

Abstract
It has been established that oxidative stress, defined as the condition when the sum of free radicals in a cell exceeds the antioxidant capacity of the cell, contributes to the pathogenesis of Parkinson’s disease. Glutathione is a ubiquitous thiol tripeptide that acts alone, or in concert with enzymes within cells to reduce superoxide radicals, hydroxyl radicals and peroxynitrites. In this review, we examine the synthesis, metabolism and functional interactions of glutathione, and discuss how this relates to protection of dopaminergic neurons from oxidative damage and its therapeutic potential in Parkinson’s disease.
Keywords: Glutathione, Glutathione S-transferase, Parkinson’s disease, Oxidative Stress, Substantia nigra
Introduction
Neurons are among the most metabolically active cells in the body, requiring the correct balance of oxygen and glucose to maintain healthy function. However, when the metabolic balance is overwhelmed and the sum of free radicals in a cell is greater than the capacity of the cell to detoxify these substances, oxidative stress is generated. Increased oxidative stress has been shown to contribute to the etiology or progression of a number of neurodegenerative diseases since the brain uses a disproportionate amount of oxygen per volume of tissue compared to other organs [1]. When free radicals of oxygen are present within the environment of the cell, they may damage lipid membranes, interfere with DNA integrity, and interrupt cellular respiration through alterations in mitochondrial complex I [2–4]. The reduction or detoxification of free radicals is handled by a number of homeostatic mechanisms, in normal physiological conditions.
Parkinson’s Disease (PD) is one of the neurological disorders affected by changes in oxidative balance. PD is a progressive neurodegenerative disease with noticeable outward symptoms generally appearing in the sixth decade of life. The most common phenotypes of this disorder include progressive deterioration of autonomic and motor functions and in some cases, cognitive decline. Although the underlying etiology of Parkinson’s disease is not completely understood [5, 6], the most common neuroanatomical pathology is the accumulation of misfolded alpha-synuclein into intracellular aggregates called Lewy Bodies, present throughout the enteric [7, 8], peripheral [9] and central nervous systems [10, 11]. Progression of the disease results in the significant loss of the dopaminergic neurons situated in the midbrain substantia nigra pars compacta.
Sources of Reactive Oxygen Species in the Substantia Nigra
The loss of dopaminergic neurons located in the substantia nigra pars compacta (A9) is the lesion most characteristic of Parkinson’s disease, although other regions of the central, peripheral and enteric nervous systems also show considerable cell loss [12–15]. Within the CNS, it is not entirely clear why the substantia nigra is so significantly affected, although this region does have a number of characteristics that make it particularly vulnerable to oxidative stress. These factors include (but are not limited to) the presence of endogenous dopamine, iron, and neuromelanin [16–18]. Additionally, the intrinsic antioxidant defenses in this structure are more vulnerable than in other brain regions due to lower levels of glutathione (GSH) [19, 20] and glutamylcysteine ligase activity [21], and higher microglial:astrocyte ratios [22, 23].
Dopamine (DA), which is the most abundant neurotransmitter in the basal ganglia [24], is synthesized in the large diameter neurons of the substantia nigra and is released from the terminals that reside within the caudate and putamen nuclei (in rodents this is called the striatum) [25]. Functionally, dopamine modulates excitatory and inhibitory synaptic transmission ensuring smooth directed movement [26]. When released from presynaptic terminals, DA is actively taken up from the synaptic cleft through a number of monoamine transporters (i.e dopamine active transporter (DAT)), where it is packaged into intracellular vesicles by vesicular monoamine transporters (VMAT) [27]. In the SNpc dopaminergic neurons, the predominant VMAT is VMAT2 [28, 29]. When DA is produced in excess of capacity and cannot be transported into the cell through the DAT or packaged internally by VMAT, it remains in free form where it can be readily oxidized to DA quinone, or form superoxides and hydrogen peroxide [30–32]. These superoxides may damage cell and organelle membranes, leading to cellular dysfunction.
Inside the cell, DA quinones react with the sulfhydryl groups of the free amino acid cysteine, cysteine found in glutathione, and other cysteine residues to covalently modify proteins [31, 32] that cause cellular toxicity and in some cases, cell death [30, 31, 33, 34]. DA quinones have also been shown to react with neuromelanin to form eumelanin [35], which is present in DA neurons of the SN. DA may also autooxidize to form hydroxyl radicals (OH·) [30, 32, 36] or after oxidation to hydrogen peroxide, may react with iron, copper, or oxygen (O2) to form hydroxyl radicals [37].
Iron metabolism is necessary for the function of some enzymes, including tyrosine hydroxylase (the rate limiting enzyme in DA biosysthesis) and for overall neuronal health [38–41]. Iron is transported into cells from the bloodstream while bound to transferrin, and stored intracellularly by binding to the protein ferritin [37]. Ferritin in the cytosol is comprised of heavy (H) and light (L) chain subunits. The H-subunit has ferroxidase activity, converting Fe2+ to Fe3+, while the L-subunit stabilizes the complex of subunits to remain in iron storage form. The ratios of H- versus L-type subunits of ferritin vary among tissues and in different cell types within the brain. These differences can affect the interactions of iron with other cellular components and make some cell types more vulnerable to oxidative stress [37, 42].
Within the CNS, the SN is the structure containing the highest level of iron [43, 44]. In a reduced state, iron (Fe2+) readily reacts with hydrogen peroxide to form hydroxyl radicals via the Fenton reaction [37, 45]. The ratio of reduced iron (Fe2+) to oxidized iron (Fe3+) is approximately 1:1 in the normal SN [46, 47]. However, in PD patients the ratio of reduced to oxidized iron in the SN has been reported to increase [48], in one report to 1:3 [49]; a dysregulation not found in other tissues or regions of the brain [49, 50]. Since numerous studies have shown that the elevated levels of reduced iron in the SN can lead to cellular toxicity [51–54], it has been suggested that iron chelation may provide some level of neuroprotection in Parkinson’s disease [55–58].
The SN contains another protein that may also contribute to oxidative stress. Neuromelanin, a brown, black insoluble substance that is formed from oxidative metabolites of dopamine and norepinephrine [59, 60], has been shown to interact with lipids, pesticides, other toxic compounds including paraquat [61, 62], and many heavy metal ions including iron [63–65]. Of the transition metals, neuromelanin binds most tightly with iron [62, 65]. Although these interactions may initially be protective [66], when this system is overwhelmed (i.e. iron is present in excess), neuromelanin may begin to catalyze the production of free radicals [67].
Glutathione: An Important Antioxidant in the Brain
Glutathione (GSH), a ubiquitous thiol tripeptide, provides protection from oxidative stress-induced damage through the reduction of reactive oxygen species (ROS). GSH acts alone or in concert with other enzymes to reduce superoxide radicals, hydroxyl radicals and peroxynitrites [3]. Additionally, GSH detoxifies xenobiotics, is a storage and transfer form for cysteine, and maintains cellular redox potential by keeping sulfhydryl proteins in a reduced state [68]. The antioxidant characteristics of GSH have been demonstrated in a number of models of oxidative stress including depletion of GSH with L-buthionine-(S,R)-sulphoximine (BSO) [69–73], ethacrynic acid [74], or reduction of GSH synthesis using antisense directed against gamma-glutamylcysteine synthetase (γGCS), hereafter referred to as glutamylcysteine ligase (GCL) (see section on GSH synthesis below) [75–78] or glutaredoxin 2 [79]. In these studies, diminished levels of GSH increase oxidative stress in whole cells as well as in mitochondrial fractions, and increase lipid peroxidation, intracellular calcium, and gamma-glutamyl transpeptidase (γGT) activity.
Several studies discussed below illustrate these points by utilizing dopaminergic systems. Depletion of GSH by BSO, an irreversible inhibitor of GCL, that does not by itself induce nigrostriatal damage in vivo [80], potentiates the amount of MPTP–induced tyrosine hydroxylase-positive (TH+) neuron death in the SNpc (48.6% cell death compared to 30.1% cell death) [69, 80]. Additionally, in conditions of increased oxidative stress such as when mesencephalic cells are placed in culture or during normal ageing in vivo, decreasing GSH levels causes neuron loss [76].
The reduction of GSH activity by ethacrynic acid (EA), an effective loop diuretic used in clinical practice [81], has also been shown to increase cell sensitivity to free radicals. Astrocytes exposed to EA and 3-morpholinosydnonimine (SIN1, a compound that generates peroxynitrates) show significant increases in lactate dehydrogenase (an indirect marker of cell death), decreased ATP levels and decreased mitochondrial membrane potential. The critical role of GSH in this system is shown by experiments where excess GSH monoester is included with EA and SIN1, and LDH activity is inhibited [74]. Treatment with a thiol ester compound (gamma-glutamylcysteinyl ethyl esther) or exogenous GSH while examining cellular models of diminished GSH levels in concert with Complex I inhibitors MPP+, MPTP, or DHBT-1, restores mitochondrial Complex I activity, inhibits cell loss and protects against cell and striatal DA loss [73, 75, 82].
Further support for the critical role of GSH as a free radical reducing agent is demonstrated in several studies that utilize siRNA knockdown of GSH modulating enzymes. Lee et al [79] used siRNA directed against glutaredoxin 2 (Grx2), a protein that is critical for controlling redox signaling in the mitochondria by modulating the interactions between the glutathione pools and protein thiols [83] to reduce Complex I activity [84]. The reduction in Grx2 results in a loss of Complex I (30%) and m-aconitase (60%) activity, decreased ferritin levels and a subsequent increase in mitochondrial iron [79]; each leading to increased oxidative stress. Similarly, a 50% reduction of GSH by antisense knockdown of GCL [77] increases cellular oxidative stress in PC12 cells. Using this paradigm, Jha et al [78] noted a reduction in mitochondrial performance using three different measures; a 70% reduction of ATP levels, a 65% reduction in mitochondrial pyruvate-dependent 5,5′-dithiobis (2-nitrobenzoic acid) (MTT), and a 60% reduction in oxygen consumption. It was also demonstrated that this effect was specific to complex I, and not complex II, III, or IV activity. Addition of dithiothreitol (a thiol reducing agent) restored complex I activity to control levels in GCL-diminished cells, suggesting that it is the oxidation of sulfhydryl groups that results in the inhibition of complex I. In PC12 cells, GSH depletion decreases TH activity and DA uptake [77]. Additionally, mitochondrial complex I activity, which is reduced in Parkinson’s disease and is critical to DA neuron survival [85, 86] is compromised when GSH levels are reduced in combination with exposure to NO and generation of peroxynitrite radicals [74, 75].
In the SN, the reduced form of GSH is an important mediator of oxidative stress. Studies of postmortem CNS of PD patients reveal lower levels of GSH in the SN (40%), but not in other regions of the brain, compared to age-matched controls. In contrast, GSH levels in the SN from patients with other neurodegenerative diseases that involve the basal ganglia, such as Multiple System Atrophy and Supranuclear Palsy, are unchanged [87].
Glutathione Synthesis in the Brain
In non-nervous system mammalian cells, GSH is abundant and can be found at concentrations of 0.5–10 mM [68]. In the brain, however, GSH levels are often found at lower (1–3 mM) concentrations [88]; a protective mechanism itself, since the GSH precursor molecules may be toxic at high concentrations [89–92]. In general, the GSH tripeptide is synthesized as the product of two successive reactions (Figure 1). This synthesis occurs in neurons and glial cells, although astrocytes synthesize GSH more effectively than neurons based on their ability to utilize a wider variety of precursor substrates [93]. Neurons synthesize GSH using glutamine, glutamate, cysteine and glycine or conjugates of these amino acids provided by the breakdown of GSH by γ-glutamyl transpeptidase (γGT). Astrocytes, however, have the ability to utilize a far greater number of substrates in the synthesis of GSH including the amino acids: glutamate, cysteine, glycine, glutamine, aspartate, asparagine, ornithine, proline and cysteine, conjugates of these amino acids as well as glutathione disulfide (GSSG), 2-oxothiadolazine-4-carboxylate (OTC), N-acetylcysteine (NAC) and GSH itself [3, 94–96], although each of these components are ultimately converted to glutamate, cysteine or glycine prior to GSH synthesis [97].
Figure 1.
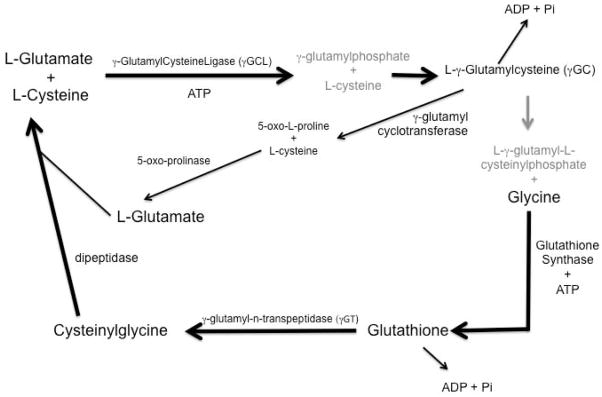
Glutathione synthesis pathway. Glutathione is synthesized from L-glutamate and L-cysteine in a 2-step reaction catalyzed in an ATP dependent manner by γ-glutamylcysteine ligase (GCL) (also referred to as γ-glutamylcysteine synthetase) and the addition of glycine by glutathione synthase. Glutathione can be recycled to its constitutive amino acids by γ-glutamyl-n-transferase and dipeptidase.
The first step in GSH synthesis is the formation of γ-glutamylcysteine (γGC) from the substrates L-glutamate and L-cysteine by glutamylcysteine ligase (GCL) (also referred to as γ-GlutamylCysteine synthetase (γGCS)) [98]. GCL is a heterodimer containing both disulfide and noncovalent bonds between its heavy and light chain subunits [99] is the enzyme in the rate-limiting step of GSH synthesis. Intracellular GSH levels are regulated by negative feedback of G by GSH [100] as well as the availability of cysteine [68]. An intermediate product of this synthesis, γ-glutamylphosphate, is produced by the reaction of L-glutamate and ATP, which reacts with L-cysteine to form L-γ-glutamylcysteine. In the second step of GSH synthesis, L-γ-glutamylcysteine and ATP combine to form L-γ–glutamyl-L-cysteinylphosphate that is combined with glycine in a reaction catalyzed by GSH synthase to form GSH [101, 102].
