

Abstract
The very amino-terminal domain of the huntingtin protein is directly located upstream of the protein’s polyglutamine tract, plays a decisive role in several important properties of this large protein and in the development of Huntington’s disease. This huntingtin 1–17 domain is on the one hand known to markedly increase polyglutamine aggregation rates and on the other hand has been shown to be involved in cellular membrane interactions. Here, we determined the high-resolution structure of huntingtin 1–17 in dodecyl phosphocholine micelles and the topology of its helical domain in oriented phosphatidylcholine bilayers. Using two-dimensional solution NMR spectroscopy the low-energy conformations of the polypeptide were identified in the presence of dodecyl phosphocholine detergent micelles. In a next step a set of four solid-state NMR angular restraints was obtained from huntingtin 1–17 labeled with 15N and 2H at selected sites. Of the micellar ensemble of helical conformations only a limited set agrees in quantitative detail with the solid-state angular restraints of huntingtin 1–17 obtained in supported planar lipid bilayers. Thereby, the solid-state NMR data were used to further refine the domain structure in phospholipid bilayers. At the same time its membrane topology was determined and different motional regimes of this membrane-associated domain were explored. The pronounced structural transitions of huntingtin 1–17 upon membrane-association result in a α-helical conformation from K6 to F17, i.e., up to the very start of the polyglutamine tract. This amphipathic helix is aligned nearly parallel to the membrane surface (tilt angle ∼77°) and is characterized by a hydrophobic ridge on one side and an alternation of cationic and anionic residues that run along the hydrophilic face of the helix. This arrangement facilitates electrostatic interactions between huntingtin 1–17 domains and possibly with the proximal polyglutamine tract.
Abbreviations used: CD, circular dichroism; DPC, dodecyl phosphocholine; HFIP, 1,1,1,3,3,3-hexafluoro-2-propanol; NES, nuclear export sequence; NOESY, nuclear Overhauser effect spectroscopy; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; TFA, trifluoroacetic acid; SDS, sodium dodecylsulfate; TFE, trifluoroethanol; TOCSY, total correlation spectroscopy
Introduction
The occurrence and onset of Huntington’s disease are correlated with structural modifications within the amino-terminal region of the huntingtin protein (1–4). Genetic analysis of this hereditary disease identified the switch from the healthy to the pathogenic state when at least one allele encodes for more than 37 CAG repeats resulting in expanded polyglutamine tracts for mutated huntingtin. Similar observations have been made with other polyglutamine-related disorders (1), and it is thought that perturbations of neuronal membranes caused by such extended polyglutamine domains are responsible for the multiple symptoms of Huntington’s disease. The pathogenesis of Huntington’s disease has been associated with mitochondrial dysfunction (5,6) and the dysregulation of calcium hemostasis (7), among other cell biological alterations.
Importantly, huntingtin or fragments thereof are involved in a number of membrane-associated processes including intracellular vesicle trafficking (8), and these proteins have been shown to be associated with the endoplasmic reticulum (ER), the Golgi-apparatus, and endosomal vesicles (7,9). Huntingtin is involved in vesicular trafficking along microtubules (10–12), the formation of autophagic vacuoles (9), and has been found to interact with F-actin through its first 14 amino- acid residues (13). It is essential for ER maintenance (14,15) and a transcriptional regulator of REST/NRSF response genes (16). The protein is found in dynamic exchange between the late ER, the cytoplasm, and the nucleus (9). Huntingtin is targeted to the ER membrane by its first 17 amino acids(In previous publications reference was made to the huntingtin 1–17 domain as N17 or Htt17.) but this localization is in competition with translocation to the nucleus that occurs when the ER interactions are weakened such as upon stress (9). Export of huntingtin from the nucleus is mediated via NESs. One of them has been identified within the same first 17 residues of its amino-terminus (17,18), whereas others using different interaction partners are located further downstream in the sequence (19). Biochemical analysis indicates that nuclear export through the huntingtin 1–17 domain occurs through interactions with exportin 1 in a Ran-dependent manner (17,18).
Notably, the cellular localization of huntingtin is heavily influenced by its first 17 amino acids (7) and to interact with membranes in vivo huntingtin requires these amino-terminal residues directly preceding the polyglutamine region (7,9). In particular, huntingtin 1–17 has been shown to be involved in huntingtin membrane interactions when at the same time this domain is known to interact with itself as well as with polyglutamines (20). Importantly, the presence of huntingtin 1–17 enhances polyglutamine aggregation (7,9,20–22), and polyglutamine has been shown to interact with itself, huntingtin 1–17 as well as with other proteins (22–25). As a consequence, the resulting perturbations in protein structure have been suggested to initiate polyglutamine oligomerization in a manner that strongly depends on the number of this amino acid making up the tract. Thereafter, the polyglutamines assemble into stable fibrillar structures, which form the nucleus for additional aggregation processes (23–25).
Although mutagenesis, cell biological, in vivo, and biophysical experiments all point to an important role of huntingtin 1–17 in the membrane targeting and interactions of huntingtin (7,9,26–30), very little data exists characterizing the huntingtin 1–17 structure in membranes. In particular, knowledge of the structure of the huntingtin 1–17 domain in membranes promises to increase our understanding not only of its interactions and properties within such environments but also to shed light on its capacity to self-associate and to interact with other proteins. This prompted us to determine the membrane structure of huntingtin 1–17 by a combination of two-dimensional (2D) solution- and solid-state NMR spectroscopies.
Whereas solution NMR spectroscopy is a well-established technique to investigate the structure and dynamics of peptides and proteins in isotropic environments, solid-state NMR is a powerful method to study peptides and proteins associated with lipid bilayers (31–34). Although solid-state NMR angular constraints have the potential for a full structure determination of membrane-associated polypeptides (35), this can become a time-consuming and expensive task. Therefore, we chose an alternative approach where we quantitatively tested a number of structural models against a set of solid-state NMR angular constraints. In a first step the qualitative agreement between the micellar and the bilayer structures was evaluated by analyzing if the solid-state NMR spectra of five 2H and 15N labeled sites spread over the sequence (cf. Methods section) match the outlines of the secondary structure elements observed in the presence of DPC. The most stringent test arose from residues, which are close to or define the ends of the huntingtin 1–17 helical domain. In a second step the angular restraints obtained from the structured region of the peptide reconstituted in oriented phospholipid bilayers were compared in a quantitative manner to the conformational ensemble observed in micellar environments (i.e., from the labeled sites 15N-Leu7, 2H3-Ala10, 15N-Phe11, and 15N-Phe17). By combining solution and solid-state NMR data in a novel manner it was thus possible not only to validate the micellar conformation in the context planar lipid bilayers and to refine the structure by additional angular restraints but at the same time to also consider peptide motions.
This work thereby complements a previous analysis where the lipid-dependence of the huntingtin 1–17 membrane interactions were investigated (30). Whereas in this prior work the membrane-association and pore-forming capacities of huntingtin 1–17 were quantitatively analyzed as a function of lipid composition; here, we describe and discuss the high-resolution NMR structure of the membrane-associated domain in quantitative detail. Although the membrane-partitioning constants and -activities have been found to be strongly dependent on lipid composition, the membrane topology of the membrane-associated huntingtin 5–17 helical domain in pure phospholipid bilayers appears to be quite stable when a set of chemical shifts and quadrupolar splittings are compared to each other as a function of lipid composition (30). Whereas in this previous work the pore-forming activities and CD-spectra were obtained from the native huntingtin 1–17 peptides also used here, much of the previous solid-state NMR data were obtained from a peptide carrying an additional proline at the amino-terminus (P-huntingtin 1–17). Importantly, when the same labeled sites are compared to each other similar solid-state spectra were obtained from the helical domains of the proline mutant when compared to the native sequence (cf. this work and reference (30). Therefore, the high-resolution structure and topology determined in this work for oriented POPC membranes are relevant also for other phospholipid membranes. Although the novel experimental approach and the data that led to the huntingtin 1–17 structure, topology, and dynamics are presented here for the first time, we have already used some of the structural coordinates to analyze these and previous data (30) in a consistent manner. Furthermore, in this work the structural and topological details of huntingtin 1–17 will be discussed in the context of regulatory posttranslational modifications and compared to other F-actin binding sequences and nuclear localization signals. Finally, the structure, topology, and dynamics of bilayer-associated huntingtin 1–17 may have important implications for the aggregation of the neighboring polyglutamine tract.
