. Semin Thromb Hemost. 1994;20:417–425. doi: 10.1055/s-2007-1001930. [DOI] [PubMed] [Google Scholar]
- 28.Phillips PG, Lum H, Malik AB, Tsan MF. Phallacidin prevents thrombin-induced increases in endothelial permeability to albumin. Am J Physiol. 1989;257:C562–C567. doi: 10.1152/ajpcell.1989.257.3.C562. [DOI] [PubMed] [Google Scholar]