GSH is also metabolized to its component peptides that are recycled to regenerate GSH. In this reaction, the ectoenzyme γ-glutamyl transpeptidase (γGT) catalyzes the transfer of the γ-glutamyl moiety from GSH or a GSH conjugate to an acceptor molecule (CysGly or CysGly conjugate) [95]. As mentioned above, glutathione disulfide (GSSG), 2-oxothiadolazine-4-carboxylate (OTC), and N-acetylcysteine (NAC) can also serve as precursors for the synthesis of GSH [95]. Alternatively, glutathione reductase (GR) uses NADPH as an electron donor to reduce GSSG and regenerate GSH [99].
Reduction of Free Radicals by Glutathione Conjugating Enzymes
Glutathione Peroxidase
Glutathione peroxidases (GPXs) are a group of 8 (GPX1-8) enzymes that are important for reducing hydrogen peroxide to water. GPX’s are selenocysteine enzymes that use GSH as a reducing agent and require selenium [103, 104] for their antioxidant function. In the brain, the selenoproteins GPX-1-3 exist as tetrameric proteins, composed of four identical subunits with each monomer having a molecular weight of 22–23 kDa, [105], while GPX4 has activity as a monomer [106]. GPX1 and 4 are found in the mitochondria, nucleus and the cytosol [107], and GPX1, also known as GSHPx, is present in both neurons and glial cells [108, 109]. Overexpression of GPx decreases the amount of neuron loss, reduces hydrogen peroxide accumulation and lipid peroxidation in neurotoxic conditions [110]. Glutathione peroxidase and catalase reduce hydrogen peroxide when acting alone, but more effectively decrease toxicity of exogenous hydrogen peroxide when acting together [111].
GPX proteins and Parkinson’s disease
An immunocytochemical study of GPX1 expression shows that dopaminergic neurons in the SNpc express low levels of this protein, while other regions not affected in PD, such the ventral tegmental area, express higher levels [108]. In addition, GPX1 immunoreactive microglia were reported in samples of SN taken from patients with PD as well as those with Dementia with Lewy Bodies Disease (DLB) [109].
In an experimental model of PD, mice that lack GPX1 under normal oxidative conditions show no apparent neuropathological lesions compared to wild-type mice. However, when challenged with MPTP, a toxin that induces oxidative stress, DA, DOPAC, and HVA levels in these mice are decreased. [112]. Additionally, microarray analysis of mRNA expression in the lateral versus medial tiers of the SN reveals downregulation of gpx1 and gpx3 in the lateral tier compared to the medial tier of the SN [113]; this is complimentary to the observation of greater cell loss in the lateral SN of PD patients [114]. Savaskan et al. have shown that GPX4 protein level is increased following a neurodegenerative lesion of the entorhinal cortex. Subsequent immunocytochemical analysis shows that the location of this increase occurs in astrocytes [115]. In cortical samples taken from PD patients, GPX3 and GPX4 protein are also elevated compared to control subjects [116].
In DA neurons of the SN, GPX4 is colocalized with neuromelanin [113]. Examination of brains in SN from PD patients shows that volume-density immunolabeling of GPX4 is reduced; however, relative to the remaining cell density, GPX4 levels are increased compared to control subjects [113]. Further study of Selenoprotein 1 (Sepp1, a transport protein and source of selenium for selenoproteins) and GPX4 in the SN and putamen shows an association between Sepp1 and GPX4 localization in the putamen of control subjects while the correlation is lost in PD patients. No correlation of Sepp1 and GPX4 immunoreactivity is seen in the SN of PD or control samples [117].
Glutathione S-Transferases
Glutathione S-Transferases (GSTs) are a class of abundant proteins [118] that function as xenobiotic metabolizing enzymes [119–121] in eukaryotes. This class of enzymes may be viewed as a cellular defense against numerous artificial and naturally occurring environmental agents. GSTs function by catalyzing the conjugation of glutathione to various electrophiles and xenobiotics. Additionally, certain GST’s have been shown to have other functions, including modulation of cell survival pathways [122, 123].
In humans, there are three distinct classes of GST’s: cytosolic, mitochondrial and microsomal. Within the brain, the cytosolic forms of GST are predominant, and will be the focus here. Each of the cytosolic GSTs is configured with different combinations of monomers that are 199–244 amino acids in length [124, 125]. The ultimate composition of the holoenzyme is critical to the function, since each of the monomers contributes half of the overall GSH:electrophile active site [126, 127]; this dimerization occurs in the presence of increased levels of oxidative stress [128].
At this time, seven classes of cytosolic GST have been identified, and are named alpha, mu, pi, sigma, theta, omega, and zeta [129]. Within each GST molecule, there are two characteristic domains: I and II. Domain I comprises the N-terminal residues (1–80) of the protein whose structure consists of a series of beta pleated sheets and alpha helices. Domain II comprises the remaining residues (81–209±11) and is also referred to as the hydrophobic site (H site) [126]. The GSH binding domain is found in Domain I and is structurally conserved in each of the isoforms [130]. Structural differences among the isoforms are found within domain II. The variable residues in this domain contribute to the array of substrate specificity found among the GSTs [130, 131]. For instance, mu and pi classes contain more polar domain II regions that enable them to react with charged substrates [132] whereas the alpha isoform contains a more hydrophobic domain II [130].
In the brain, the active GSTs are composed of dimers containing alpha, mu, or pi class GST monomers [133–137]. GSTmu is the most highly expressed isoform in the brain in terms of abundance, followed by GSTpi, then GST alpha [135]. Cellular localization studies show that GSTpi and GSTmu are expressed in both neurons and astrocytes [135, 138, 139]. Interestingly, in the SN, the structure most affected in the CNS of PD patients, only GSTpi, but not GSTmu, is found in the A9 DA neurons [135]; a finding that may provide a clue why these neurons are particularly sensitive to oxidative stress. The distribution of GSTs in the brain appears to also be age dependent [136]. GSTpi is the only GST isoform that appears to be expressed in human fetal brain [136, 140], while the alpha, mu and pi classes are found in adult [136]. Unlike other components of the glutathione system, GSTs have also been implicated in signal transduction regulation, specifically in pathways involved in mediation of cell death. Mechanistically, GSTmu has been shown to inhibit apoptosis signal-regulating kinase 1 (ASK1) [141] while GSTpi has been shown to inhibit JNK signaling [128, 142–144].
ASK1, which is activated upon oxidative challenge [145–147], facilitates the downstream activation of JNK and the stress activated protein kinase p38 [148, 149]. GSTmu inhibits ASK1 signaling through a protein-protein interaction [141]. Furthermore, following stress, downstream activation of p38 requires the dissociation of GSTmu from ASK1, suggesting that GSTmu regulates the signal cascade mediated by ASK1 [141, 148].
GSTpi has two distinct functions related to oxidative stress [123]. Under non-stressed conditions, GSTpi exists predominantly in a monomeric form [128]. In this conformation, sequences at the C-terminus of GSTpi have been shown to bind to JNK; which inhibits the interaction of JNK with cJUN and reduces downstream apoptotic signaling [142, 144, 150]. As oxidative stress increases within the cell, GSTpi subunits form dimers. This interferes with the C-terminus interaction with JNK, allowing progression of cell death signaling [128]. The GSH and GST systems collaborate to maintain oxidative homeostasis. When the cellular environment is stress free (i.e. there are low levels of free radicals), GSH levels alone are sufficient to maintain redox balance [151] and monomeric GSTpi bind JNK [128]. As the number of free radicals within the cell increases, GSTpi dimerizes and in this conformation interacts with GSH to more efficiently reduce these free radicals [152], bringing the cell back to homeostasis.
As stated above, GSTpi is the only GST isoform found in the A9 DA neurons [135]; a finding that may provide a clue why these neurons are particularly sensitive to oxidative stress. Suggested by evidence of a decrease in total GSH [20, 153] and increase in oxidative stress in postmortem analysis of PD brains [154–157], alterations in structure or sequence (polymorphisms) of GSTs (particularly GSTmu and GSTpi) may contribute to the disease susceptibility and progression of PD. A number of studies have examined the correlation of GST sequence polymorphisms with PD (Table 1) and with one exception, no associations were found (although see DePalma et al, 1998 [158].
Table 1.
GST polymorphisms and Parkinson’s Disease
| GST isoform | association with PD | population | locus | Amino acid change | effect | Reference |
|---|---|---|---|---|---|---|
| GST O1 | PD Risk | Caucasian | rs4925 | Ala140Asp | p = 0.034 | [254] |
| PD Risk | Japanese | rs4925 | Ala140Asp | none | [255] | |
| PD Risk | Japanese | rs11191972 | C to T | none | [255] | |
| PD Risk with smoking | California | none | [256] | |||
| PD Risk with smoking | California | rs4925 | Ala140Asp | none | [256] | |
| PD Risk with smoking | California | rs2297235 | Chr10: 10861 A to G | none | [256] | |
| Age of Onset | USA | rs4925 | Ala140Asp | none | [257] | |
| PD Risk | Japanese | rs4925 | Ala140Asp | none | [258] | |
| PD Risk, | ||||||
| Age of Onset | Australian | rs4925 | Ala140Asp | none | [259] | |
| PD Risk, Age of Onset | Australian | rs4925 | Ala140Asp | none | [259] | |
| GST O2 | PD Risk | Caucasian | rs156697 | Asn142Asp | none | [254] |
| PD Risk | Japanese | rs156697 | Asn142Asp | none | [255] | |
| PD Risk | Japanese | rs2297235 | -183A to G | none | [255] | |
| none | California | none | [256] | |||
| PD Risk, Age of Onset | Australian | rs156697 | Asn142Asp | none | [259] | |
| Age of Onset | USA | rs2297235 | Chr10: 10861 A to G | none | [257] | |
| GST M1 | PD Risk | East Indian | null | null | none | [260] |
| PD Risk | Japanese | null | null | none | [255] | |
| PD Risk | Chilean | null | null | p = 0.0092 | [261] | |
| none | Portuguese | null | null | none | [262] | |
| age of onset | Greek A53T | null | null | none | [159] | |
| PD Risk, synergy with CYP2D6 | French | null | null | none | [263] | |
| PD Risk | USA | null | none | [264] | ||
| PD Risk | Japanese | null | null | none | [265] | |
| PD Risk, Age of Onset | Swedish | null | p=.03 for earlier age of onset | [266] | ||
| PD Risk | UK | null | none | [267] | ||
| PD Risk with exposure to pesticides | Australian | null | none | [268] | ||
| GST M3 | age of onset | Greek A53T | deletion | intron 6 3-bp deletion | [159] | |
| GST A4 | PD Risk | Italian | mutations | sequence 7 exons | none | [269] |
| GST T1 | PD Risk | East Indian | null | null | none | [260] |
| PD Risk | Japanese | null | null | none | [255] | |
| none | Japanese | null | null | none | [270] | |
| age of onset | Greek A53T | null | null | [159] | ||
| GST P1 | PD Risk | East Indian | rs1695 | Ile105Val | none | [260] |
| PD Risk | Japanese | rs1695 | Ile105Val | none | [255] | |
| none | Japanese | rs1695 | Ile104Val | none | 18327668 | |
| PD Risk | Portuguese | rs1695 | Ile105Val | odds ratio = 2.0 | [262] | |
| PD Risk with smoking | California | rs947894 | Ile105Val | none | [256] | |
| PD Risk with smoking | California | rs1799811 | Ala114VAl | none | [256] | |
| PD Risk with smoking | California | rs1799811 | Ala114VAl | none | [256] | |
| none | UK | rs947894 | Ile105Val | none | [271] | |
| age of onset and exposure to pesticides | UK | rs1799811 | Ala114VAl | p=.04 | [271] | |
| age of onset | Greek A53T | rs1695 | Ile105Val | p=.02 | [159] | |
| age of onset | Greek A53T | rs1799811 | Ala114Val | p=.05 | [159] | |
| PD Risk | USA | Ile104Val | none | [264] | ||
| PD Risk | USA | Ala113Val | none | [264] | ||
| PD Risk and exposure to pesticides | Australian | rs1799811 | Ala114Val | p=0.009 | [268] | |
| GST Z1 | age of onset | Greek A53T | Lys32Glu | Lys32Glu | [159] | |
| age of onset | Greek A53T | Arg42Gly | Arg42Gly | [159] | ||
| PD Risk | Australian | Glu32Lys | none | [272] | ||
| PD Risk | Australian | Gly42Arg | none | [272] | ||
| PD Risk and exposure to pesticides | Australian | Arg42Gly | none | [268] |
Although polymorphisms in GSTs alone do not appear to correlate with PD etiology, they do appear to have some effect on other aspects of PD, including age of onset and interactions with environmental agents. For example, Golbe et al reported that age of onset in individuals carrying the A53T alpha-synuclein (PARK1) mutation is positively associated with a GSTpi G-for-A nucleotide substitution at position 313 [159], while McCormick et al identified polymorphisms in GSTpi that correlate with increased risk of PD after exposure to pesticides [160]. Additionally, DePalma et al reported a marked increase in association of PD with a GSTT1*0 polymorphism when correlated with other PD risk factors, such as rural living and well water consumption [158]. The lack of a consistent correlation between GST polymorphisms and PD does not rule out a significant role for this enzyme in the etiology of PD, as there could be other regulatory influences on these proteins, including those controlling synthesis and breakdown.
GSTpi and animal models of Parkinson’s Disease
The role of GSTs in the protection of DA neurons has been examined using several models of Parkinson’s disease. Several groups have mutated the Parkin gene in Drosophila, and find subtle but significant loss of neurons in the protocerebral posterior lateral (PPL) DA neuron cluster [161] as well as an increase in oxidative stress [162] and a downregulation of GSTO1, which has high thiol transferase activity [163]. Examination of these mutants reveals that when GSTS1 is deleted the subtle loss of DA neurons in the PPL is enhanced. Conversely, overexpression of GSTS1 ameliorates this neurodegeneration [161]. When GSTO1, which has two distinct alleles, GSTO1A and GSTO1B, is deleted, there is an increased sensitivity to the xenobiotic paraquat [162, 164]. The re-expression of the GSTO1A in the null mutant eliminates the sensitivity of the PPL DA neurons to paraquat and suppresses phospho-JNK activity, which is implicated in apoptosis [163].
A number of polymorphisms in the leucine rich repeat kinase 2 (LRRK2) gene have been shown to confer PD in humans [165–167]. The most common polymorphisms in the LRRK2 gene are the G2019S and R1441C alelles, which affect kinase signaling [168] and GTPase activity [169], respectively. Recently, Chen et al have demonstrated that expression of the G2019S polymorphic LRRK2 gene in SH-SY5Y cells induces apoptotic cell death and that overexpression of GSTP1 reduces this apoptosis. The mechanism for this induction is a G2019S-induced hypermethylation of the GSTP1 promoter that can be reduced by addition of NAC or catalase [170].