Materials and Methods
Deuterated TFE (d3) was purchased from Sigma Aldrich (Steinheim, Germany). Deuterated DPC (d38) was from Cambridge Isotope Laboratories (Andover, MA). Amino-acid derivatives and other reagents for peptide synthesis were from Novabiochem-Merck (Darmstadt, Germany). All lipids were purchased from Avanti Polar Lipids (Alabaster, AL).
Peptide synthesis of specifically labeled huntingtin 1–17 peptides
The peptides [15N-Phe11]-huntingtin 1–17, [15N-Leu7, 2H3-Ala10]-huntingtin 1–17, and [15N-Phe17, 2H3-Ala2]-huntingtin 1–17 were prepared by automated solid-phase peptide synthesis using a Millipore 9050 synthesizer, the Fmoc (9-fluorenylmethyloxycarbonyl) chemistry and purified by semipreparative HPLC as described previously (36). The sequence of huntingtin 1–17 is MATLEKLMKAFESLKSF and was prepared with an amidated carboxy-terminus. Therefore, at neutral pH the peptide carries a net charge of about +1. At the positions shown in bold the natural abundance Fmoc-protected amino acid derivatives (Bachem, Heidelberg, Germany and Applied Biosystems, Weiterstadt, Germany) were replaced with their isotopically enriched analogs (eurisotope, Paris, France) one or two at a time. The identity and high purity of the product (>90%) was confirmed by NMR spectroscopy and MALDI mass spectrometry. After lyophilisation, the TFA counter ions were exchanged twice in 5% acetate (v/v). Before functional assays or NMR measurements all peptides were exposed to HFIP/TFA for disaggregation as described previously (37).
CD spectropolarimetry
CD spectropolarimetry was performed in 50 mM sodium phosphate buffer, 75 mM sodium chloride, pH 7.3 following protocols described in (36). The dilution factor was recalculated after each experiment. Four scans from 250 to 195 nm with a scan speed of 20 nm/min were averaged and the CD signal of the solvent or buffer was subtracted subsequently. Analysis of spectra was performed using the spectra analyzer (Jasco, Tokyo, Japan) and the Origin software (Northhampton, MA). The secondary structure of the peptide was determined by using the spectral fitting methods CONTIN/LL, CDSSTR, and SELCON in the CDpro package from which the average was calculated (38).
NMR spectroscopy of huntingtin 1–17 in micellar environments
For 2D solution NMR analysis the disaggregated peptide was dissolved in the presence of 100 mM DPC-d38 micelles at a concentration of 0.5 mM in 50 mM sodium phosphate buffer, 0.001% (w/v) sodium azide, 10% (v/v) D2O pH 6.0. Alternatively, the peptide solution was prepared in 50% TFE-d3, 40% phosphate buffered saline and 10% D2O (v/v/v). For both solutions homonuclear (1H-1H) two-dimensional TOCSY and NOESY spectra were recorded at 298 K on a Bruker Avance NMR spectrometer operating at a 1H frequency of 600 MHz (Bruker Biospin, Rheinstetten, Germany) and being equipped with a 5 mm BBI 1H/2H probe head with z-gradient. The mixing times were 80 ms for TOCSY, and 150 ms and 200 ms for NOESY spectra, respectively. NOESY- and TOCSY-spectra were collected using gradient pulses and the DIPSI-2 spin lock, respectively, as well as the excitation sculpting technique for water suppression (39). For experiments with DPC micelles the spectral width was chosen to be 7212 Hz and 8–16 transients for 512 t1-increments with 4096 complex data points for each free-induction decay. The spectral width for experiments in TFE was set to 6602 Hz and 512 t1-increments with 8–16 transients for 2048 complex data points were collected. The relaxation delay was 2 s in all experiments. Before Fourier transformation, a squared sine bell function was applied in both dimensions, as well as linear prediction in t1. All spectra were processed using NMRPIPE/NMRDraw (40) and analyzed by NMRVIEW (41).
Structure calculation
The structure calculations were performed using Xplor-NIH 2.17 (42). NOE intensities from the 200 ms NOESY spectrum were classified into strong, medium, and weak, corresponding to upper distance restraints of 2.8, 3.4, and 5.0 Å, respectively. 200 structures were calculated, starting from an extended structure, using a simulated annealing protocol with the topology and parameter sets topallhdg and parallhdg, respectively. During high temperature dynamics 10,000 steps were taken per cycle at 1000 K with a subsequent decrease of temperature by 6000 steps in the cooling step to 100 K and subsequently refined by molecular dynamics in explicit water (43). Finally, 20 conformers with the lowest energy and no NOE distance violation >0.5Å were calculated to generate the ensemble of the peptide’s structure. The quality of the ensemble was validated using the PSVS tool (44). The pairwise root mean-square deviation (RMSD) was calculated as well as all graphical representations were generated using the program MOLMOL 2K.2 (45). The average structure of both ensembles, resembling the structure of huntingtin 1–17 in TFE or DPC micelles, was generated using the average.inp Xplor-NIH script, and subsequently energy minimized by a final water refinement step. Both coordinate files of the ensembles are accessible through the Protein Data Base (PDB) database (2LD0 and 2LD2); the chemical shift information is available through the Biological Magnetic Resonance Bank (BMRB) database, accession nos. 17642 for huntingtin 1–17 in 50% (v/v) TFE or 17644 in the presence of DPC micelles, respectively.
Paramagnetic line broadening experiments
To investigate the topology of huntingtin 1–17 in micelles 0.5 mM disaggregated peptide and 200 mM DPC-d38 were dissolved in 90% water/10% (v/v) D2O. Spin-labeled 16-doxyl stearic acid (Sigma Aldrich, Steinheim, Germany) was solubilized in methanol-d4 and added to the NMR sample at final concentrations of 1.0 and 3.5 mM. MnCl2 was dissolved in water, sterilized by filtration, and subsequently added to the sample at concentrations of 0.2 and 0.5 mM. For all experiments, homonuclear 2D TOCSY spectra at an isotropic mixing time of 60 ms at 313 K were recorded. Reference spectra lacking the spin-labels were recorded at identical conditions. The line broadening of the fingerprint HN-Hα region was taken into consideration for data analysis.
Solid-state NMR spectroscopy
Samples for solid-state NMR spectroscopy were prepared by dissolving 7 mg of huntingtin 1–17 peptide in 100% formic acid and 105 mg of POPC lipid in HFIP. Both solutions were mixed and carefully applied onto 25 ultrathin cover glasses (8 × 22 mm; Paul Marienfeld GmbH & Co. KG, Lauda-Königshofen, Germany) as described previously (46).
Solid-state NMR spectra were recorded on a Bruker Avance wide-bore NMR spectrometer operating at 9.4 T. A commercial double-resonance solid-state NMR probe modified with flattened coils of dimensions 15 × 4 × 9 mm was used (47). Proton-decoupled 15N solid-state NMR spectra were acquired using a cross-polarization sequence and processed as described previously (48). NH4Cl (40.0 ppm) was used as an external reference corresponding to 0 ppm for liquid NH3. An exponential apodization function corresponding to a line broadening of 50 Hz was applied before Fourier transformation.
Deuterium solid-state NMR spectra were recorded using a quadrupolar echo pulse sequence (49) with parameters detailed in (50). The spectra were referenced relative to 2H2O (0 Hz). An exponential apodization function corresponding to a line broadening of 300 Hz was applied before Fourier transformation.