Smeyne et al [135] examined the effects of loss of GSTpi on MPP+ or MPTP-induced dopaminergic neuron death. Using primary cultured dopaminergic cells harvested from the SN of MPTP resistant Swiss-Webster mice [171], inhibition of GST by ethacrynic acid or siRNA directed against GSTpi increases the amount of MPP+-induced DA neuron death to levels that mimic neurons from an MPTP-sensitive C57BL/6 strain. Similarly, when MPTP is administered to Swiss-Webster mice carrying a null mutation of GSTpi [172] SNpc DA neuron loss increases to levels seen in the MPTP-sensitive C57Bl/6 strain [135]. Administration of MPTP to mice lacking GSTpi also shows cellular ubiquitination and increased susceptibility to ubiquitin proteosome system (UPS) damage and inactivation [173]. Shi et al [174] demonstrates that overexpression of GSTP1 in Neuro2A cells as well as in primary cortical neurons confers protection from oxidative stress induced by addition of rotenone.
GSH Transport in the Brain: Multidrug Resistance Proteins (MDRP) and the Blood Brain Barrier (BBB)
Free radicals that have been reduced by thiol conjugation are transported out of the brain through multidrug resistance proteins (MDRP) at the gliovascular interface [175–178]. MDRPs are a family of ATP-binding cassette (ABC) transporters that actively transport electrophilic substances across cell membranes [179]. This large family of proteins (MDR1 (also known as P-glycoprotein), MRP1-MRP6, MRP8, BSEP, and BCRP), is encoded by 10 genes (ABCB1, ABCC1-ABCC6, ABCC11, ABCB11, and ABCG2) [180, 181]. Each member of the ABC transporter family has a unique pattern of expression in tissue, which may vary between and within species [182–184]. In particular, the localization of transporters is specific to the luminal or basolateral membrane of endothelial cells and at astrocytic endfeet of the BBB [181, 183, 185]. In addition to the BBB, these ABC transporters are also found at the blood-cerebrospinal fluid interface, which forms both a physical and enzymatic barrier between the tight ependymal cell junctions of the choroid plexus and cerebrospinal fluid [186, 187], and also expresses proteins critical to modulating ROS detoxification. The transport of free radical conjugates through MDRPs across the BBB is both ATP and sodium dependent [175, 176].
P-glycoprotein (Mdr1), Mrp1, Mrp2 and Mrp5 are the most widely studied of the transporters in the brain. These transmembrane proteins alter their conformation by utilizing ATP to translocate substances, including xenobiotics conjugated with GSH, across membranes [188–193]. Traditionally, it was believed that the expression of these transporters was found solely on capillary endothelial cells [194–196]; however, confocal studies suggest that some of these transporters may be localized to astrocytes [197].
A number of experiments demonstrate that the expression of these efflux transport proteins are correlated to intracellular oxidative stress levels [176, 198–202]. For example, Hong et al [176] demonstrated that BSO depletion of GSH increases P-gp levels, and conversely, treatment with the antioxidant NAC decreases its expression. In hepatocytes, P-gp transports MPP+ into the cell [203], suggesting that this pore contributes to xenobiotic sensitivity. P-gps can also transport the anti-Parkinsonian drug budipine [204], suggesting that it may also play a protective role in PD. Ziemann et al [198] demonstrated that addition of H2O2 to hepatocytes increases expression of mdr1, while Deng et al [205] showed a similar effect on the P-gp protein following genotoxic stress-inducing DNA damage. In models of the BBB, oxidative stress generated by hydrogen peroxide [202] and GSH depletion [176, 201] result in increased expression of P-gp. Each of these studies demonstrates the cellular coordination of ROS detoxification between GSH or GST regulation and cellular efflux. In conditions of high ROS, where free GSH levels are lower due to binding of electrophiles, MDRP expression is increased. Conversely, when cells are at homeostasis, free GSH levels are elevated and MDRP levels decrease.
MDRPs and GSTs often act in concert to remove free radicals as well as xenobiotic compounds from cells. Nitrogen monoxide (nitric oxide, NO) has been shown to induce iron release from activated macrophages during the cytotoxic defense against tumor cells [206]. Although necessary for cell signaling and enzyme activity, NO and iron are toxic when free and in excess concentrations within the cell [207–209]. Within the cell, free iron can complex with intracellular NO and GSH to form dinitrosyl-dithiol-iron complexes (DNICs) [206, 210]. A recent study has demonstrated the selective relationship between GSTP1, DNIC’s and MRP1 [211]. In the presence of NO or iNOS and hyperexpressed MRP1, cells transfected with GSTP1, but not GSTA1 or GSTM1, have reduced iron efflux compared to cells transfected with the control vector. Cells with MRP1 hyperexpression and GSTP1 expression are also resistant to the cytotoxic effects of S-nitroso-glutathione (GSNO), which is a NO generator, demonstrating the critical role for GSTP1 and MRP1 as a mechanism for free radical detoxification. Investigation of DNICs, GSTp, and MRP1 as an effective storage and release mechanism for NO and iron in the SN may be useful to discover the elements at work in the balance of homeostasis versus oxidative stress leading to neurodegeneration.
Numerous studies demonstrate alterations in MDRPs in Parkinson’s disease. Expression of MDR1 in blood vessels is reduced in the striatum of PD patients compared to control subjects [212]. Additionally, studies that examined the uptake of [(11)C]-verapamil, a compound that is usually extruded from the brain by P-gp, demonstrate a significant elevation of [(11)C]-verapamil in PD patients compared to controls [213, 214] suggesting that P-gp dysfunction contributes to PD pathogenesis [213, 214]. Polymorphisms in MDRPs have also been examined as they relate to PD risk. Westerlund [215] showed that a 1236C/T, but not 2677G/T/A or 3435C/T increases the risk of PD in a Swedish population. The lack of a direct effect of the 3435T is supported by Funke et al [216] and Furano et al [217]. The 3435T polymorphism, however, is implicated as a contributing risk factor in the development of Parkinson’s disease when combined with exposure to pesticides [218, 219].
GST Expression as a Biomarker for Parkinson’s Disease
One of the major obstacles in interrupting the process of pathogenesis in Parkinson’s disease is the inability to detect pathology (i.e. SNpc dopaminergic neuron loss, striatal dopamine loss) prior to onset of observable symptoms (tremor). This has lead to a search for a presymptomatic Parkinson’s disease biomarker. According to the NCI Dictionary of Cancer terms, a biomarker is “a biological molecule found in blood, other body fluids, or tissues that is a sign of a normal or abnormal process, or of a condition or disease”. Numerous functional tests (olfaction, gut motility) as well as imaging modalities (PET, fMRI) have been examined to identify persons with Parkinson’s disease prior to symptom onset [220]. However, these are often time reliant on personal narrative or are quite expensive and/or invasive.
Since GSTs are modulated by a number of actions that induce oxidative stress, it is thought that measurement of this class of proteins may allow identification of individuals where this process is aberrant. The observation that certain GSTs are expressed in brain regions affected in Parkinson’s disease and change expression in models of PD [135, 139, 174] make these particularly interesting enzymes for study. Werner et al used a proteomic method to examine protein expression in the substantia nigra of PD and age-matched controls and find increased expression of both GSTmu and GSTpi isoforms in PD patients [221]. Shi et al [174, 222] examined synaptosomal fractions isolated from the frontal lobes of PD patients and find significantly increased levels of GSTpi protein. Maarouf et al [223], also using a proteomic analysis, examined postmortem ventricular cerebrospinal fluid from PD patients and find a significant increase in GSTpi. Recently, Korff et al [224] measured changes in GSTpi protein in blood, comparing GSTpi levels at baseline and at various times after addition of MPP+, as an inducer of oxidative stress. They demonstrated that 4 hours after MPP+, GSTpi is significantly increased in the white blood cells of PD patients compared to control subjects, while no changes are seen when examining whole blood, plasma or the red cell fraction. These studies suggest that GSTpi and potentially other members of this and other anti-oxidant families may be viable biomarkers for PD.
GSH and Precursor Delivery as a Therapy for Parkinson’s Disease
As discussed earlier, increased levels of oxidative stress and decreased levels of GSH have been described in a number of PD models [69, 73, 74, 76, 77, 80], as well as in the SN of PD patients [6, 19, 20, 85]. Based on these findings, it has been suggested that restoring the level of GSH in brains of Parkinson’s disease patients may be a promising therapy to protect the affected DA neurons from further injury [225]. A number of therapeutic compounds have been examined, including GSH alone (via delivery in liposomes and nanoparticles), co-drugs, such as GSH:L-Dopa or GSH:DA conjugates, as well as GSH analogues, and other hybrid compounds. Optimally, successful candidates should be stable during gastrointestinal digestion, undergo bioconversion to constituent compounds that are transported into the brain, navigate to the desired site of action, and protect against the oxidative damage. Additionally, for effective treatment with these GSH analogues, they should be characterized by limited γ-GT metabolism, while also maintaining their reducing ability [226].
Several examples of these types of compounds have been generated and tested. Minimal improvement was reported in studies examining effect of GSH infusion on the amelioration of PD symptoms. Sechi et al [227] used iv infusion of GSH into nine patients with early untreated PD and found that during infusion patients showed a 42% decline in disability compared to vehicle treated controls. This effect lasted 2–4 months after discontinuation of the infusions. However, in a study that examined 21 patients, intravenous administration of GSH produced no significant improvement [228]. Another chemical method has also been used to increase GSH levels in the brain. Lee et al [229] coupled a hydrogen sulfide (H2S)-releasing molecule to L-dopa. When injected intravenously, this compound reached the brain and increased dopamine levels by 2.2-fold and GSH by 1.4-fold.
The lack of significant improvement seen following administration of GSH leads to questions regarding the delivery and transport of GSH into the brain. A number of molecules have been used to improve transport through the BBB. Liposomes are artificially prepared vesicles with outer lipid bilayers that encapsulate aqueous materials, including GSH. These liposomes have low toxicity, do not induce an immune reaction and are protected from enzymatic digestion and metabolism [230]. Based on their solubility, liposomes can easily pass through cell membranes including the BBB [231–233]. Although Liposomal-GSH has not been used in human trials, Zeevalk et al show that these conjugates are 100-fold more effective at replenishing GSH levels and preventing degeneration in paraquat + maneb treated mesencephalic cultures than GSH alone. Additionally, the liposomal-GSH is not a substrate for GST’s or γ-GT [234].
Nanoparticles provide another method for improving GSH transport into the brain. These are nanometer-scale polymeric substances with a structural and functional organization that delivers compounds that are encased by, or linked to the surface of the nanoparticle. Nanoparticles provide high capacity drug loading while maintaining resistance to enzymatic degradation, allowing prolonged drug delivery in the plasma with low toxicity side effects [226, 235]. One such nanoparticle, linking GSH and chitosan, maintains improved stability of GSH in oxidative conditions more effectively than free GSH [236]. Another type of nanoparticle used for drug-delivery across the BBB is dendrimers, a tree-like polymer, approximately 5nm in size, to which a number of functional groups can be attached [237]. These dendrimers are used to deliver the anti-inflammatory and anti-oxidant agent N-acetyl-l-cysteine (NAC, a reducing agent like GSH) to LPS stimulated BV-2 microglial cell cultures. The presence of these NAC-containing dendrimers is significantly more effective than NAC alone in reducing H2O2 (68% vs 41%) and TNF-α (77% vs 44%) [238].
Co-drug delivery of compounds, including GSH, links two different compounds that have similar or different modes of action in order to synergize their actions in the brain [239]. A number of these co-drugs have been developed as a potential treatment for Parkinson’s disease. The most frequent cofactor for these co-drugs is l-dopa. L-dopa has been conjugated to a number of other agents including entacapone (a COMT inhibitor marketed under the trade name Stalevo®) [240], cysteine [241], N-acetyl cysteine [242], l-Methionine [241], lipoic acid [243], caffeic acid and carnosine [244]. Co-drugs have been made that directly link GSH and L-dopa [245, 246]. Functionally, it is hypothesized that by joining L-dopa to GSH the exogenous GSH can be directed to the specific neurons within the SNpc that are affected in PD. A second application for co-drug development is to better target the protein of interest (in this case GSH) through the BBB, while protecting the protein from enzymatic degradation so that it can be released in a functional state once in the brain. To achieve this goal, More et al developed a series of metabolically stable urea analogues of glutathione [247], that are protected from 3GT cleavage [248].
Other recently generated codrugs include flavanoid compounds that enhance the uptake of cystine/cysteine by uncoupling their uptake from the cystine/glutamate antiporter, X(c)(−). Flavanoids are plant polyphenols with free radical scavenging capacity. Amino acid moities were added to flavanol compounds to test their effectiveness as neuroprotectants in conditions of glutamate toxicity [249]. Conjugation of the flavanoid epicatechin (EC) with cysteine, cysteamine-EC, increases cell survival and GSH level in a dose-dependent manner [250]. Support for the hypothesis that neuroprotection occurs through increasing cystine/cysteine availability is provided by experiments demonstrating the loss of this effect following BSO treatment or cystine depletion [250].
In addition to co-drugs, glutathione analogues called UPF peptides, each with anti-oxidative capacities, have been synthesized [251]. These compounds link GSH molecules to tyrosine derivatives by an amide bond and have better hydroxyl radical scavenging properties than glutathione alone. Two of these UPF proteins, UPF1 (4-methoxy-L-tyrosinyl-γ-L-glutamyl-L-cysteinyl-glycine) and UPF17 (4-methoxy-L-tyrosinyl-α-L-glutamyl-L-cysteinyl-glycine) are shown to increase free radical scavenging by 500-fold [252]. UPF1 is resistant to γGT activity and increased CuZnSOD activity, suggesting that UPF1 has antioxidant activity independent from GSH [253]. Although not yet tested in animals, or PD patients, these conjugated proteins may provide novel peptide based therapies to slow or ameliorate disease progression.
Conclusions
The cause of Parkinson’s disease is multifactorial, thought to have genetic and environmental etiologies. In each case, the mechanism underlying the disease includes alterations in pathways that increase oxidative stress. In this review, we have discussed the role of glutathione as well as facets of glutathione metabolism and function, which can be summarized in Figure 2. Further understanding of the mechanisms by which glutathione interacts with free radicals should lead to new approaches for slowing or ameliorating progression of this and other neurodegenerative diseases.
Figure 2.

Schematic representation of glutathione synthesis and catabolism in the substantia nigra. Glutathione (GSH) synthesis occurs in astrocytes (green) and dopaminergic (DA) neurons (blue). GSH is synthesized from L-glutamate (Glu) and L-cysteine (Cys) by γ-glutamylcysteine ligase (GCL) and the addition of glycine by glutathione synthase. Once generated, the oxidized form of GSH (GSSG) can be recycled to reduced GSH by glutathione reductase (GR) and NADPH. Additionally, GSH and/or its conjugates can be recycled by γ-glutamyl transpeptidase (γGT). GSH reduces ROS generated by a number of agents that are transported through the dopamine transporter (DAT), including MPTP, MPP+, and rotenone, that block mitochondrial Complex I. GSH can also reduce direct redox agents such as paraquat (PQ) or DA adducts (DA quinone) and inflammatory cytokines released from microglia (pink). GSH maintenance and clearance of conjugated electrophiles requires energy in the form of ATP and NADPH. Hydrogen peroxide (H2O2) is reduced by glutathione peroxidase (GPx) to water using GSH. In DA neurons, the reduction of free radicals is catalyzed by conjugation of GSH to an electrophile by glutathione S-transferase pi (GSTp). Conjugated adducts are transported from the brain parenchyma through MDRPs, including Mrp1 and Mrp5 through the basolateral membrane into capillary endothelial cells. Once in these cells, other MDRPs, including P-gp and Mrp2 transport these to the bloodstream for excretion.
Highlighs.
Glutathione, a thiol tripeptide, is an important antioxidant in the brain.
GSH is critical for protecting DA neurons in the SNpc from free radicals damage.
GPx and GSTs catalyze the reduction of electrophiles using GSH
GST mutations combined with environmental insults correlate with increased PD risk
Maintaining GSH levels may provide a therapeutic treatment for PD
Abbreviations
- ABC
ATP-binding cassette transporter
- ASK1
apoptosis signal-regulating kinase 1
- BBB
Blood Brain Barrier
- BSO
L-buthionine-(S,R)-sulphoximine
- COMT
Catechol-O-methyltransferase
- DA
Dopamine
- DAT
Dopamine transporter
- DHBT-1
7-(2-aminoethyl)-3,4-dihydro-5-hydroxy-2H-1,4-benzothiazine-3-carboxylic acid
- DOPAC
3,4-Dihydroxyphenylacetic acid
- γGCS
γ-glutamylcysteine synthetase
- γGC
γ-glutamylcysteine
- γGT
γ-glutamyl-n-transpeptidase
- GCL
glutamylcysteine ligase
- GPX
Glutathione Peroxidase
- GSH
Glutathione
- GSSG
glutathione disulfide
- GST
Glutathione S-Transferase
- HVA
Homovanillac Acid
- JNK
c-Jun N-terminal kinase
- LDH
Lactate dehydrogenase
- LPS
Lippopolysaccharide
- MDRP
Multidrug Resistance Protein
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MTT
mitochondrial pyruvate-dependent 59,5-Dithiobis (2-nitrobenzoic acid)
- NAC
N-acetylcysteine
- OTC
2-oxothiadolazine-4-carboxylate
- PD
Parkinson’s disease
- Pgp
P-glycoprotein
- ROS
Reactive oxygen species
- SIN1
3-morpholinosydnonimine
- SNP
Single nucleotide polymorphism
- SNpc
Substantia nigra pars compacta
- TH
Tyrosine hydroxylase
- VMAT2
Vesicular monoamine transporter 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michelle Smeyne, Email: Michelle.smeye@stjude.org, Department of Developmental Neurobiology, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105, 901-595-3066.
Richard Jay Smeyne, Email: Richard.smeyne@stjude.org, Department of Developmental Neurobiology, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105, 901-595-2830.
References
- 1.Raichle ME, Gusnard DA. Appraising the brain’s energy budget. Proc Natl Acad Sci U S A. 2002;99:10237–10239. doi: 10.1073/pnas.172399499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Signal. 2003;5:597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- 3.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 4.Spencer JP, Jenner A, Aruoma OI, Evans PJ, Kaur H, Dexter DT, Jenner P, Lees AJ, Marsden DC, Halliwell B. Intense oxidative DNA damage promoted by L-dopa and its metabolites. Implications for neurodegenerative disease. FEBS Letters. 1994;353:246–250. doi: 10.1016/0014-5793(94)01056-0. [DOI] [PubMed] [Google Scholar]
- 5.Owen AD, Schapira AH, Jenner P, Marsden CD. Oxidative stress and Parkinson’s disease. Annals of the New York Academy of Sciences. 1996;786:217–223. doi: 10.1111/j.1749-6632.1996.tb39064.x. [DOI] [PubMed] [Google Scholar]
- 6.Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann Neurol. 1992;32(Suppl):S82–87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- 7.Cersosimo MG, Benarroch EE. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol Dis. 2012;46:559–564. doi: 10.1016/j.nbd.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Sastre M, Bohl JR, de Vos RA, Del Tredici K. Parkinson’s disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007;113:421–429. doi: 10.1007/s00401-007-0193-x. [DOI] [PubMed] [Google Scholar]
- 10.Lee SJ, Lim HS, Masliah E, Lee HJ. Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci Res. 2011;70:339–348. doi: 10.1016/j.neures.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breydo L, Wu JW, Uversky VN. Alpha-synuclein misfolding and Parkinson’s disease. Biochim Biophys Acta. 2012;1822:261–285. doi: 10.1016/j.bbadis.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Jellinger KA. Post mortem studies in Parkinson’s disease--is it possible to detect brain areas for specific symptoms? J Neural Transm Suppl. 1999;56:1–29. doi: 10.1007/978-3-7091-6360-3_1. [DOI] [PubMed] [Google Scholar]
- 13.Bohnen NI, Albin RL. The cholinergic system and Parkinson disease. Behav Brain Res. 2011;221:564–573. doi: 10.1016/j.bbr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grinberg LT, Rueb U, Alho AT, Heinsen H. Brainstem pathology and non-motor symptoms in PD. J Neurol Sci. 2010;289:81–88. doi: 10.1016/j.jns.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 15.Singaram C, Ashraf W, Gaumnitz EA, Torbey C, Sengupta A, Pfeiffer R, Quigley EM. Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet. 1995;346:861–864. doi: 10.1016/s0140-6736(95)92707-7. [DOI] [PubMed] [Google Scholar]
- 16.Ben-Shachar D, Youdim MB. Iron, melanin and dopamine interaction: relevance to Parkinson’s disease. Progress In Neuro-Psychopharmacology And Biological Psychiatry. 1993;17:139–150. doi: 10.1016/0278-5846(93)90038-t. [DOI] [PubMed] [Google Scholar]
- 17.Gerlach M, Double KL, Ben-Shachar D, Zecca L, Youdim MB, Riederer P. Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson’s disease. Neurotox Res. 2003;5:35–44. doi: 10.1007/BF03033371. [DOI] [PubMed] [Google Scholar]
- 18.Gotz ME, Double K, Gerlach M, Youdim MB, Riederer P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann N Y Acad Sci. 2004;1012:193–208. doi: 10.1196/annals.1306.017. [DOI] [PubMed] [Google Scholar]
- 19.Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- 20.Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia [see comments] Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 21.Kang Y, Viswanath V, Jha N, Qiao X, Mo JQ, Andersen JK. Brain gamma-glutamyl cysteine synthetase (GCS) mRNA expression patterns correlate with regional-specific enzyme activities and glutathione levels. J Neurosci Res. 1999;58:436–441. [PubMed] [Google Scholar]
- 22.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smeyne M, Jiao Y, Shepherd KR, Smeyne RJ. Glia cell number modulates sensitivity to MPTP in mice. Glia. 2005;52:144–152. doi: 10.1002/glia.20233. [DOI] [PubMed] [Google Scholar]
- 24.Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:925–964. [PubMed] [Google Scholar]
- 25.Carpenter MB. Human Neuroanatomy. Baltimore: Williams and Wilkins Company; 1976. [Google Scholar]
- 26.Hauber W. Involvement of basal ganglia transmitter systems in movement initiation. Prog Neurobiol. 1998;56:507–540. doi: 10.1016/s0301-0082(98)00041-0. [DOI] [PubMed] [Google Scholar]
- 27.Gainetdinov RR, Caron MG. Monoamine transporters: from genes to behavior. Annu Rev Pharmacol Toxicol. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Hernandez T, Barroso-Chinea P, De La Cruz Muros I, Del Mar Perez-Delgado M, Rodriguez M. Expression of dopamine and vesicular monoamine transporters and differential vulnerability of mesostriatal dopaminergic neurons. J Comp Neurol. 2004;479:198–215. doi: 10.1002/cne.20323. [DOI] [PubMed] [Google Scholar]
- 29.Harrington KA, Augood SJ, Kingsbury AE, Foster OJ, Emson PC. Dopamine transporter (Dat) and synaptic vesicle amine transporter (VMAT2) gene expression in the substantia nigra of control and Parkinson’s disease. Brain Res Mol Brain Res. 1996;36:157–162. doi: 10.1016/0169-328x(95)00278-z. [DOI] [PubMed] [Google Scholar]
- 30.Hastings TG, Lewis DA, Zigmond MJ. Reactive dopamine metabolites and neurotoxicity: implications for Parkinson’s disease. Adv Exp Med Biol. 1996;387:97–106. doi: 10.1007/978-1-4757-9480-9_13. [DOI] [PubMed] [Google Scholar]
- 31.Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- 32.Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Res. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 33.Asanuma M, Miyazaki I, Ogawa N. Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox Res. 2003;5:165–176. doi: 10.1007/BF03033137. [DOI] [PubMed] [Google Scholar]
- 34.Rabinovic AD, Hastings TG. Role of endogenous glutathione in the oxidation of dopamine. J Neurochem. 1998;71:2071–2078. doi: 10.1046/j.1471-4159.1998.71052071.x. [DOI] [PubMed] [Google Scholar]
- 35.Carstam R, Brinck C, Hindemith-Augustsson A, Rorsman H, Rosengren E. The neuromelanin of the human substantia nigra. Biochim Biophys Acta. 1991;1097:152–160. doi: 10.1016/0925-4439(91)90100-n. [DOI] [PubMed] [Google Scholar]
- 36.Hastings TG, Zigmond MJ. Identification of catechol-protein conjugates in neostriatal slices incubated with [3H]dopamine: impact of ascorbic acid and glutathione. J Neurochem. 1994;63:1126–1132. doi: 10.1046/j.1471-4159.1994.63031126.x. [DOI] [PubMed] [Google Scholar]
- 37.Mounsey RB, Teismann P. Chelators in the treatment of iron accumulation in Parkinson’s disease. Int J Cell Biol. 2012:983245. doi: 10.1155/2012/983245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu XM, Moller SG. Iron-sulfur clusters: biogenesis, molecular mechanisms, and their functional significance. Antioxid Redox Signal. 2011;15:271–307. doi: 10.1089/ars.2010.3259. [DOI] [PubMed] [Google Scholar]
- 39.Levi S, Rovida E. The role of iron in mitochondrial function. Biochim Biophys Acta. 2009;1790:629–636. doi: 10.1016/j.bbagen.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Nagatsu T. Tyrosine hydroxylase: human isoforms, structure and regulation in physiology and pathology. Essays Biochem. 1995;30:15–35. [PubMed] [Google Scholar]
- 41.Ponting CP. Domain homologues of dopamine beta-hydroxylase and ferric reductase: roles for iron metabolism in neurodegenerative disorders? Hum Mol Genet. 2001;10:1853–1858. doi: 10.1093/hmg/10.17.1853. [DOI] [PubMed] [Google Scholar]
- 42.Gerlach M, Ben-Shachar D, Riederer P, Youdim MB. Altered brain metabolism of iron as a cause of neurodegenerative diseases? J Neurochem. 1994;63:793–807. doi: 10.1046/j.1471-4159.1994.63030793.x. [DOI] [PubMed] [Google Scholar]
- 43.Sofic E, Paulus W, Jellinger K, Riederer P, Youdim M. Selective increase in iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem. 1991;56:978–982. doi: 10.1111/j.1471-4159.1991.tb02017.x. [DOI] [PubMed] [Google Scholar]
- 44.Haacke EM, Ayaz M, Khan A, Manova ES, Krishnamurthy B, Gollapalli L, Ciulla C, Kim I, Petersen F, Kirsch W. Establishing a baseline phase behavior in magnetic resonance imaging to determine normal vs. abnormal iron content in the brain. J Magn Reson Imaging. 2007;26:256–264. doi: 10.1002/jmri.22987. [DOI] [PubMed] [Google Scholar]
- 45.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 46.Ide-Ektessabi A, Kawakami T, Watt F. Distribution and chemical state analysis of iron in the Parkinsonian substantia nigra using synchrotron radiation micro beams. Nuclear Instruments and Methods in Physics Research B. 2004;213:590–594. [Google Scholar]
- 47.Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, Youdim MB. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm. 1988;74:199–205. doi: 10.1007/BF01244786. [DOI] [PubMed] [Google Scholar]
- 48.Faucheux BA, Martin ME, Beaumont C, Hunot S, Hauw JJ, Agid Y, Hirsch EC. Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J Neurochem. 2002;83:320–330. doi: 10.1046/j.1471-4159.2002.01118.x. [DOI] [PubMed] [Google Scholar]
- 49.Youdim MB, Stephenson G, Ben Shachar D. Ironing iron out in Parkinson’s disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann N Y Acad Sci. 2004;1012:306–325. doi: 10.1196/annals.1306.025. [DOI] [PubMed] [Google Scholar]
- 50.Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- 51.Peng J, Peng L, Stevenson FF, Doctrow SR, Andersen JK. Iron and paraquat as synergistic environmental risk factors in sporadic Parkinson’s disease accelerate age-related neurodegeneration. J Neurosci. 2007;27:6914–6922. doi: 10.1523/JNEUROSCI.1569-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaasch JA, Lockman PR, Geldenhuys WJ, Allen DD, Van der Schyf CJ. Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem Res. 2007;32:1196–1208. doi: 10.1007/s11064-007-9290-4. [DOI] [PubMed] [Google Scholar]