Proton-decoupled 31P solid-state NMR spectra were recorded using a (90°-τ-180°-τ-acquisition) Hahn echo pulse sequence (51) with phase-cycling (52). The 1H B1 field was 40 kHz, the 90° pulse length 2.5 μs, the echo delay (τ) 40 μs, and the spectral width 40 kHz. For acquisition 256 scans of 2048 data points were recorded with a repetition time of 3 s. The spectra were referenced relative to 85% phosphoric acid (0 ppm). An exponential apodization function corresponding to a line broadening of 20 Hz was applied before Fourier transformation.
Calculation of orientational restraints from the solid-state NMR spectra
To evaluate the peptide orientations that agree with the experimental spectra, a coordinate system was defined with the tilt angle being the angle between the long axis of the helix and the membrane normal, and a pitch angle between the membrane normal and the line within the arbitrary plane of peptide helical wheel projection (cf. Fig. 5 d for angle definitions).
Figure 5.
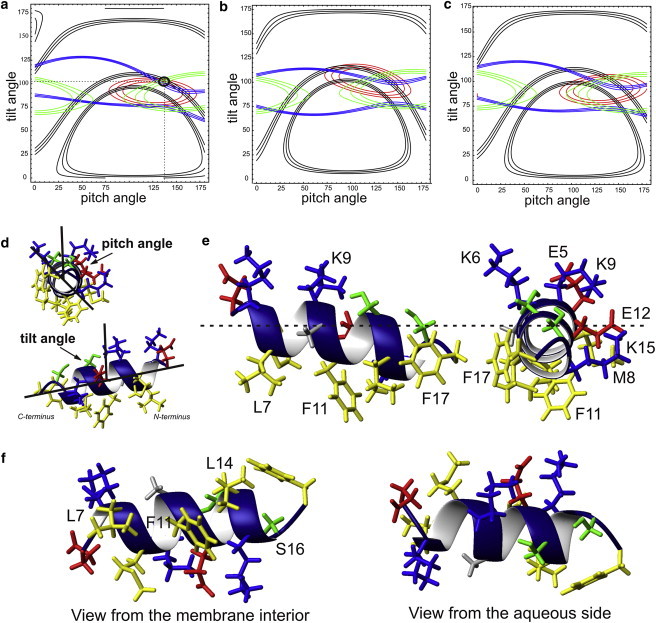
Angular restrictions obtained from solid-state NMR spectra of huntingtin 1–17 reconstituted in oriented POPC bilayers. (a) The possible alignments of the low-energy conformer 3 obtained by solution NMR in the presence of DPC micelles, structure calculation, and refinement are represented by their helical tilt and the rotational pitch angles. The solid black lines represent angular pairs that agree with the experimental 2H quadrupolar splitting obtained from 2H3-Ala-10 (11 ± 2.5 kHz), the 15N chemical shifts of 15N-Leu-7 (red; 71.5 ± 2.5 ppm), 15N-Phe-11 (green; 78.5 ± 3 ppm), and 15N-Phe-17 (blue; 88 ± 1.7 ppm) (cf. Fig. 4, a, c, e, and g). The error bars correspond to the line width at half-height. The tilt/rotational pitch angular pair is circled where all experimental data agree. In (b) the related analysis performed with the lowest energy structure 1 and in (c) with the average of the 20 low-energy conformations are depicted. Peptide dynamics were taken into account by wobbling motions of the helix (10° Gaussian distribution) as well as pitch angle fluctuations around the helix long axis (18°). (d) Exhibits the pitch angle and tilt angle definitions with some complimentary views to those depicted in (e) and (f). (e) Structural details of residues 5–17 are shown when viewed from the side or from the carboy-terminus, or (f) when viewed from the membrane interior (left) or the aqueous side (right). The hydrophobic residues are shown in yellow, alanine in gray, serines in green, glutamates in red, and lysines in blue.
If not indicated otherwise, the calculations were performed using the 15N chemical shift main tensor elements (56, 81, 223) ppm (53) and 74 kHz for the maximum quadrupolar splitting for the alanine 2H3C-group (54), respectively. By successively changing the tilt and pitch angles of the peptides (50 × 50 steps) the three-dimensional topological space was systematically screened and the corresponding 15N chemical shift and quadrupolar splitting calculated (50). Wagging and azimuthal fluctuations were assumed to be independent and were taken into account by averaging the resonance values on the ensemble of orientations with corresponding Gaussian distributions. The motional regimes tested here have been shown previously to best fit the solid-state NMR data of amphipathic helices of similar dimensions (e.g. (55)). Contour plots (cf. Fig. 5, a–c), mark the angular restrictions that agree with the experimental results.
Results
To find optimal conditions for high-resolution structural investigations and at the same time to obtain insight into the secondary structure preferences of the huntingtin 1–17 domain a number of sample conditions were screened using CD spectroscopy. Whereas in aqueous buffer the peptide adopts mostly random coil conformations with a helix content of only 10%, this value increases to ∼70% in a more hydrophobic environment like TFE/water mixtures, or upon stepwise addition of SDS or DPC detergents to aqueous buffer (Fig. 1). The critical micelle concentrations of these detergents are 2 mM and 1 mM, respectively (56). Notably, the critical micellar concentration of SDS is strongly dependent on the presence of electrolytes and the value indicated here represents the experimental conditions of 50–100 mM salt used in this work.
Figure 1.
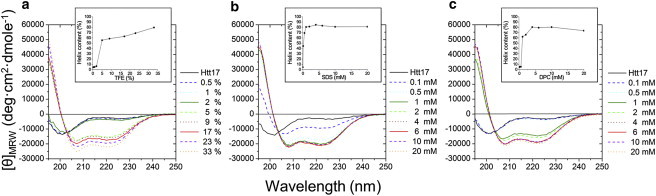
CD spectra of 50 μM huntingtin 1–17 in the presence of increasing amounts of TFE (a), SDS (b), and DPC micelles (c). The insets show the α-helix content calculated from the CDPro-package using three different algorithms.
At modest detergent concentrations the helical content of huntingtin 1–17 obtained in the presence of DPC or SDS agrees well with the limiting values obtained upon titration of phospholipid membranes (30). The molecular structure of the DPC polar group resembles closely the headgroup of phosphatidylcholines, a lipid abundant in eukaryotic membranes. Therefore, the NMR structural analysis was performed in the presence of DPC and validated in oriented POPC bilayers where the fatty acyl composition represents well the hydrophobic thickness and the saturated-unsaturated fatty acyl chain composition of biological membranes.
NMR structures of huntingtin 1–17 in micellar environments
To obtain a detailed view on the huntingtin 1–17 structure in membrane environments the peptide was investigated in the presence of 100 mM deuterated DPC using 2D 1H/1H-NMR spectroscopy (Fig. S1, a and b in the Supporting Material). The CD spectroscopic analysis is indicative that the conformational transition from random coil to helical is complete at this detergent concentration (Fig. 1 c). The NOE assignment and distance constraints are graphically summarized in Fig. S1 c and the statistics of the structure calculations in Table S1. From these experimental data 100 structures were calculated by simulated annealing, followed by refinement in explicit water and their energies evaluated. Using standard protocols an averaged structure was obtained from the 20 lowest energy structures, which was further energy minimized. In the presence of detergent micelles huntingtin 1–17 forms an amphipathic α-helix from residue 6 to 17, where the conformation of the first five residues is characterized by a large RMSD (Fig. 2 a). Notably, within the helical domain the hydrophobic side chains all orient toward the core of the DPC membrane when at the same time the charged residues accumulate on the opposite face.
Figure 2.