- 53.Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, Giaveri G, Arosio P, Santambrogio P, Fariello RG, Karatekin E, Kleinman MH, Turro N, Hornykiewicz O, Zucca FA. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A. 2004;101:9843–9848. doi: 10.1073/pnas.0403495101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou ZD, Lan YH, Tan EK, Lim TM. Iron species-mediated dopamine oxidation, proteasome inhibition, and dopaminergic cell demise: implications for iron-related dopaminergic neuron degeneration. Free Radic Biol Med. 2010;49:1856–1871. doi: 10.1016/j.freeradbiomed.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 55.Chiueh CC, Andoh T, Lai AR, Lai E, Krishna G. Neuroprotective strategies in Parkinson’s disease: protection against progressive nigral damage induced by free radicals. Neurotox Res. 2000;2:293–310. doi: 10.1007/BF03033799. [DOI] [PubMed] [Google Scholar]
- 56.Xu Q, Kanthasamy AG, Reddy MB. Neuroprotective effect of the natural iron chelator, phytic acid in a cell culture model of Parkinson’s disease. Toxicology. 2008;245:101–108. doi: 10.1016/j.tox.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 57.Li X, Jankovic J, Le W. Iron chelation and neuroprotection in neurodegenerative diseases. J Neural Transm. 2011;118:473–477. doi: 10.1007/s00702-010-0518-0. [DOI] [PubMed] [Google Scholar]
- 58.Sian-Hulsmann J, Mandel S, Youdim MB, Riederer P. The relevance of iron in the pathogenesis of Parkinson’s disease. J Neurochem. 2011;118:939–957. doi: 10.1111/j.1471-4159.2010.07132.x. [DOI] [PubMed] [Google Scholar]
- 59.Sulzer D, Bogulavsky J, Larsen KE, Behr G, Karatekin E, Kleinman MH, Turro N, Krantz D, Edwards RH, Greene LA, Zecca L. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci U S A. 2000;97:11869–11874. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zucca FA, Giaveri G, Gallorini M, Albertini A, Toscani M, Pezzoli G, Lucius R, Wilms H, Sulzer D, Ito S, Wakamatsu K, Zecca L. The neuromelanin of human substantia nigra: physiological and pathogenic aspects. Pigment Cell Res. 2004;17:610–617. doi: 10.1111/j.1600-0749.2004.00201.x. [DOI] [PubMed] [Google Scholar]
- 61.Lindquist NG, Larsson BS, Lyden-Sokolowski A. Autoradiography of [14C]paraquat or [14C]diquat in frogs and mice: accumulation in neuromelanin. Neurosci Lett. 1988;93:1–6. doi: 10.1016/0304-3940(88)90002-x. [DOI] [PubMed] [Google Scholar]
- 62.Zecca L, Tampellini D, Gerlach M, Riederer P, Fariello RG, Sulzer D. Substantia nigra neuromelanin: structure, synthesis, and molecular behaviour. Mol Pathol. 2001;54:414–418. [PMC free article] [PubMed] [Google Scholar]
- 63.Swartz HM, Sarna T, Zecca L. Modulation by neuromelanin of the availability and reactivity of metal ions. Ann Neurol. 1992;32(Suppl):S69–75. doi: 10.1002/ana.410320712. [DOI] [PubMed] [Google Scholar]
- 64.Zecca L, Shima T, Stroppolo A, Goj C, Battiston GA, Gerbasi R, Sarna T, Swartz HM. Interaction of neuromelanin and iron in substantia nigra and other areas of human brain. Neuroscience. 1996;73:407–415. doi: 10.1016/0306-4522(96)00047-4. [DOI] [PubMed] [Google Scholar]
- 65.Zecca L, Tampellini D, Gatti A, Crippa R, Eisner M, Sulzer D, Ito S, Fariello R, Gallorini M. The neuromelanin of human substantia nigra and its interaction with metals. J Neural Transm. 2002;109:663–672. doi: 10.1007/s007020200055. [DOI] [PubMed] [Google Scholar]
- 66.Zucca FA, Bellei C, Giannelli S, Terreni MR, Gallorini M, Rizzio E, Pezzoli G, Albertini A, Zecca L. Neuromelanin and iron in human locus coeruleus and substantia nigra during aging: consequences for neuronal vulnerability. J Neural Transm. 2006;113:757–767. doi: 10.1007/s00702-006-0453-2. [DOI] [PubMed] [Google Scholar]
- 67.Enochs WS, Sarna T, Zecca L, Riley PA, Swartz HM. The roles of neuromelanin, binding of metal ions, and oxidative cytotoxicity in the pathogenesis of Parkinson’s disease: a hypothesis. J Neural Transm Park Dis Dement Sect. 1994;7:83–100. doi: 10.1007/BF02260963. [DOI] [PubMed] [Google Scholar]
- 68.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 69.Wullner U, Loschmann PA, Schulz JB, Schmid A, Dringen R, Eblen F, Turski L, Klockgether T. Glutathione depletion potentiates MPTP and MPP+ toxicity in nigral dopaminergic neurones. Neuroreport. 1996;7:921–923. doi: 10.1097/00001756-199603220-00018. [DOI] [PubMed] [Google Scholar]
- 70.Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena MA. Glutathione depletion switches nitric oxide neurotrophic effects to cell death in midbrain cultures: implications for Parkinson’s disease. J Neurochem. 2001;79:1183–1195. doi: 10.1046/j.1471-4159.2001.00635.x. [DOI] [PubMed] [Google Scholar]
- 71.Andersen JK, Mo JQ, Hom DG, Lee FY, Harnish P, Hamill RW, McNeill TH. Effect of buthionine sulfoximine, a synthesis inhibitor of the antioxidant glutathione, on the murine nigrostriatal neurons. J Neurochem. 1996;67:2164–2171. doi: 10.1046/j.1471-4159.1996.67052164.x. [DOI] [PubMed] [Google Scholar]
- 72.Ibi M, Sawada H, Kume T, Katsuki H, Kaneko S, Shimohama S, Akaike A. Depletion of intracellular glutathione increases susceptibility to nitric oxide in mesencephalic dopaminergic neurons. J Neurochem. 1999;73:1696–1703. doi: 10.1046/j.1471-4159.1999.731696.x. [DOI] [PubMed] [Google Scholar]
- 73.Chinta SJ, Andersen JK. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson’s disease. Free Radic Biol Med. 2006;41:1442–1448. doi: 10.1016/j.freeradbiomed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 74.Muyderman H, Nilsson M, Sims NR. Highly selective and prolonged depletion of mitochondrial glutathione in astrocytes markedly increases sensitivity to peroxynitrite. J Neurosci. 2004;24:8019–8028. doi: 10.1523/JNEUROSCI.1103-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chinta SJ, Kumar JM, Zhang H, Forman HJ, Andersen JK. Up-regulation of gamma-glutamyl transpeptidase activity following glutathione depletion has a compensatory rather than an inhibitory effect on mitochondrial complex I activity: implications for Parkinson’s disease. Free Radic Biol Med. 2006;40:1557–1563. doi: 10.1016/j.freeradbiomed.2005.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chinta SJ, Kumar MJ, Hsu M, Rajagopalan S, Kaur D, Rane A, Nicholls DG, Choi J, Andersen JK. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci. 2007;27:13997–14006. doi: 10.1523/JNEUROSCI.3885-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jurma OP, Hom DG, Andersen JK. Decreased glutathione results in calcium-mediated cell death in PC12. Free Radic Biol Med. 1997;23:1055–1066. doi: 10.1016/s0891-5849(97)00134-2. [DOI] [PubMed] [Google Scholar]
- 78.Jha N, Jurma O, Lalli G, Liu Y, Pettus EH, Greenamyre JT, Liu RM, Forman HJ, Andersen JK. Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson’s disease. J Biol Chem. 2000;275:26096–26101. doi: 10.1074/jbc.M000120200. [DOI] [PubMed] [Google Scholar]
- 79.Lee DW, Kaur D, Chinta SJ, Rajagopalan S, Andersen JK. A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: implications for Parkinson’s disease. Antioxid Redox Signal. 2009;11:2083–2094. doi: 10.1089/ars.2009.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toffa S, Kunikowska GM, Zeng BY, Jenner P, Marsden CD. Glutathione depletion in rat brain does not cause nigrostriatal pathway degeneration. J Neural Transm. 1997;104:67–75. doi: 10.1007/BF01271295. [DOI] [PubMed] [Google Scholar]
- 81.Somberg JC, Molnar J. The pleiotropic effects of ethacrynic Acid. Am J Ther. 2009;16:102–104. doi: 10.1097/MJT.0b013e3181961264. [DOI] [PubMed] [Google Scholar]
- 82.Li H, Shen XM, Dryhurst G. Brain mitochondria catalyze the oxidation of 7-(2-aminoethyl)-3,4-dihydro-5-hydroxy-2H-1,4-benzothiazine-3-carboxylic acid (DHBT-1) to intermediates that irreversibly inhibit complex I and scavenge glutathione: potential relevance to the pathogenesis of Parkinson’s disease. J Neurochem. 1998;71:2049–2062. doi: 10.1046/j.1471-4159.1998.71052049.x. [DOI] [PubMed] [Google Scholar]
- 83.Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 84.Karunakaran S, Saeed U, Ramakrishnan S, Koumar RC, Ravindranath V. Constitutive expression and functional characterization of mitochondrial glutaredoxin (Grx2) in mouse and human brain. Brain Res. 2007;1185:8–17. doi: 10.1016/j.brainres.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 85.Jenner P. Altered mitochondrial function, iron metabolism and glutathione levels in Parkinson’s disease. Acta Neurol Scand Suppl. 1993;146:6–13. [PubMed] [Google Scholar]
- 86.Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson’s disease. IUBMB Life. 2001;52:135–141. doi: 10.1080/15216540152845939. [DOI] [PubMed] [Google Scholar]
- 87.Sian J, Dexter DT, Lees AJ, Daniel S, Jenner P, Marsden CD. Glutathione- related enzymes in brain in Parkinson’s disease. Ann Neurol. 1994;36:356–361. doi: 10.1002/ana.410360306. [DOI] [PubMed] [Google Scholar]
- 88.Cooper AJL. The molecular and genetic basis of neurological disease. Newton, MA: Butterworth-Heinemann; 1997. Glutathione in the brain: disorder of glutathione metabolism; pp. 1195–1230. [Google Scholar]
- 89.Yudkoff M, Daikhin Y, Nissim I, Horyn O, Luhovyy B, Lazarow A. Brain amino acid requirements and toxicity: the example of leucine. J Nutr. 2005;135:1531S–1538S. doi: 10.1093/jn/135.6.1531S. [DOI] [PubMed] [Google Scholar]
- 90.Calabrese V, Ragusa N, Antico A, Mangiameli S, Rizza V. Cysteine-induced enhancement of lipid peroxidation in substantia nigra: comparative effect with exogenous administration of reduced glutathione. Drugs Exp Clin Res. 1997;23:25–31. [PubMed] [Google Scholar]
- 91.Schubert D, Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21:7455–7462. doi: 10.1523/JNEUROSCI.21-19-07455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang XF, Cynader MS. Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J Neurosci. 2001;21:3322–3331. doi: 10.1523/JNEUROSCI.21-10-03322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 94.Dringen R, Hamprecht B. Glutathione content as an indicator for the presence of metabolic pathways of amino acids in astroglial cultures. J Neurochem. 1996;67:1375–1382. doi: 10.1046/j.1471-4159.1996.67041375.x. [DOI] [PubMed] [Google Scholar]
- 95.Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dringen R, Kranich O, Loschmann PA, Hamprecht B. Use of dipeptides for the synthesis of glutathione by astroglia-rich primary cultures. J Neurochem. 1997;69:868–874. doi: 10.1046/j.1471-4159.1997.69020868.x. [DOI] [PubMed] [Google Scholar]
- 97.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 98.Huang CS, Moore WR, Meister A. On the active site thiol of gamma- glutamylcysteine synthetase: relationships to catalysis, inhibition, and regulation. Proc Natl Acad Sci U S A. 1988;85:2464–2468. doi: 10.1073/pnas.85.8.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 100.Richman PG, Meister A. Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J Biol Chem. 1975;250:1422–1426. [PubMed] [Google Scholar]
- 101.Griffith OW, Mulcahy RT. The enzymes of glutathione synthesis: gamma-glutamylcysteine synthetase. Adv Enzymol Relat Areas Mol Biol. 1999;73:209–267. doi: 10.1002/9780470123195.ch7. [DOI] [PubMed] [Google Scholar]
- 102.Trotta PP, Platzer KE, Haschemeyer RH, Meister A. Glutamine-binding subunit of glutamate synthase and partial reactions catalyzed by this glutamine amidotransferase. Proc Natl Acad Sci U S A. 1974;71:4607–4611. doi: 10.1073/pnas.71.11.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–590. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- 104.Flohe L, Gunzler WA, Schock HH. Glutathione peroxidase: a selenoenzyme. FEBS Lett. 1973;32:132–134. doi: 10.1016/0014-5793(73)80755-0. [DOI] [PubMed] [Google Scholar]
- 105.Epp O, Ladenstein R, Wendel A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur J Biochem. 1983;133:51–69. doi: 10.1111/j.1432-1033.1983.tb07429.x. [DOI] [PubMed] [Google Scholar]
- 106.Brown KM, Pickard K, Nicol F, Beckett GJ, Duthie GG, Arthur JR. Effects of organic and inorganic selenium supplementation on selenoenzyme activity in blood lymphocytes, granulocytes, platelets and erythrocytes. Clin Sci (Lond) 2000;98:593–599. [PubMed] [Google Scholar]
- 107.Margis R, Dunand C, Teixeira FK, Margis-Pinheiro M. Glutathione peroxidase family - an evolutionary overview. Febs J. 2008;275:3959–3970. doi: 10.1111/j.1742-4658.2008.06542.x. [DOI] [PubMed] [Google Scholar]
- 108.Trepanier G, Furling D, Puymirat J, Mirault ME. Immunocytochemical localization of seleno-glutathione peroxidase in the adult mouse brain. Neuroscience. 1996;75:231–243. doi: 10.1016/0306-4522(96)00222-9. [DOI] [PubMed] [Google Scholar]
- 109.Power JH, Blumbergs PC. Cellular glutathione peroxidase in human brain: cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:63–73. doi: 10.1007/s00401-008-0438-3. [DOI] [PubMed] [Google Scholar]
- 110.Wang H, Cheng E, Brooke S, Chang P, Sapolsky R. Over-expression of antioxidant enzymes protects cultured hippocampal and cortical neurons from necrotic insults. J Neurochem. 2003;87:1527–1534. doi: 10.1046/j.1471-4159.2003.02123.x. [DOI] [PubMed] [Google Scholar]
- 111.Dringen R, Hamprecht B. Involvement of glutathione peroxidase and catalase in the disposal of exogenous hydrogen peroxide by cultured astroglial cells. Brain Res. 1997;759:67–75. doi: 10.1016/s0006-8993(97)00233-3. [DOI] [PubMed] [Google Scholar]
- 112.Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci. 2000;20:1–7. doi: 10.1523/JNEUROSCI.20-01-00001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bellinger FP, Bellinger MT, Seale LA, Takemoto AS, Raman AV, Miki T, Manning-Bog AB, Berry MJ, White LR, Ross GW. Glutathione Peroxidase 4 is associated with Neuromelanin in Substantia Nigra and Dystrophic Axons in Putamen of Parkinson’s brain. Mol Neurodegener. 2011;6:8. doi: 10.1186/1750-1326-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1991;54:388–396. doi: 10.1136/jnnp.54.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Savaskan NE, Borchert A, Brauer AU, Kuhn H. Role for glutathione peroxidase-4 in brain development and neuronal apoptosis: specific induction of enzyme expression in reactive astrocytes following brain injury. Free Radic Biol Med. 2007;43:191–201. doi: 10.1016/j.freeradbiomed.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 116.Blackinton J, Kumaran R, van der Brug MP, Ahmad R, Olson L, Galter D, Lees A, Bandopadhyay R, Cookson MR. Post-transcriptional regulation of mRNA associated with DJ-1 in sporadic Parkinson disease. Neurosci Lett. 2009;452:8–11. doi: 10.1016/j.neulet.2008.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bellinger FP, Raman AV, Rueli RH, Bellinger MT, Dewing AS, Seale LA, Andres MA, Uyehara-Lock JH, White LR, Ross GW, Berry MJ. Changes in Selenoprotein P in Substantia Nigra and Putamen in Parkinson’s Disease. J Parkinsons Dis. 2012;2:115–126. doi: 10.3233/JPD-2012-11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boyer TD. The glutathione S-transferases: an update. Hepatology. 1989;9:486–496. doi: 10.1002/hep.1840090324. [DOI] [PubMed] [Google Scholar]
- 119.Philbert MA, Beiswanger CM, Manson MM, Green JA, Novak RF, Primiano T, Reuhl KR, Lowndes HE. Glutathione S-transferases and gamma-glutamyl transpeptidase in the rat nervous systems: a basis for differential susceptibility to neurotoxicants. Neurotoxicology. 1995;16:349–362. [PubMed] [Google Scholar]
- 120.Abel EL, Bammler TK, Eaton DL. Biotransformation of methyl parathion by glutathione S-transferases. Toxicol Sci. 2004;79:224–232. doi: 10.1093/toxsci/kfh118. [DOI] [PubMed] [Google Scholar]
- 121.Drukarch B, van Muiswinkel FL. Drug treatment of Parkinson’s disease. Time for phase II. Biochem Pharmacol. 2000;59:1023–1031. doi: 10.1016/s0006-2952(99)00340-8. [DOI] [PubMed] [Google Scholar]
- 122.Tew KD, Townsend DM. Glutathione-S-Transferases As Determinants of Cell Survival and Death. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2012.4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tew KD, Townsend DM. Regulatory functions of glutathione S-transferase P1–1 unrelated to detoxification. Drug Metab Rev. 2011;43:179–193. doi: 10.3109/03602532.2011.552912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 125.Wilce MC, Parker MW. Structure and function of glutathione S-transferases. Biochim Biophys Acta. 1994;1205:1–18. doi: 10.1016/0167-4838(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 126.Dirr H, Reinemer P, Huber R. X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur J Biochem. 1994;220:645–661. doi: 10.1111/j.1432-1033.1994.tb18666.x. [DOI] [PubMed] [Google Scholar]
- 127.Ketterer B, Christodoulides LG. Enzymology of cytosolic glutathione S-transferases. Adv Pharmacol. 1994;27:37–69. doi: 10.1016/s1054-3589(08)61029-7. [DOI] [PubMed] [Google Scholar]
- 128.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Regulation of JNK signaling by GSTp. Embo J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Higgins LG, Hayes JD. Mechanisms of induction of cytosolic and microsomal glutathione transferase (GST) genes by xenobiotics and pro-inflammatory agents. Drug Metab Rev. 2011;43:92–137. doi: 10.3109/03602532.2011.567391. [DOI] [PubMed] [Google Scholar]
- 130.Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 131.Babbitt PC. Reengineering the glutathione S-transferase scaffold: a rational design strategy pays off. Proc Natl Acad Sci U S A. 2000;97:10298–10300. doi: 10.1073/pnas.97.19.10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Awasthi YC, Singh SV, Shen RS, Abell CW, Gessner W, Brossi A. MPTP metabolites inhibit rat brain glutathione S-transferases. Neurosci Lett. 1987;81:159–164. doi: 10.1016/0304-3940(87)90358-2. [DOI] [PubMed] [Google Scholar]
- 133.Salinas AE, Wong MG. Glutathione S-transferases--a review. Curr Med Chem. 1999;6:279–309. [PubMed] [Google Scholar]
- 134.Shang W, Liu WH, Zhao XH, Sun QJ, Bi JZ, Chi ZF. Expressions of glutathione S-transferase alpha, mu, and pi in brains of medically intractable epileptic patients. BMC Neurosci. 2008;9:67. doi: 10.1186/1471-2202-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Smeyne M, Boyd J, Raviie Shepherd K, Jiao Y, Pond BB, Hatler M, Wolf R, Henderson C, Smeyne RJ. GSTpi expression mediates dopaminergic neuron sensitivity in experimental parkinsonism. Proc Natl Acad Sci U S A. 2007;104:1977–1982. doi: 10.1073/pnas.0610978104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Carder PJ, Hume R, Fryer AA, Strange RC, Lauder J, Bell JE. Glutathione S-transferase in human brain. Neuropathol Appl Neurobiol. 1990;16:293–303. doi: 10.1111/j.1365-2990.1990.tb01264.x. [DOI] [PubMed] [Google Scholar]
- 137.Tchaikovskaya T, Fraifeld V, Urphanishvili T, Andorfer JH, Davies P, Listowsky I. Glutathione S-transferase hGSTM3 and ageing-associated neurodegeneration: relationship to Alzheimer’s disease. Mech Ageing Dev. 2005;126:309–315. doi: 10.1016/j.mad.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 138.Abramovitz M, Homma H, Ishigaki S, Tansey F, Cammer W, Listowsky I. Characterization and localization of glutathione-S-transferases in rat brain and binding of hormones, neurotransmitters, and drugs. J Neurochem. 1988;50:50–57. doi: 10.1111/j.1471-4159.1988.tb13228.x. [DOI] [PubMed] [Google Scholar]
- 139.Castro-Caldas M, Neves Carvalho A, Peixeiro I, Rodrigues E, Lechner MC, Gama MJ. GSTpi expression in MPTP-induced dopaminergic neurodegeneration of C57BL/6 mouse midbrain and striatum. J Mol Neurosci. 2009;38:114–127. doi: 10.1007/s12031-008-9141-z. [DOI] [PubMed] [Google Scholar]
- 140.van Lieshout EM, Knapen MF, Lange WP, Steegers EA, Peters WH. Localization of glutathione S-transferases alpha and pi in human embryonic tissues at 8 weeks gestational age. Hum Reprod. 1998;13:1380–1386. doi: 10.1093/humrep/13.5.1380. [DOI] [PubMed] [Google Scholar]
- 141.Cho SG, Lee YH, Park HS, Ryoo K, Kang KW, Park J, Eom SJ, Kim MJ, Chang TS, Choi SY, Shim J, Kim Y, Dong MS, Lee MJ, Kim SG, Ichijo H, Choi EJ. Glutathione S-transferase mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276:12749–12755. doi: 10.1074/jbc.M005561200. [DOI] [PubMed] [Google Scholar]
- 142.Adler V, Pincus MR. Effector peptides from glutathione-S-transferase-pi affect the activation of jun by jun-N-terminal kinase. Ann Clin Lab Sci. 2004;34:35–46. [PubMed] [Google Scholar]
- 143.Wang T, Arifoglu P, Ronai Z, Tew KD. Glutathione S-transferase P1–1 (GSTP1–1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 144.Castro-Caldas M, Carvalho AN, Rodrigues E, Henderson C, Wolf CR, Gama MJ. Glutathione S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase activation in the nigrostriatal pathway. Mol Neurobiol. 2012;45:466–477. doi: 10.1007/s12035-012-8266-9. [DOI] [PubMed] [Google Scholar]
- 145.Vasieva O. The many faces of glutathione transferase pi. Curr Mol Med. 2011;11:129–139. doi: 10.2174/156652411794859278. [DOI] [PubMed] [Google Scholar]
- 146.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 147.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. Embo J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- 149.Soga M, Matsuzawa A, Ichijo H. Oxidative Stress-Induced Diseases via the ASK1 Signaling Pathway. Int J Cell Biol. 2012;2012:439587. doi: 10.1155/2012/439587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Asakura T, Hashizume Y, Tashiro K, Searashi Y, Ohkawa K, Nishihira J, Sakai M, Shibasaki T. Suppression of GST-P by treatment with glutathione-doxorubicin conjugate induces potent apoptosis in rat hepatoma cells. Int J Cancer. 2001;94:171–177. doi: 10.1002/ijc.1465. [DOI] [PubMed] [Google Scholar]
- 151.Circu ML, Aw TY. Glutathione and apoptosis. Free Radic Res. 2008;42:689–706. doi: 10.1080/10715760802317663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Coles B, Wilson I, Wardman P, Hinson JA, Nelson SD, Ketterer B. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: a stopped-flow kinetic study. Arch Biochem Biophys. 1988;264:253–260. doi: 10.1016/0003-9861(88)90592-9. [DOI] [PubMed] [Google Scholar]
- 153.Sofic E, Lange KW, Jellinger K, Riederer P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett. 1992;142:128–130. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- 154.Kumar H, Lim HW, More SV, Kim BW, Koppula S, Kim IS, Choi DK. The role of free radicals in the aging brain and Parkinson’s disease: convergence and parallelism. Int J Mol Sci. 2012;13:10478–10504. doi: 10.3390/ijms130810478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- 156.Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- 157.Dexter D, Carter C, Agid F, Agid Y, Lees AJ, Jenner P, Marsden CD. Lipid peroxidation as cause of nigral cell death in Parkinson’s disease. Lancet. 1986;2:639–640. doi: 10.1016/s0140-6736(86)92471-2. [DOI] [PubMed] [Google Scholar]
- 158.De Palma G, Mozzoni P, Mutti A, Calzetti S, Negrotti A. Case-control study of interactions between genetic and environmental factors in Parkinson’s disease. Lancet. 1998;352:1986–1987. doi: 10.1016/s0140-6736(05)61332-3. [DOI] [PubMed] [Google Scholar]
- 159.Golbe LI, Di Iorio G, Markopoulou K, Athanassiadou A, Papapetropoulos S, Watts RL, Vance JM, Bonifati V, Williams TA, Spychala JR, Stenroos ES, Johnson WG. Glutathione S-transferase polymorphisms and onset age in alpha-synuclein A53T mutant Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2007;144:254–258. doi: 10.1002/ajmg.b.30450. [DOI] [PubMed] [Google Scholar]
- 160.McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson’s disease: Selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 161.Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 163.Kim K, Kim SH, Kim J, Kim H, Yim J. Glutathione s-transferase omega 1 activity is sufficient to suppress neurodegeneration in a Drosophila model of Parkinson disease. J Biol Chem. 2012;287:6628–6641. doi: 10.1074/jbc.M111.291179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet. 2005;14:799–811. doi: 10.1093/hmg/ddi074. [DOI] [PubMed] [Google Scholar]
- 165.Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dachsel JC, Farrer MJ. LRRK2 and Parkinson disease. Arch Neurol. 2010;67:542–547. doi: 10.1001/archneurol.2010.79. [DOI] [PubMed] [Google Scholar]
- 168.Reichling LJ, Riddle SM. Leucine-rich repeat kinase 2 mutants I2020T and G2019S exhibit altered kinase inhibitor sensitivity. Biochem Biophys Res Commun. 2009;384:255–258. doi: 10.1016/j.bbrc.2009.04.098. [DOI] [PubMed] [Google Scholar]
- 169.Li Y, Dunn L, Greggio E, Krumm B, Jackson GS, Cookson MR, Lewis PA, Deng J. The R1441C mutation alters the folding properties of the ROC domain of LRRK2. Biochim Biophys Acta. 2009;1792:1194–1197. doi: 10.1016/j.bbadis.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chen J, Liou A, Zhang L, Weng Z, Gao Y, Cao G, Zigmond MJ. GST P1, a novel downstream regulator of LRRK2, G2019S-induced neuronal cell death. Front Biosci (Elite Ed) 2012;4:2365–2377. doi: 10.2741/e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hamre K, Tharp R, Poon K, Xiong X, Smeyne RJ. Differential strain susceptibility following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration acts in an autosomal dominant fashion: quantitative analysis in seven strains of Mus musculus. Brain Res. 1999;828:91–103. doi: 10.1016/s0006-8993(99)01273-1. [DOI] [PubMed] [Google Scholar]
- 172.Henderson CJ, Smith AG, Ure J, Brown K, Bacon EJ, Wolf CR. Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc Natl Acad Sci U S A. 1998;95:5275–5280. doi: 10.1073/pnas.95.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Carvalho AN, Marques C, Rodrigues E, Henderson CJ, Wolf CR, Pereira P, Gama MJ. Ubiquitin-Proteasome System Impairment and MPTP-Induced Oxidative Stress in the Brain of C57BL/6 Wild-type and GSTP Knockout Mice. Mol Neurobiol. 2012 doi: 10.1007/s12035-012-8368-4. [DOI] [PubMed] [Google Scholar]
- 174.Shi M, Bradner J, Bammler TK, Eaton DL, Zhang J, Ye Z, Wilson AM, Montine TJ, Pan C, Zhang J. Identification of glutathione S-transferase pi as a protein involved in Parkinson disease progression. Am J Pathol. 2009;175:54–65. doi: 10.2353/ajpath.2009.081019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kannan R, Mittur A, Bao Y, Tsuruo T, Kaplowitz N. GSH transport in immortalized mouse brain endothelial cells: evidence for apical localization of a sodium-dependent GSH transporter. J Neurochem. 1999;73:390–399. doi: 10.1046/j.1471-4159.1999.0730390.x. [DOI] [PubMed] [Google Scholar]