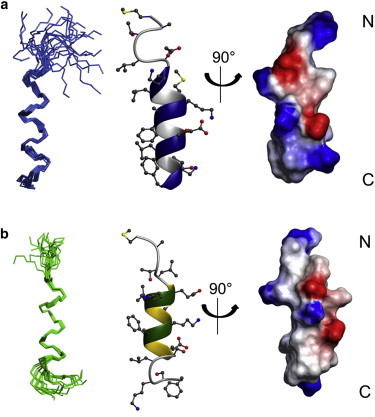
Three-dimensional structures of huntingtin 1–17 in 100 mM DPC micelles or TFE/buffer 50/50 (v/v). (a) The ensemble of the 20 best structures is shown (left) and reveals a more rigid carboxy-terminus for the conformation of huntingtin 1–17 in the presence of 100 mM DPC micelles compared to the flexible termini in TFE/buffer 50/50 v/v (b). The ribbon representation of the averaged structure (center) shows the α-helical conformation from K6 to F17 in the presence of 100 mM DPC micelles (a) and from residue E5 to E12 in TFE/buffer 50/50 v/v (b). Notably in the presence of DPC micelles the hydrophobic side chains are all oriented along one side of huntingtin 1–17, shown for the electrostatic surface potential with unipolar, negatively and positively charged amino-acid residue side chains in white, red, and blue, respectively (right).
In a next step the topology of DPC-associated huntingtin 1–17 was investigated by the addition of paramagnetic relaxation reagents (Fig. 3). The residual amplitude in the presence of 0.2 and 0.5 mM of the water soluble Mn2+ ion or of 1 and 3.5 mM of the hydrophobic 16-doxyl-stearate are shown in Figs. 3, a and b, respectively. Clearly, the amino-terminus and hydrophilic side chains of the helical region are mostly affected by the Mn2+ ions, whereas the hydrophobic residues of the helix strongly interact with the doxyl group deeply buried in the core of the micelles. The helical oscillation is most obvious when the ratio of the residual amplitude in the presence of hydrophilic/hydrophobic quencher is shown (Fig. 3 c). Although these data are only qualitative in nature, they suggest that the amphipathic huntingtin 1–17 domain localizes along the micellar interface.
Figure 3.
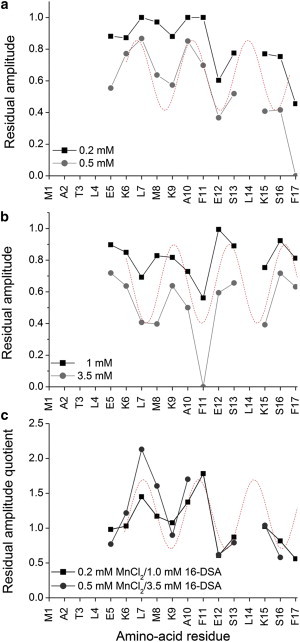
Paramagnetic line broadening caused by the addition of Mn2+ (a) or 16-doxylstearic acid (b) to 0.5 mM huntingtin 1–17 in 200 mM DPC-d38. The calculation of the quotient of the residual amplitudes (c) indicates that the amino-acid side-chains Leu7, Met8, Ala10, and Phe11 face the hydrophobic environment of the micelles. In these cases, the residual amplitude is small in the presence of 16-DSA but elevated in the presence of Mn2+. The inverse correlation holds for the hydrophilic face of the helix. The dotted trace shows a simulated α-helical wave. Where data points are missing the TOCSY HN-Hα crosspeaks are absent (residues 1–4) or cannot be resolved (residue 14).
For comparison the huntingtin 1–17 structure was determined in 50% (v/v) TFE in water (Fig. S1, d–f and Table S1). In this environment the helical part of huntingtin 1–17 extends from residues 5 to 12 and the termini exhibit conformations with high RMSD (Fig. 2 b). Furthermore, although the Ramanchandran angles of the central residues occupy the α-helical region the helical pitch is smaller than in the presence of DPC micelles and ridges of negatively and positively side chains form parallel to the helix long axis (Fig. 2 b). Clearly the TFE/water mixture represents only the average hydrophobicity of the membrane but not its interfacial properties and, as will be reported below, already the outlines of the helix disagree with the solid-state NMR measurements in supported phospholipid bilayers. On the other hand, the DPC membranes provide a membrane system mimicking quite well the interface of a phosphatidylcholine membrane, when at the same time being accessible to solution NMR techniques. However, also for this micellar system differences persist when its curvature and molecular packing are compared to phospholipid bilayers. Therefore, it is important that the micellar data are validated in bilayer environments. Consequently, we tested if the solid-state NMR orientational restraints, which were obtained from the selectively labeled sites of huntingtin 1–17, agree with the structural coordinates that result from the NMR analysis in DPC micellar environments.
Topological and structural analysis of bilayer-associated huntingtin 1–17 by oriented solid-state NMR spectroscopy
To gain insight into the interactions of huntingtin 1–17 with phospholipid bilayers several polypeptide sequences were prepared by solid-phase peptide synthesis and reconstituted into uniaxially oriented POPC phospholipid bilayers. To perform a topological and structural analysis of huntingtin 1–17 a labeling scheme was applied, where selected positions were enriched close to 100% with 15N or 2H isotopes. Three different types of solid-state NMR spectra were recorded from these samples (Fig. 4). First, due to the unique properties of the 15N amide chemical shift tensor, 15N chemical shift measurement of backbone-labeled polypeptides provide a direct measure of the approximate tilt angle of the helical domain (57). Whereas transmembrane helical alignments are characterized by 15N chemical shifts in the 200 ppm region, peptides that orient along the membrane surface exhibit values <100 ppm. Second, when combined with 2H solid-state NMR investigations of methyl deuterated alanines accurate rotational pitch and tilt angle information is obtained (33,50). Alternatively, the helical tilt and rotational pitch angles of helical peptides have been established from a series of deuterated alanines (55,58). In addition, oriented solid-state NMR measurements test if residues that are part of the helical structure in DPC micelles remain helical also in planar lipid bilayers, because highly mobile (random coil) regions resonate at the isotropic chemical shift positions and in the case of 15N are also characterized by inefficient cross-polarization (59). Third, the 31P NMR spectra of these samples are an indicator of the alignment of the phospholipid headgroup as well as the membrane macroscopic phase properties (60) and such control experiments were systematically recorded from all samples.
Figure 4.
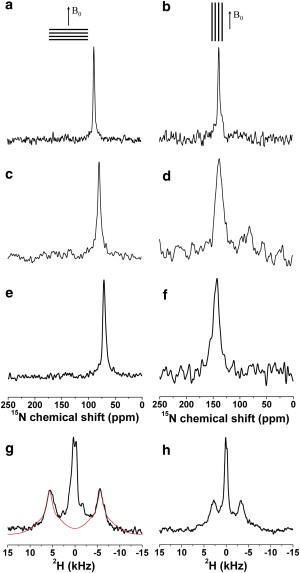
Proton-decoupled 15N- (a–f) as well as 2H quadrupolar-echo solid-state NMR spectra (g and h) of 2.5 mol % huntingtin 1–17 peptides reconstituted into uniaxially oriented POPC bilayers. Huntingtin 1–17 was labeled with 15N at position Phe17 (a,b), Phe11 (c,d), Leu7 (e,f), or with 2H3 at Ala10 (g,h). The glass plate normal was aligned parallel (a, c, e, g) or perpendicular to Bo (b, d, f, h), as illustrated on the top of each column. Panel g also shows a spectral simulation for a sample mosaicity with Gaussian distribution of 3°. The line broadening applied during this simulation of the 2H spectrum was 1.5 kHz.
Fig. 4, a, c, and e, shows proton-decoupled 15N solid-state NMR spectra of huntingtin 1–17 labeled with 15N at Phe17 (Fig. 4 a), Phe11 (Fig. 4 c), and Leu7 (Fig. 4 e) with anisotropic 15N chemical shifts of 88.2 ± 0.9 ppm, 78.9 ± 1.5 ppm, and 71.2 ± 1.7 ppm, respectively. The data indicate a nearly perpendicular orientation of the 15N-1H amide bonds relative to the bilayer normal, and consequently, an alignment of the peptide helix parallel to the membrane surface (33). The solid-state NMR data are indicative of an interfacial localization of huntingtin 1–17 in POPC bilayers, similar to the one that has been observed in DPC micelles (Fig. 3). The 2H NMR spectrum shown in Fig. 4 g exhibits a quadrupolar splitting of 11 kHz, a value that aligns the Cα–Cβ bond of Ala10 at an instantaneous angle of either 49° or 61° relative to the bilayer normal (depending on the sign of the quadrupolar splitting). In contrast, a much smaller quadrupolar interaction is observed for 2H3-Ala2 (not shown) in agreement with the nonhelical structure observed for the most amino-terminal residues in DPC micelles (Fig. 2 a). Therefore, the spectra of the 15N and the 2H3-Ala10 labeled sites show a well-structured polypeptide in the presence of the lipid bilayer environment even at the outermost positions of the helical domain (Fig. 4), suggesting that the helical conformation of residues 6 to 17 found in micellar environments is also present when huntingtin 1–17 is associated with phospholipid bilayers. In contrast, the solid-state NMR data obtained in phospholipid bilayers is in disagreement with the helical outlines observed in the TFE/water environment (residues 5–12; Fig. 2 b).
The solid-state NMR measurements from the labeled positions 7, 10, 11, and 17 (Fig. 4, a, c, e, and g), restrict the possible helix orientations relative to the membrane normal (33,61). Therefore, these experiments also permit a more detailed evaluation of structural models of huntingtin (residues 6–17), including the conformational ensemble of huntingtin 1–17 found in micellar environments (Fig. 2 a). The topological analyses shown in Fig. 5, a–c, illustrate the simulations of the 15N chemical shifts and 2H quadrupolar splittings of the helical part of the huntingtin 1–17 DPC micellar structure for all possible peptide alignments in the membrane. Whenever the calculated and the experimental values agree (including experimental uncertainties), the combination of tilt and orientational pitch angles is highlighted in the graph (Fig. 5, a–c). The restriction plots that arise in this manner for three 15N (Leu7, Phe11, Phe17) and the deuterium labels (Ala10) are outlined in Fig. 5, a–c, by red, green, blue, and black traces, respectively. To take into account the dynamics of peptides that interact with liquid disordered bilayers, wagging motions of the peptide as well as azimuthal movements around the helix long axis were systematically taken into account (Figs. 5, a–c, and Fig. S2). The effects of such motions on the 15N chemical shift anisotropy are presented in Fig. S3.
When the averaged structure of the 20 low-energy conformations (Fig. 2 a) is considered there is no perfect match with all four solid-state NMR topological constraints (Fig. 5 c). Notably, the structural models shown in Fig. 2 represent an ensemble of low-energy conformations that agree with the experimental NMR constraints in micellar or TFE/water environments. However, solution NMR structures are based on a limited number and precision of geometrical constraints, and adding complementary information from solid-state NMR further refines the conformational analysis (62). In addition, some of these conformers are expected to better represent the situation in phospholipid bilayers, thereby reflecting differences in planarity as well as the detailed molecular structures when the bilayer is compared to the curved micellar interfaces. Therefore, we also tested the agreement with solid-state NMR data of individual conformers of the ensemble, where the numbering of individual structures represents the resulting energy from the molecular dynamics calculations.
Notably, the lowest energy structure 1, like several others, does not match well the solid-state NMR orientational restraints from supported phospholipid bilayers (Fig. 5 b), but a perfect fit is obtained for conformations 3 or 20 where all four orientational restraints intersect at tilt/rotational pitch angles around 103°/137° (Fig. 5 a). When taking into account experimental errors, uncertainties in the tensor values, or the dynamics of the peptide, the very details of the topological analysis vary in a narrow range, but importantly, good agreement between individual structures and all four solid-state NMR restraints is maintained (cf. Fig. S2 and Fig. S3). Therefore, of all the models tested the membrane structure of huntingtin 1–17 is best described by the conformer 3 where the helical part is oriented at tilt/rotational pitch angles of 103°±5°/137°±5°. The corresponding structural arrangement of huntingtin 5–17 is illustrated in Fig. 5, e and f, and the conformational differences between structures 1, 3, and 20 are visualized in Fig. S4. By far the largest deviation was observed for Phe17 when conformer 1 (no match, Fig. 5 b) and 3 (good match, Fig. 5 a) are compared to each other and where the alignment difference of the NH vector is ∼20° (Fig. S4 a). Fig. S4 and the error estimates from Fig. S2 thereby also provide an indication of the limited range of alternative conformational models that can describe the membrane-structure of huntingtin 1–17.
Additional insight into the structural and dynamic properties of membrane-associated peptides was obtained when the same samples used to record the spectra shown in Fig. 4, a, c, e, and g were tilted by 90°. In such an arrangement the membrane normal is perpendicular to the magnetic field direction (Bo). Thus, rotational diffusion positions an in-plane oriented helix at any alignment relative to the magnetic field direction, including parallel and perpendicular to Bo and all possible orientations in between. It is therefore expected that the resulting NMR spectra exhibit a broad assembly of resonances where all orientations are represented and add up to a circular powder pattern line shape (63). However, if rotational diffusion is fast when compared to the 15N chemical shift anisotropy or the 2H quadrupolar interactions, respectively, an averaged spectrum is obtained.
The proton-decoupled 15N solid-state NMR spectrum of the 15N labeled amide at position 17 at a 90°-sample alignment shows a single 15N resonance at 138 ppm (Fig. 4 b), which indicates that the peptide diffuses fast around the membrane normal when compared to the 10−4 s timescale of the 15N chemical shift anisotropy. The latter covers a range from ∼60 to 230 ppm (53), which is almost two orders of magnitude increased when compared to the line width at half-height of the observed resonance (±3 ppm). Notably, the chemical shift value of the tilted sample confirms the in-plane alignment of the helical domain (57,64). When the amide position of Leu7 was investigated in the tilted arrangement a 15N chemical shift of 144 ppm was observed (Fig. 4 f), which is indicative of fast rotational averaging around the membrane normal also for this position. A related behavior was also found for 15N-Phe11 (138 ppm, Fig. 4 d). The chemical shift positions obtained from the 0° and 90° aligned samples allow one to calculate the isotropic chemical shift positions one by one from the three 15N labeled sites. They are all within 120 ± 2 ppm suggesting that the wide range of tensor variations taken into account for the calculation of systematic errors (Fig. S2) also cover the differences between individual sites.
Fast rotational averaging at even faster timescales becomes obvious when the [2H3-Ala10]-huntingtin 1–17 peptide was investigated by deuterium solid-state NMR spectroscopy (Fig. 4 h). In the case of the 2H3-alanine label a reduction of the quadrupolar splitting by −1/2 is in agreement with fast motional averaging around the membrane normal at an order of magnitude decreased timescale (10−5 s) (63).
Discussion
Despite intensive research and although the hereditary modifications of huntingtin have been clearly identified and correlated with Huntington’s disease (65) the biological function and the molecular mechanisms of how the pathogenesis develops remain obscure. On the one hand, this is due to the large number of interaction partners of this protein and the lack of high-resolution structural information. On the other hand, the situation is further complicated by the localization of huntingtin in a great number of organelles including the cytoplasm, the nucleus, the plasma membrane, ER, Golgi apparatus, endocytic vesicles, and mitochondria (5–7,9,26,66,67), which makes the unambiguous identification of the biological functions of huntingtin or of the disease-promoting biochemical processes difficult.
The amino-terminal domain of huntingtin encompassing the first 17 amino acids (huntingtin 1–17) has proven tightly linked to polyglutamine aggregation and the resulting toxicity of the protein (7,9,22,29). In this work, we show that huntingtin 1–17 is capable of undergoing a structural transition from random coil in aqueous solution to α-helical conformations in the presence of micelles or isotropic membrane-mimetic solvents (Figs. 1 and 2). Notably, the increase in helicity saturates at modest detergent concentrations of 1–4 mM or reaches similarly high values at TFE concentrations of as little as 17% (v/v) suggesting that a nucleation core for the structural transition exists already in aqueous solution (20–22). Consequently, only a small energetic barrier needs to be overcome during the structural transitions (Fig. 1). Indeed, helix forming propensities have become evident also in aqueous solution by CD- (Fig. 1 and (9)) and solution NMR spectroscopies where transient helical conformations have been detected involving residues 5 to 7 (22), in molecular dynamics simulations (21) as well as in the x-ray crystallographic analysis of a MBP-huntingtin 1–17-polyglutamine fusion protein (68). The biological relevance of the α-helical conformation of huntingtin 1–17 is strengthened further by the results of mutagenesis experiments (9).