- 176.Hong H, Lu Y, Ji ZN, Liu GQ. Up-regulation of P-glycoprotein expression by glutathione depletion-induced oxidative stress in rat brain microvessel endothelial cells. J Neurochem. 2006;98:1465–1473. doi: 10.1111/j.1471-4159.2006.03993.x. [DOI] [PubMed] [Google Scholar]
- 177.Tew KD, Boyd JT, Chen ZJ, Davis W, Jr, Fazilev F, Findlay V, Gate L, Ile K, Soulika A, Townsend DM. Glutathione and ABC transporters as determinants of sensitivity to oxidative and nitrosative stress. J Nutr. 2004;134:3205S–3206S. doi: 10.1093/jn/134.11.3205S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hirrlinger J, Konig J, Keppler D, Lindenau J, Schulz JB, Dringen R. The multidrug resistance protein MRP1 mediates the release of glutathione disulfide from rat astrocytes during oxidative stress. J Neurochem. 2001;76:627–636. doi: 10.1046/j.1471-4159.2001.00101.x. [DOI] [PubMed] [Google Scholar]
- 179.Sharom FJ. The P-glycoprotein multidrug transporter. Essays Biochem. 2011;50:161–178. doi: 10.1042/bse0500161. [DOI] [PubMed] [Google Scholar]
- 180.Scotto KW. Transcriptional regulation of ABC drug transporters. Oncogene. 2003;22:7496–7511. doi: 10.1038/sj.onc.1206950. [DOI] [PubMed] [Google Scholar]
- 181.Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hermann DM, Kilic E, Spudich A, Kramer SD, Wunderli-Allenspach H, Bassetti CL. Role of drug efflux carriers in the healthy and diseased brain. Ann Neurol. 2006;60:489–498. doi: 10.1002/ana.21012. [DOI] [PubMed] [Google Scholar]
- 183.Soontornmalai A, Vlaming ML, Fritschy JM. Differential, strain-specific cellular and subcellular distribution of multidrug transporters in murine choroid plexus and blood-brain barrier. Neuroscience. 2006;138:159–169. doi: 10.1016/j.neuroscience.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 184.Lee G, Dallas S, Hong M, Bendayan R. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev. 2001;53:569–596. [PubMed] [Google Scholar]
- 185.Scherrmann JM. Expression and function of multidrug resistance transporters at the blood-brain barriers. Expert Opin Drug Metab Toxicol. 2005;1:233–246. doi: 10.1517/17425255.1.2.233. [DOI] [PubMed] [Google Scholar]
- 186.Strazielle N, Ghersi-Egea JF. Demonstration of a coupled metabolism-efflux process at the choroid plexus as a mechanism of brain protection toward xenobiotics. J Neurosci. 1999;19:6275–6289. doi: 10.1523/JNEUROSCI.19-15-06275.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ghersi-Egea JF, Strazielle N, Murat A, Jouvet A, Buenerd A, Belin MF. Brain protection at the blood-cerebrospinal fluid interface involves a glutathione-dependent metabolic barrier mechanism. J Cereb Blood Flow Metab. 2006;26:1165–1175. doi: 10.1038/sj.jcbfm.9600267. [DOI] [PubMed] [Google Scholar]
- 188.Sauna ZE, Ambudkar SV. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther. 2007;6:13–23. doi: 10.1158/1535-7163.MCT-06-0155. [DOI] [PubMed] [Google Scholar]
- 189.Bellarosa C, Bortolussi G, Tiribelli C. The role of ABC transporters in protecting cells from bilirubin toxicity. Curr Pharm Des. 2009;15:2884–2892. doi: 10.2174/138161209789058246. [DOI] [PubMed] [Google Scholar]
- 190.Zaman GJ, Lankelma J, van Tellingen O, Beijnen J, Dekker H, Paulusma C, Oude Elferink RP, Baas F, Borst P. Role of glutathione in the export of compounds from cells by the multidrug-resistance-associated protein. Proc Natl Acad Sci U S A. 1995;92:7690–7694. doi: 10.1073/pnas.92.17.7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bauer B, Hartz AM, Lucking JR, Yang X, Pollack GM, Miller DS. Coordinated nuclear receptor regulation of the efflux transporter, Mrp2, and the phase-II metabolizing enzyme, GSTpi, at the blood-brain barrier. J Cereb Blood Flow Metab. 2008;28:1222–1234. doi: 10.1038/jcbfm.2008.16. [DOI] [PubMed] [Google Scholar]
- 192.Lee G, Bendayan R. Functional expression and localization of P-glycoprotein in the central nervous system: relevance to the pathogenesis and treatment of neurological disorders. Pharm Res. 2004;21:1313–1330. doi: 10.1023/b:pham.0000036905.82914.8e. [DOI] [PubMed] [Google Scholar]
- 193.Golden PL, Pollack GM. Blood-brain barrier efflux transport. J Pharm Sci. 2003;92:1739–1753. doi: 10.1002/jps.10424. [DOI] [PubMed] [Google Scholar]
- 194.Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, Piwnica-Worms D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc Natl Acad Sci U S A. 1999;96:3900–3905. doi: 10.1073/pnas.96.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Decleves X, Regina A, Laplanche JL, Roux F, Boval B, Launay JM, Scherrmann JM. Functional expression of P-glycoprotein and multidrug resistance-associated protein (Mrp1) in primary cultures of rat astrocytes. J Neurosci Res. 2000;60:594–601. doi: 10.1002/(SICI)1097-4547(20000601)60:5<594::AID-JNR4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 197.Golden PL, Pardridge WM. P-Glycoprotein on astrocyte foot processes of unfixed isolated human brain capillaries. Brain Res. 1999;819:143–146. doi: 10.1016/s0006-8993(98)01305-5. [DOI] [PubMed] [Google Scholar]
- 198.Ziemann C, Burkle A, Kahl GF, Hirsch-Ernst KI. Reactive oxygen species participate in mdr1b mRNA and P-glycoprotein overexpression in primary rat hepatocyte cultures. Carcinogenesis. 1999;20:407–414. doi: 10.1093/carcin/20.3.407. [DOI] [PubMed] [Google Scholar]
- 199.Hirsch-Ernst KI, Kietzmann T, Ziemann C, Jungermann K, Kahl GF. Physiological oxygen tensions modulate expression of the mdr1b multidrug-resistance gene in primary rat hepatocyte cultures. Biochem J. 2000;350(Pt 2):443–451. doi: 10.1042/0264-6021:3500443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Thevenod F, Friedmann JM, Katsen AD, Hauser IA. Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem. 2000;275:1887–1896. doi: 10.1074/jbc.275.3.1887. [DOI] [PubMed] [Google Scholar]
- 201.Wu J, Ji H, Wang YY, Wang Y, Li YQ, Li WG, Long Y, Xia YZ, Hong H. Glutathione depletion upregulates P-glycoprotein expression at the blood-brain barrier in rats. J Pharm Pharmacol. 2009;61:819–824. doi: 10.1211/jpp/61.06.0016. [DOI] [PubMed] [Google Scholar]
- 202.Felix RA, Barrand MA. P-glycoprotein expression in rat brain endothelial cells: evidence for regulation by transient oxidative stress. J Neurochem. 2002;80:64–72. doi: 10.1046/j.0022-3042.2001.00660.x. [DOI] [PubMed] [Google Scholar]
- 203.Martel F, Martins MJ, Hipolito-Reis C, Azevedo I. Inward transport of [3H]-1-methyl-4-phenylpyridinium in rat isolated hepatocytes: putative involvement of a P-glycoprotein transporter. Br J Pharmacol. 1996;119:1519–1524. doi: 10.1111/j.1476-5381.1996.tb16067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Uhr M, Ebinger M, Rosenhagen MC, Grauer MT. The anti-Parkinson drug budipine is exported actively out of the brain by P-glycoprotein in mice. Neurosci Lett. 2005;383:73–76. doi: 10.1016/j.neulet.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 205.Deng L, Lin-Lee YC, Claret FX, Kuo MT. 2-acetylaminofluorene up-regulates rat mdr1b expression through generating reactive oxygen species that activate NF-kappa B pathway. J Biol Chem. 2001;276:413–420. doi: 10.1074/jbc.M004551200. [DOI] [PubMed] [Google Scholar]
- 206.Watts RN, Hawkins C, Ponka P, Richardson DR. Nitrogen monoxide (NO)-mediated iron release from cells is linked to NO-induced glutathione efflux via multidrug resistance-associated protein 1. Proc Natl Acad Sci U S A. 2006;103:7670–7675. doi: 10.1073/pnas.0602515103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Youdim MB, Riederer P. The role of iron in senescence of dopaminergic neurons in Parkinson’s disease. J Neural Transm Suppl. 1993;40:57–67. [PubMed] [Google Scholar]
- 208.Aquilano K, Baldelli S, Cardaci S, Rotilio G, Ciriolo MR. Nitric oxide is the primary mediator of cytotoxicity induced by GSH depletion in neuronal cells. J Cell Sci. 2011;124:1043–1054. doi: 10.1242/jcs.077149. [DOI] [PubMed] [Google Scholar]
- 209.Madathil KS, KSS, Haobam R, Varghese M, Rajamma U, Mohanakumar KP. Nitric oxide synthase inhibitors protect against rotenone-induced, oxidative stress mediated parkinsonism in rats. Neurochem Int. 2013 doi: 10.1016/j.neuint.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 210.Suryo Rahmanto Y, Kalinowski DS, Lane DJ, Lok HC, Richardson V, Richardson DR. Nitrogen monoxide (NO) storage and transport by dinitrosyl-dithiol-iron complexes: long-lived NO that is trafficked by interacting proteins. J Biol Chem. 2012;287:6960–6968. doi: 10.1074/jbc.R111.329847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lok HC, Suryo Rahmanto Y, Hawkins CL, Kalinowski DS, Morrow CS, Townsend AJ, Ponka P, Richardson DR. Nitric oxide storage and transport in cells are mediated by glutathione S-transferase P1–1 and multidrug resistance protein 1 via dinitrosyl iron complexes. J Biol Chem. 2012;287:607–618. doi: 10.1074/jbc.M111.310987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Westerlund M, Belin AC, Olson L, Galter D. Expression of multi-drug resistance 1 mRNA in human and rodent tissues: reduced levels in Parkinson patients. Cell Tissue Res. 2008;334:179–185. doi: 10.1007/s00441-008-0686-5. [DOI] [PubMed] [Google Scholar]
- 213.Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, Hendrikse NH. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 214.Bartels AL, Willemsen AT, Kortekaas R, de Jong BM, de Vries R, de Klerk O, van Oostrom JC, Portman A, Leenders KL. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm. 2008;115:1001–1009. doi: 10.1007/s00702-008-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Westerlund M, Belin AC, Anvret A, Hakansson A, Nissbrandt H, Lind C, Sydow O, Olson L, Galter D. Association of a polymorphism in the ABCB1 gene with Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:422–424. doi: 10.1016/j.parkreldis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 216.Funke C, Soehn AS, Tomiuk J, Riess O, Berg D. Genetic analysis of coding SNPs in blood-brain barrier transporter MDR1 in European Parkinson’s disease patients. J Neural Transm. 2009;116:443–450. doi: 10.1007/s00702-009-0196-y. [DOI] [PubMed] [Google Scholar]
- 217.Furuno T, Landi MT, Ceroni M, Caporaso N, Bernucci I, Nappi G, Martignoni E, Schaeffeler E, Eichelbaum M, Schwab M, Zanger UM. Expression polymorphism of the blood-brain barrier component P-glycoprotein (MDR1) in relation to Parkinson’s disease. Pharmacogenetics. 2002;12:529–534. doi: 10.1097/00008571-200210000-00004. [DOI] [PubMed] [Google Scholar]
- 218.Zschiedrich K, Konig IR, Bruggemann N, Kock N, Kasten M, Leenders KL, Kostic V, Vieregge P, Ziegler A, Klein C, Lohmann K. MDR1 variants and risk of Parkinson disease. Association with pesticide exposure? J Neurol. 2009;256:115–120. doi: 10.1007/s00415-009-0089-x. [DOI] [PubMed] [Google Scholar]
- 219.Drozdzik M, Bialecka M, Mysliwiec K, Honczarenko K, Stankiewicz J, Sych Z. Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease. Pharmacogenetics. 2003;13:259–263. doi: 10.1097/01.fpc.0000054087.48725.d9. [DOI] [PubMed] [Google Scholar]
- 220.Sherer TB. Biomarkers for Parkinson’s disease. Sci Transl Med. 2011;3:79ps14. doi: 10.1126/scitranslmed.3002488. [DOI] [PubMed] [Google Scholar]
- 221.Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008;6:8. doi: 10.1186/1477-5956-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ano Y, Sakudo A, Kimata T, Uraki R, Sugiura K, Onodera T. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci Lett. 2010;469:39–43. doi: 10.1016/j.neulet.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 223.Maarouf CL, Beach TG, Adler CH, Shill HA, Sabbagh MN, Wu T, Walker DG, Kokjohn TA, Roher AE. Cerebrospinal fluid biomarkers of neuropathologically diagnosed Parkinson’s disease subjects. Neurol Res. 2012;34:669–676. doi: 10.1179/1743132812Y.0000000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Korff A, Pfeiffer B, Smeyne M, Kocak M, Pfeiffer RF, Smeyne RJ. Alterations in glutathione S-transferase pi expression following exposure to MPP(+)-induced oxidative stress in the blood of Parkinson’s disease patients. Parkinsonism Relat Disord. 2011 doi: 10.1016/j.parkreldis.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Abdel-Salam OM. Drugs used to treat Parkinson’s disease, present status and future directions. CNS Neurol Disord Drug Targets. 2008;7:321–342. doi: 10.2174/187152708786441867. [DOI] [PubMed] [Google Scholar]
- 226.Cacciatore I, Baldassarre L, Fornasari E, Mollica A, Pinnen F. Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid Med Cell Longev. 2012;2012:240146. doi: 10.1155/2012/240146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sechi G, Deledda MG, Bua G, Satta WM, Deiana GA, Pes GM, Rosati G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 1996;20:1159–1170. doi: 10.1016/s0278-5846(96)00103-0. [DOI] [PubMed] [Google Scholar]