In the past the high curvature of micellar systems has been thought to be responsible for subtle conformational alterations, especially at the termini of peptides (cf. e.g. (69)). It is therefore interesting to note that the solid-state NMR data obtained in POPC bilayers agree with a high mobility of the amino-terminus (not shown), and confirm the participation of residues 7, 10, 11, and 17 in the α-helical domain (Fig. 4). The solid-state and solution NMR data also agree well with the CD line shape analysis of huntingtin 1–17 where up to ∼70% of the peptides adopt helical conformations in the presence of lipid bilayers (30) or in the presence of detergent micelles (Fig. 1). Notably, the solid-state NMR topological analysis confirms a α-helical structure at position 7, i.e., close to the first helical residue in micellar environments, and of the very carboxy-terminal amino acid (Fig. 4, a and e). This carboxy-terminal helical conformation is probably stabilized by interactions between the negative end of the helix dipole and the lysine at position 15, similar to previous observations made with other polypeptides (e.g. (70)).
Furthermore, by comparing the solution and solid-state NMR data in detail it is possible to select a small subset of conformations from the solution NMR conformational ensemble that fits best the orientational restraints derived from supported bilayers. It should be emphasized that the high-resolution NMR structure of huntingtin 1–17 (Fig. 2 a) is of similar quality than those obtained for other peptides of similar size in micellar environments. However, the solid-state NMR approach has been shown to be a sensitive indicator of angular deviations, which under favorable conditions can detect differences as small as 1° (33,50). Therefore, perfect agreement between multiple orientational restraints requires a coordinate file that closely matches the bilayer structure (cf. Fig. S4), and combining both NMR approaches results in a much refined structural analysis.
The ensemble of data presented in this work indicates that when bound to lipid bilayers huntingtin 1–17 adopts an amphipathic helical structure encompassing residues 6 to 17 and that this helical domain is oriented at 103°±5°/137°±5° (tilt/pitch angles), i.e., close to parallel to the membrane surface (Fig. 5 a). Oriented solid-state NMR spectroscopy indicates that similar alignments of the P-huntingtin 1–17 helical domain are obtained in POPE/POPG 3:1 or POPC/POPS 3:1 membranes (30). Therefore, the structural results presented here appear valid also for other phospholipid membranes. Moreover, the solution NMR structure (Fig. 2 a) and its topology observed by paramagnetic relaxation experiments in micellar environments (Fig. 3) are in good agreement with the precise bilayer topology determined by solid-state NMR spectroscopy (Fig. 5 a).
These structural and topological details have important implications for some of the biological activities that have been associated with huntingtin and with the amino-terminal domain in particular. First, posttranslational modifications within huntingtin 1–17 have been identified and these have important regulatory effects on the activities and cellular localization of the protein (71,72). The amino-terminal domain encompasses residues T3, S13, and S16, which can be phosphorylated, as well as three lysines and the amino-terminus that can be acetylated, ubiquitinated, or SUMOylated. In membrane-associated huntingtin 1–17 all of these sites but K16 are exposed to the aqueous phase (Fig. 5 e). Indeed, SUMOylation occurs preferentially at positions 6 and 9 and much less at position 16 (72). In a related manner polyubiquitination of huntingtin 1–17 is favored by introducing a negative charge at serine 13 either by S13D mutagenesis or by phosphorylation (72). Whereas changing the amino-acid composition and charge at this position can modulate enzymatic activities, such modifications have also been shown to decrease the membrane-association of huntingtin 1–17 (C. Aisenbrey and B. Bechinger, unpublished; H. Lashuel, personal communication), which in turn increases the enzymatic accessibility of ubiquitination sites (72). Furthermore, such modifications result in the accumulation of the protein inside the nucleus (28,71,73).
Second, recently a nuclear export signal was identified encompassing L4, L7, F11, and L14 of huntingtin (17,18). These form a well-defined hydrophobic ridge in our structure (cf. Fig. 5 f) and are essential for NES activity. This is in contrast to a number of residues on the hydrophilic surface (T3, K6, K9, S13, or S16), which in the presence of L4 do not affect the NES activity (9,18). However, in an L4A mutant the hydrophobic contribution can be restored by S16L suggesting that interaction with exportin also involves this residue (17). Indeed, within the structural variety of leucine-rich nuclear export signals helix or helix-loop conformations (Fig. S5) have been found in relative abundance in the context of the ternary nuclear export complexes of exportin/CRM1 and Ran-GTP (74). Therefore, the membrane conformation of huntingtin 1–17 comes close to fit the hydrophobic pocket of the binding site but probably requires some conformational alternations at the carboxy-terminal region to better position S16 (Fig. 5, e and f, and Fig. S5). Despite this structural resemblance, membrane-association of huntingtin 1–17 occludes its NES interactions site (Fig. 5, e and f), and thereby contributes to the regulation of huntingtin localization and activity.
Third, huntingtin 1–14 has been identified to interact with F-actin at 2 μM affinity (13). We have therefore compared the structures of other known F-actin binders with that of huntingtin 1–17. Indeed, gelsolin has the capacity to bind F-actin and to form an amphipathic helix with properties related to the huntingtin 1–17 structure (Fig. S5).
Finally, the amphipathic helical conformation of huntingtin 6–17 (Figs. 2 a and 5) assures the reversible interaction of the protein with biological membranes and thereby results in a high local concentration of the polyglutamines at the membrane surface. Thus, membrane association potentially enhances protein-protein interactions and aggregation. In this context it is important to note that docking experiments using the DOT 2.0 software (75) reveal the potential of huntingtin 6–17 to associate into antiparallel helical dimers where residues K9-E12, and K6-S16 from different polypeptide chains face each other (not shown). Furthermore, the structural details in conjunction with the topological arrangement of huntingtin 1–17 indicate that its very carboxy-terminus is located at the membrane interface (Fig. 5, e and f), thus that the polyglutamines can remain in contact with the water phase and interact with each other.
Additional domains for membrane interaction of huntingtin have been identified in a region proximal to residues 229–249 (76). This site is further apart from the polyglutamine tract and can reinforce the membrane association of huntingtin by electrostatic interactions. Furthermore, a recent publication indicates that amyloidogenesis of huntingtin can be further enhanced by residues 105–138 localized downstream of the polyglutamine domain (77).
Upon fibril formation the polyglutamine tract undergoes a transition into a β-strand conformation (78,79), when at the same time much of the α-helical conformation of huntingtin 1–17 persists (79). This observation is suggestive that the increase in polyglutamine aggregation due to the presence of the huntingtin 1–17 domain (7,9,20–22) is a result of accelerated aggregation kinetics and/or a stabilization of aggregation intermediates (80) rather than of differences in the structure of the polyglutamine fibrils that ultimately form. Therefore, during the formation of polyglutamine oligomers the helical domain may fulfill two functions, namely anchoring huntingtin to the membrane thereby serving as a nucleation scaffold for the aggregation process (20).
Conclusions
By combining 2D solution and solid-state NMR spectroscopies, we determined the structure and topology of the huntingtin amino-terminal domain in membranes. The polypeptide adopts an amphipathic α-helical structure between residues 6 and 17, i.e., right to the start of the polyglutamine tract. The helix is oriented close to parallel to the membrane surface (at an angle of ∼13°) and can thereby act as a reversible membrane anchor for huntingtin. Furthermore, this amphipathic helix has the potential to interact with its likes or with other proteins where mutagenesis has identified the huntingtin amino-terminus as a key element. Therefore, the formation of an amphipathic helical structure can have important consequences for the regulation of the biological activities of this protein as well as the development of Huntington’s disease.
Acknowledgments
We thank Delphine Hatey for technical assistance during peptide synthesis and purification. We acknowledge the helpful discussions with Sebastiaan Werten, Fabrice Klein, Yvon Trottier, and Sudheendra U. S. when initiating the project.
We are most grateful for financial support by an early discovery initiative of CHDI, a foundation dedicated to research on Huntington’s Disease. We are indepted to the Agence Nationale de la Recherche (project LabEx Chemistry of Complex Systems), the RTRA International Center of Frontier Research in Chemistry, the University of Strasbourg, the CNRS, and the Région Alsace as well as the Deutsche Forschungsgemeinschaft (DFG) for supporting M.M. when contributing to the manuscript.
Supporting Material
References
- 1.Ross C.A. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 2.Rubinsztein D.C., Leggo J., Hayden M.R. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am. J. Hum. Genet. 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- 3.Sathasivam K., Amaechi I., Bates G. Identification of an HD patient with a (CAG)180 repeat expansion and the propagation of highly expanded CAG repeats in lambda phage. Hum. Genet. 1997;99:692–695. doi: 10.1007/s004390050432. [DOI] [PubMed] [Google Scholar]
- 4.Andresen J.M., Gayán J., Wexler N.S., US-Venezuela Collaborative Research Group. HD MAPS Collaborative Research Group The relationship between CAG repeat length and age of onset differs for Huntington’s disease patients with juvenile onset or adult onset. Ann. Hum. Genet. 2007;71:295–301. doi: 10.1111/j.1469-1809.2006.00335.x. [DOI] [PubMed] [Google Scholar]
- 5.Panov A.V., Gutekunst C.A., Greenamyre J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 6.Choo Y.S., Johnson G.V., Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 7.Rockabrand E., Slepko N., Thompson L.M. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 2007;16:61–77. doi: 10.1093/hmg/ddl440. [DOI] [PubMed] [Google Scholar]
- 8.Qin Z.H., Wang Y., DiFiglia M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 2004;24:269–281. doi: 10.1523/JNEUROSCI.1409-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atwal R.S., Xia J., Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 2007;16:2600–2615. doi: 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- 10.Lee W.C.M., Yoshihara M., Littleton J.T. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. USA. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunawardena S., Her L.S., Goldstein L.S.B. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- 12.Gauthier L.R., Charrin B.C., Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Angeli S., Shao J., Diamond M.I. F-actin binding regions on the androgen receptor and huntingtin increase aggregation and alter aggregate characteristics. PLoS ONE. 2010;5:e9053. doi: 10.1371/journal.pone.0009053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilditch-Maguire P., Trettel F., MacDonald M.E. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum. Mol. Genet. 2000;9:2789–2797. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- 15.Reiner A., Dragatsis I., Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol. Neurobiol. 2003;28:259–276. doi: 10.1385/MN:28:3:259. [DOI] [PubMed] [Google Scholar]
- 16.Zuccato C., Tartari M., Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 17.Maiuri T., Woloshansky T., Truant R. The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal. Hum. Mol. Genet. 2013;22:1383–1394. doi: 10.1093/hmg/dds554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng Z.Q., Li A.M., Diamond M.I. An N-terminal nuclear export signal regulates trafficking and aggregation of Huntingtin (Htt) protein exon 1. J. Biol. Chem. 2013;288:6063–6071. doi: 10.1074/jbc.M112.413575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desmond C.R., Atwal R.S., Truant R. Identification of a karyopherin β1/β2 proline-tyrosine nuclear localization signal in huntingtin protein. J. Biol. Chem. 2012;287:39626–39633. doi: 10.1074/jbc.M112.412379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tam S., Spiess C., Frydman J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelley N.W., Huang X., Pande V.S. The predicted structure of the headpiece of the Huntingtin protein and its implications on Huntingtin aggregation. J. Mol. Biol. 2009;388:919–927. doi: 10.1016/j.jmb.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thakur A.K., Jayaraman M., Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N-terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ignatova Z., Thakur A.K., Gierasch L.M. In-cell aggregation of a polyglutamine-containing chimera is a multistep process initiated by the flanking sequence. J. Biol. Chem. 2007;282:36736–36743. doi: 10.1074/jbc.M703682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellisdon A.M., Thomas B., Bottomley S.P. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J. Biol. Chem. 2006;281:16888–16896. doi: 10.1074/jbc.M601470200. [DOI] [PubMed] [Google Scholar]
- 25.Bulone D., Masino L., Pastore A. The interplay between PolyQ and protein context delays aggregation by forming a reservoir of protofibrils. PLoS ONE. 2006;1:e111. doi: 10.1371/journal.pone.0000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka Y., Igarashi S., Ellerby L.M. Progressive phenotype and nuclear accumulation of an amino-terminal cleavage fragment in a transgenic mouse model with inducible expression of full-length mutant huntingtin. Neurobiol. Dis. 2006;21:381–391. doi: 10.1016/j.nbd.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 27.Ratovitski T., Chighladze E., Ross C.A. Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant huntingtin. J. Biol. Chem. 2011;286:12578–12589. doi: 10.1074/jbc.M110.185348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atwal R.S., Desmond C.R., Truant R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 2011;7:453–460. doi: 10.1038/nchembio.582. [DOI] [PubMed] [Google Scholar]
- 29.Ratovitski T., Gucek M., Ross C.A. Mutant huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J. Biol. Chem. 2009;284:10855–10867. doi: 10.1074/jbc.M804813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michalek M., Salnikov E.S., Bechinger B. Membrane interactions of the amphipathic amino-terminus of huntingtin. Biochemistry. 2013;52:847–858. doi: 10.1021/bi301325q. [DOI] [PubMed] [Google Scholar]
- 31.Ramamoorthy A. NMR structural studies on membrane proteins. Biochim. Biophys. Acta. 2007;1768:2947–2948. doi: 10.1016/j.bbamem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Ader C., Schneider R., Baldus M. Magic-angle-spinning NMR spectroscopy applied to small molecules and peptides in lipid -bilayers. Biochem. Soc. Trans. 2007;35:991–995. doi: 10.1042/BST0350991. [DOI] [PubMed] [Google Scholar]
- 33.Bechinger B., Resende J.M., Aisenbrey C. The structural and topological analysis of membrane-associated polypeptides by oriented solid-state NMR spectroscopy: established concepts and novel developments. Biophys. Chem. 2011;153:115–125. doi: 10.1016/j.bpc.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 34.Dürr U.H.N., Gildenberg M., Ramamoorthy A. The magic of bicelles lights up membrane protein structure. Chem. Rev. 2012;112:6054–6074. doi: 10.1021/cr300061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cross T.A. Solid-state nuclear magnetic resonance characterization of gramicidin channel structure. Methods Enzymol. 1997;289:672–696. doi: 10.1016/s0076-6879(97)89070-2. [DOI] [PubMed] [Google Scholar]
- 36.Verly R.M., Moraes C.M., Bechinger B. Structure and membrane interactions of the antibiotic peptide dermadistinctin k by solution and oriented 15 N and 31 P solid-state NMR spectroscopy. Biophys. J. 2009;96:2194–2202. doi: 10.1016/j.bpj.2008.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Nuallain B., Thakur A.K., Wetzel R. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 2006;413:34–74. doi: 10.1016/S0076-6879(06)13003-7. [DOI] [PubMed] [Google Scholar]
- 38.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 39.Hwang T.L., Shaka A.J. Water suppression that works - excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. A. 1995;112:275–279. [Google Scholar]