- 228.Hauser RA, Lyons KE, McClain T, Carter S, Perlmutter D. Randomized, double-blind, pilot evaluation of intravenous glutathione in Parkinson’s disease. Mov Disord. 2009;24:979–983. doi: 10.1002/mds.22401. [DOI] [PubMed] [Google Scholar]
- 229.Lee M, Tazzari V, Giustarini D, Rossi R, Sparatore A, Del Soldato P, McGeer E, McGeer PL. Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: potential for treating Parkinson disease. J Biol Chem. 2010;285:17318–17328. doi: 10.1074/jbc.M110.115261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Chen C, Han D, Cai C, Tang X. An overview of liposome lyophilization and its future potential. J Control Release. 2010;142:299–311. doi: 10.1016/j.jconrel.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 231.Brasnjevic I, Steinbusch HW, Schmitz C, Martinez-Martinez P. Delivery of peptide and protein drugs over the blood-brain barrier. Prog Neurobiol. 2009;87:212–251. doi: 10.1016/j.pneurobio.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 232.Chen Y, Dalwadi G, Benson HA. Drug delivery across the blood-brain barrier. Curr Drug Deliv. 2004;1:361–376. doi: 10.2174/1567201043334542. [DOI] [PubMed] [Google Scholar]
- 233.Schnyder A, Huwyler J. Drug transport to brain with targeted liposomes. NeuroRx. 2005;2:99–107. doi: 10.1602/neurorx.2.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zeevalk GD, Bernard LP, Guilford FT. Liposomal-glutathione provides maintenance of intracellular glutathione and neuroprotection in mesencephalic neuronal cells. Neurochem Res. 2010;35:1575–1587. doi: 10.1007/s11064-010-0217-0. [DOI] [PubMed] [Google Scholar]
- 235.De Jong WH, Borm PJ. Drug delivery and nanoparticles:applications and hazards. Int J Nanomedicine. 2008;3:133–149. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Koo SH, Lee JS, Kim GH, Lee HG. Preparation, characteristics, and stability of glutathione-loaded nanoparticles. J Agric Food Chem. 2011;59:11264–11269. doi: 10.1021/jf2024648. [DOI] [PubMed] [Google Scholar]
- 237.Wolinsky JB, Grinstaff MW. Therapeutic and diagnostic applications of dendrimers for cancer treatment. Adv Drug Deliv Rev. 2008;60:1037–1055. doi: 10.1016/j.addr.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 238.Wang B, Navath RS, Romero R, Kannan S, Kannan R. Anti-inflammatory and anti-oxidant activity of anionic dendrimer-N-acetyl cysteine conjugates in activated microglial cells. Int J Pharm. 2009;377:159–168. doi: 10.1016/j.ijpharm.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Das N, Dhanawat M, Dash B, Nagarwal RC, Shrivastava SK. Codrug: an efficient approach for drug optimization. Eur J Pharm Sci. 2010;41:571–588. doi: 10.1016/j.ejps.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 240.Seeberger LC, Hauser RA. Levodopa/carbidopa/entacapone in Parkinson’s disease. Expert Rev Neurother. 2009;9:929–940. doi: 10.1586/ern.09.64. [DOI] [PubMed] [Google Scholar]
- 241.Pinnen F, Cacciatore I, Cornacchia C, Sozio P, Cerasa LS, Iannitelli A, Nasuti C, Cantalamessa F, Sekar D, Gabbianelli R, Falcioni ML, Di Stefano A. Codrugs linking L-dopa and sulfur-containing antioxidants: new pharmacological tools against Parkinson’s disease. J Med Chem. 2009;52:559–563. doi: 10.1021/jm801266x. [DOI] [PubMed] [Google Scholar]
- 242.Sharma A, Kaur P, Kumar V, Gill KD. Attenuation of 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine induced nigrostriatal toxicity in mice by N-acetyl cysteine. Cell Mol Biol. 2007;53:48–55. [PubMed] [Google Scholar]
- 243.Di Stefano A, Sozio P, Cocco A, Iannitelli A, Santucci E, Costa M, Pecci L, Nasuti C, Cantalamessa F, Pinnen F. L-dopa- and dopamine-(R)-alpha-lipoic acid conjugates as multifunctional codrugs with antioxidant properties. J Med Chem. 2006;49:1486–1493. doi: 10.1021/jm051145p. [DOI] [PubMed] [Google Scholar]
- 244.Sozio P, Iannitelli A, Cerasa LS, Cacciatore I, Cornacchia C, Giorgioni G, Ricciutelli M, Nasuti C, Cantalamessa F, Di Stefano A. New L-dopa codrugs as potential antiparkinson agents. Arch Pharm (Weinheim) 2008;341:412–417. doi: 10.1002/ardp.200700228. [DOI] [PubMed] [Google Scholar]
- 245.Pinnen F, Cacciatore I, Cornacchia C, Mollica A, Sozio P, Cerasa LS, Iannitelli A, Fontana A, Nasuti C, Di Stefano A. CNS delivery of L-dopa by a new hybrid glutathione-methionine peptidomimetic prodrug. Amino Acids. 2012;42:261–269. doi: 10.1007/s00726-010-0804-z. [DOI] [PubMed] [Google Scholar]
- 246.Pinnen F, Cacciatore I, Cornacchia C, Sozio P, Iannitelli A, Costa M, Pecci L, Nasuti C, Cantalamessa F, Di Stefano A. Synthesis and study of L-dopa-glutathione codrugs as new anti-Parkinson agents with free radical scavenging properties. J Med Chem. 2007;50:2506–2515. doi: 10.1021/jm070037v. [DOI] [PubMed] [Google Scholar]
- 247.More SS, Vince R. Design, synthesis and biological evaluation of glutathione peptidomimetics as components of anti-Parkinson prodrugs. J Med Chem. 2008;51:4581–4588. doi: 10.1021/jm800239v. [DOI] [PubMed] [Google Scholar]
- 248.Vince R, Brownell J, Akella LB. Synthesis and activity of gamma-(L-gamma-azaglutamyl)-S-(p-bromobenzyl)-L-cysteinylglycine: a metabolically stable inhibitor of glyoxalase I. Bioorg Med Chem Lett. 1999;9:853–856. doi: 10.1016/s0960-894x(99)00097-9. [DOI] [PubMed] [Google Scholar]
- 249.Torres JL, Lozano C, Maher P. Conjugation of catechins with cysteine generates antioxidant compounds with enhanced neuroprotective activity. Phytochemistry. 2005;66:2032–2037. doi: 10.1016/j.phytochem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 250.Maher P, Lewerenz J, Lozano C, Torres JL. A novel approach to enhancing cellular glutathione levels. J Neurochem. 2008;107:690–700. doi: 10.1111/j.1471-4159.2008.05620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Ehrlich K, Viirlaid S, Mahlapuu R, Saar K, Kullisaar T, Zilmer M, Langel U, Soomets U. Design, synthesis and properties of novel powerful antioxidants, glutathione analogues. Free Radic Res. 2007;41:779–787. doi: 10.1080/10715760701348611. [DOI] [PubMed] [Google Scholar]
- 252.Ehrlich K, Ida K, Mahlapuu R, Kairane C, Oit I, Zilmer M, Soomets U. Characterization of UPF peptides, members of the glutathione analogues library, on the basis of their effects on oxidative stress-related enzymes. Free Radic Res. 2009;43:572–580. doi: 10.1080/10715760902918691. [DOI] [PubMed] [Google Scholar]
- 253.Kairane C, Mahlapuu R, Ehrlich K, Kilk K, Zilmer M, Soomets U. Diverse Effects of Glutathione and UPF Peptides on Antioxidant Defense System in Human Erythroleukemia Cells K562. Int J Pept. 2012;2012:124163. doi: 10.1155/2012/124163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Allen M, Zou F, Chai HS, Younkin CS, Miles R, Nair AA, Crook JE, Pankratz VS, Carrasquillo MM, Rowley CN, Nguyen T, Ma L, Malphrus KG, Bisceglio G, Ortolaza AI, Palusak R, Middha S, Maharjan S, Georgescu C, Schultz D, Rakhshan F, Kolbert CP, Jen J, Sando SB, Aasly JO, Barcikowska M, Uitti RJ, Wszolek ZK, Ross OA, Petersen RC, Graff-Radford NR, Dickson DW, Younkin SG, Ertekin-Taner N. Glutathione S-transferase omega genes in Alzheimer and Parkinson disease risk, age-at-diagnosis and brain gene expression: an association study with mechanistic implications. Mol Neurodegener. 2012;7:13. doi: 10.1186/1750-1326-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kiyohara C, Miyake Y, Koyanagi M, Fujimoto T, Shirasawa S, Tanaka K, Fukushima W, Sasaki S, Tsuboi Y, Yamada T, Oeda T, Miki T, Kawamura N, Sakae N, Fukuyama H, Hirota Y, Nagai M. GST polymorphisms, interaction with smoking and pesticide use, and risk for Parkinson’s disease in a Japanese population. Parkinsonism Relat Disord. 2010;16:447–452. doi: 10.1016/j.parkreldis.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 256.Wahner AD, Glatt CE, Bronstein JM, Ritz B. Glutathione S-transferase mu, omega, pi, and theta class variants and smoking in Parkinson’s disease. Neurosci Lett. 2007;413:274–278. doi: 10.1016/j.neulet.2006.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Li YJ, Scott WK, Zhang L, Lin PI, Oliveira SA, Skelly T, Doraiswamy MP, Welsh-Bohmer KA, Martin ER, Haines JL, Pericak-Vance MA, Vance JM. Revealing the role of glutathione S-transferase omega in age-at-onset of Alzheimer and Parkinson diseases. Neurobiol Aging. 2006;27:1087–1093. doi: 10.1016/j.neurobiolaging.2005.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Nishimura M, Kuno S, Kaji R, Yasuno K, Kawakami H. Glutathione-S-transferase- 1 and interleukin-1beta gene polymorphisms in Japanese patients with Parkinson’s disease. Mov Disord. 2005;20:901–902. doi: 10.1002/mds.20477. [DOI] [PubMed] [Google Scholar]
- 259.Whitbread AK, Mellick GD, Silburn PA, Le Couteur DG, Board PG. Glutathione transferase Omega class polymorphisms in Parkinson disease. Neurology. 2004;62:1910–1911. doi: 10.1212/01.wnl.0000125282.09308.b1. [DOI] [PubMed] [Google Scholar]
- 260.Biswas A, Sadhukhan T, Bose K, Ghosh P, Giri AK, Das SK, Ray K, Ray J. Role of glutathione S-transferase T1, M1 and P1 polymorphisms in Indian Parkinson’s disease patients. Parkinsonism Relat Disord. 2012;18:664–665. doi: 10.1016/j.parkreldis.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 261.Perez-Pastene C, Graumann R, Diaz-Grez F, Miranda M, Venegas P, Godoy OT, Layson L, Villagra R, Matamala JM, Herrera L, Segura-Aguilar J. Association of GST M1 null polymorphism with Parkinson’s disease in a Chilean population with a strong Amerindian genetic component. Neurosci Lett. 2007;418:181–185. doi: 10.1016/j.neulet.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 262.Vilar R, Coelho H, Rodrigues E, Gama MJ, Rivera I, Taioli E, Lechner MC. Association of A313 G polymorphism (GSTP1*B) in the glutathione-S-transferase P1 gene with sporadic Parkinson’s disease. Eur J Neurol. 2007;14:156–161. doi: 10.1111/j.1468-1331.2006.01590.x. [DOI] [PubMed] [Google Scholar]
- 263.Santt O, Baranova H, Albuisson E, Bignon YJ, Lucotte G. Interaction between GSTM1-null and CYP2D6-deficient alleles in the pathogenesis of Parkinson’s disease. Eur J Neurol. 2004;11:247–251. doi: 10.1046/j.1468-1331.2003.00756.x. [DOI] [PubMed] [Google Scholar]
- 264.Kelada SN, Stapleton PL, Farin FM, Bammler TK, Eaton DL, Smith-Weller T, Franklin GM, Swanson PD, Longstreth WT, Jr, Checkoway H. Glutathione S-transferase M1, T1, and P1 polymorphisms and Parkinson’s disease. Neurosci Lett. 2003;337:5–8. doi: 10.1016/s0304-3940(02)01286-7. [DOI] [PubMed] [Google Scholar]
- 265.Harada S, Fujii C, Hayashi A, Ohkoshi N. An association between idiopathic Parkinson’s disease and polymorphisms of phase II detoxification enzymes: glutathione S-transferase M1 and quinone oxidoreductase 1 and 2. Biochem Biophys Res Commun. 2001;288:887–892. doi: 10.1006/bbrc.2001.5868. [DOI] [PubMed] [Google Scholar]
- 266.Ahmadi A, Fredrikson M, Jerregard H, Akerback A, Fall PA, Rannug A, Axelson O, Soderkvist P. GSTM1 and mEPHX polymorphisms in Parkinson’s disease and age of onset. Biochem Biophys Res Commun. 2000;269:676–680. doi: 10.1006/bbrc.2000.2338. [DOI] [PubMed] [Google Scholar]
- 267.Nicholl DJ, Bennett P, Hiller L, Bonifati V, Vanacore N, Fabbrini G, Marconi R, Colosimo C, Lamberti P, Stocchi F, Bonuccelli U, Vieregge P, Ramsden DB, Meco G, Williams AC. A study of five candidate genes in Parkinson’s disease and related neurodegenerative disorders. European Study Group on Atypical Parkinsonism. Neurology. 1999;53:1415–1421. doi: 10.1212/wnl.53.7.1415. [DOI] [PubMed] [Google Scholar]
- 268.Menegon A, Board PG, Blackburn AC, Mellick GD, Le Couteur DG. Parkinson’s disease, pesticides, and glutathione transferase polymorphisms. Lancet. 1998;352:1344–1346. doi: 10.1016/s0140-6736(98)03453-9. [DOI] [PubMed] [Google Scholar]
- 269.Coppede F, Armani C, Bidia DD, Petrozzi L, Bonuccelli U, Migliore L. Molecular implications of the human glutathione transferase A-4 gene (hGSTA4) polymorphisms in neurodegenerative diseases. Mutat Res. 2005;579:107–114. doi: 10.1016/j.mrfmmm.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 270.Singh M, Khan AJ, Shah PP, Shukla R, Khanna VK, Parmar D. Polymorphism in environment responsive genes and association with Parkinson disease. Mol Cell Biochem. 2008;312:131–138. doi: 10.1007/s11010-008-9728-2. [DOI] [PubMed] [Google Scholar]
- 271.Wilk JB, Tobin JE, Suchowersky O, Shill HA, Klein C, Wooten GF, Lew MF, Mark MH, Guttman M, Watts RL, Singer C, Growdon JH, Latourelle JC, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Berg CJ, Sun M, Goldwurm S, Pezzoli G, Racette BA, Perlmutter JS, Parsian A, Baker KB, Giroux ML, Litvan I, Pramstaller PP, Nicholson G, Burn DJ, Chinnery PF, Vieregge P, Slevin JT, Cambi F, MacDonald ME, Gusella JF, Myers RH, Golbe LI. Herbicide exposure modifies GSTP1 haplotype association to Parkinson onset age: the GenePD Study. Neurology. 2006;67:2206–2210. doi: 10.1212/01.wnl.0000249149.22407.d1. [DOI] [PubMed] [Google Scholar]
- 272.Taylor MC, Board PG, Blackburn AC, Mellick GD, Le Couteur DG. Zeta class glutathione transferase polymorphisms and Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2001;70:407. doi: 10.1136/jnnp.70.3.407. [DOI] [PMC free article] [PubMed] [Google Scholar]