- 40.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 41.Johnson B.A., Blevins R.A. NMR View: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 42.Schwieters C.D., Kuszewski J.J., Clore G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 43.Linge J.P., Williams M.A., Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 44.Laskowski R.A., Rullmannn J.A., Thornton J.M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 45.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- 46.Aisenbrey C., Bertani P., Bechinger B. Solid-state NMR investigations of membrane-associated antimicrobial peptides. In: Guiliani A., Rinaldi A.C., editors. Antimicrobial Peptides. Humana Press; Springer, N.Y.: 2010. pp. 209–233. [DOI] [PubMed] [Google Scholar]
- 47.Bechinger B., Opella S.J. Flat-coil probe for NMR spectroscopy of oriented membrane samples. J. Magn. Reson. 1991;95:585–588. [Google Scholar]
- 48.Salnikov E.S., Aisenbrey C., Bechinger B. Structure and alignment of the membrane-associated antimicrobial peptide arenicin by oriented solid-state NMR spectroscopy. Biochemistry. 2011;50:3784–3795. doi: 10.1021/bi1018732. [DOI] [PubMed] [Google Scholar]
- 49.Davis J.H., Jeffrey K.R., Higgs T.P. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 1976;42:390–394. [Google Scholar]
- 50.Aisenbrey C., Bechinger B. Tilt and rotational pitch angles of membrane-inserted polypeptides from combined 15N and 2H solid-state NMR spectroscopy. Biochemistry. 2004;43:10502–10512. doi: 10.1021/bi049409h. [DOI] [PubMed] [Google Scholar]
- 51.Hahn E.L. Spin echoes. Phys. Rev. 1950;80:580–594. [Google Scholar]
- 52.Rance M., Byrd R.A. Obtaining high-fidelity spin-1/2 powder spectra in anisotropic media: phase-cycled Hahn echo spectroscopy. J. Magn. Reson. 1983;52:221–240. [Google Scholar]
- 53.Salnikov E., Bertani P., Bechinger B. Analysis of the amide (15)N chemical shift tensor of the C(alpha) tetrasubstituted constituent of membrane-active peptaibols, the alpha-aminoisobutyric acid residue, compared to those of di- and tri-substituted proteinogenic amino acid residues. J. Biomol. NMR. 2009;45:373–387. doi: 10.1007/s10858-009-9380-5. [DOI] [PubMed] [Google Scholar]
- 54.Batchelder L.S., Niu H., Torchia D.A. Methyl reorientation in polycrystalline amino acids and peptides: a 2H NMR spin lattice relaxation study. J. Am. Chem. Soc. 1983;105:2228–2231. [Google Scholar]
- 55.Strandberg E., Esteban-Martín S., Ulrich A.S. Orientation and dynamics of peptides in membranes calculated from 2H-NMR data. Biophys. J. 2009;96:3223–3232. doi: 10.1016/j.bpj.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aisenbrey C., Michalek M., Bechinger B. Solid-state NMR approaches to study protein structure and protein-lipid interactions. In: Kleinschmidt J.H., editor. Lipid-Protein Interactions: Methods and Protocols. Springer; New York: 2013. pp. 357–387. [DOI] [PubMed] [Google Scholar]
- 57.Bechinger B., Sizun C. Alignment and structural analysis of membrane polypeptides by 15N and 31P solid-state NMR spectroscopy. Concepts Magn. Reson. 2003;18A:130–145. [Google Scholar]
- 58.Vostrikov V.V., Koeppe R.E., 2nd Response of GWALP transmembrane peptides to changes in the tryptophan anchor positions. Biochemistry. 2011;50:7522–7535. doi: 10.1021/bi2006459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aisenbrey C., Prongidi-Fix L., Bechinger B. Side chain resonances in static oriented proton-decoupled 15N solid-state NMR spectra of membrane proteins. J. Am. Chem. Soc. 2009;131:6340–6341. doi: 10.1021/ja900677b. [DOI] [PubMed] [Google Scholar]
- 60.Bechinger B., Kinder R., Schinzel S. Peptide structural analysis by solid-state NMR spectroscopy. Biopolymers. 1999;51:174–190. doi: 10.1002/(SICI)1097-0282(1999)51:3<174::AID-BIP2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 61.Resende J.M., Mendonca Moraes C., Bechinger B. Membrane structure and conformational changes during bilayer-association of the antibiotic heterodimeric peptide distinction by oriented solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2009;106:16639–16644. doi: 10.1073/pnas.0905069106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi L., Traaseth N.J., Veglia G. A refinement protocol to determine structure, topology, and depth of insertion of membrane proteins using hybrid solution and solid-state NMR restraints. J. Biomol. NMR. 2009;44:195–205. doi: 10.1007/s10858-009-9328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aisenbrey C., Bechinger B. Investigations of polypeptide rotational diffusion in aligned membranes by 2H and 15N solid-state NMR spectroscopy. J. Am. Chem. Soc. 2004;126:16676–16683. doi: 10.1021/ja0468675. [DOI] [PubMed] [Google Scholar]
- 64.Prongidi-Fix L., Bertani P., Bechinger B. The membrane alignment of helical peptides from non-oriented 15N chemical shift solid-state NMR spectroscopy. J. Am. Chem. Soc. 2007;129:8430–8431. doi: 10.1021/ja072668k. [DOI] [PubMed] [Google Scholar]
- 65.Zuccato C., Valenza M., Cattaneo E.V. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- 66.DiFiglia M., Sapp E., Aronin N. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 67.Gutekunst C.A., Li S.H., Hersch S.M. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J. Neurosci. 1998;18:7674–7686. doi: 10.1523/JNEUROSCI.18-19-07674.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim M.W., Chelliah Y., Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17:1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mattila K., Kinder R., Bechinger B. The alignment of a voltage sensing peptide in DPC micelles and in oriented lipid bilayers by NMR and molecular modeling. Biophys. J. 1999;77:2102–2113. doi: 10.1016/S0006-3495(99)77051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Resende J.M., Moraes C.M., Bechinger B. Solution NMR structures of the antimicrobial peptides phylloseptin-1, -2, and -3 and biological activity: the role of charges and hydrogen bonding interactions in stabilizing helix conformations. Peptides. 2008;29:1633–1644. doi: 10.1016/j.peptides.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 71.Thompson L.M., Aiken C.T., Steffan J.S. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steffan J.S., Agrawal N., Marsh J.L. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 73.Gu X.F., Greiner E.R., Yang X.W. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu D.R., Grishin N.V., Chook Y.M. NESdb: a database of NES-containing CRM1 cargoes. Mol. Biol. Cell. 2012;23:3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mandell J.G., Roberts V.A., Ten Eyck L.F. Protein docking using continuum electrostatics and geometric fit. Protein Eng. 2001;14:105–113. doi: 10.1093/protein/14.2.105. [DOI] [PubMed] [Google Scholar]
- 76.Kegel K.B., Sapp E., DiFiglia M. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 2009;110:1585–1597. doi: 10.1111/j.1471-4159.2009.06255.x. [DOI] [PubMed] [Google Scholar]
- 77.Wang Z.M., Lashuel H.A. Discovery of a novel aggregation domain in the huntingtin protein: implications for the mechanisms of Htt aggregation and toxicity. Angew. Chem. Int. Ed. Engl. 2013;52:562–567. doi: 10.1002/anie.201206561. [DOI] [PubMed] [Google Scholar]
- 78.Schneider R., Schumacher M.C., Baldus M. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J. Mol. Biol. 2011;412:121–136. doi: 10.1016/j.jmb.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 79.Sivanandam V.N., Jayaraman M., van der Wel P.C. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 2011;133:4558–4566. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Côté S., Wei G.H., Mousseau N. All-atom stability and oligomerization simulations of polyglutamine nanotubes with and without the 17-amino-acid N-terminal fragment of the Huntingtin protein. J. Phys. Chem. B. 2012;116:12168–12179. doi: 10.1021/jp306661c. [DOI] [PubMed] [Google Scholar]
- 81.Salnikov E., Aisenbrey C., Bechinger B. Solid-state NMR approaches to measure topological equilibria and dynamics of membrane polypeptides. Biochim. Biophys. Acta. 2010;1798:258–265. doi: 10.1016/j.bbamem.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 82.Monecke T., Güttler T., Ficner R. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- 83.Xian W.J., Vegners R., Braunlin W.H. Spectroscopic studies of a phosphoinositide-binding peptide from gelsolin: behavior in solutions of mixed solvent and anionic micelles. Biophys. J. 1995;69:2695–2702. doi: 10.1016/S0006-3495(95)80140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.