

Abstract
Oxidation via Cu2+/ascorbate of recombinant human interferon beta-1a (IFNβ1a) leads to highly immunogenic aggregates, however it is unknown which amino acids are modified and how covalent aggregates are formed. In the present work we mapped oxidized and cross-linked amino acid residues in aggregated IFNβ1a, formed via Cu2+/ascorbate catalyzed oxidation. Size exclusion chromatography (SEC) was used to confirm extensive aggregation of oxidized IFNβ1a. Circular dichroism and intrinsic fluorescence spectroscopy indicated substantial loss of secondary and tertiary structure, respectively. Derivatization with 4-(aminomethyl) benzenesulfonic acid was used to demonstrate, by fluorescence in combination with SEC, the presence of tyrosine (Tyr) oxidation products. High performance liquid chromatography coupled to electrospray ionization mass spectrometry of reduced, alkylated and digested protein was employed to localize chemical degradation products. Oxidation products of methionine, histidine, phenylalanine (Phe), tryptophan and Tyr residues were identified throughout the primary sequence. Covalent crosslinks via 1,4- or 1,6-type addition between primary amines and DOCH (2-amino-3-(3,4-dioxocyclohexa-1,5-dien-1-yl) propanoic acid, an oxidation product of Phe and Tyr) were detected. There was no evidence of disulfide bridge, Schiff base, or dityrosine formation. The chemical cross-links identified in this work are most likely responsible for the formation of covalent aggregates of IFNβ1a induced by oxidation, which have previously been shown to be highly immunogenic.
Keywords: IFNβ1a, metal catalyzed oxidation, covalent aggregation, tyrosine oxidation products, immunogenicity, Cu2+/ascorbate
Introduction
Recombinant human interferon beta (IFNβ), a cytokine with anti-inflammatory and tumor-suppressor functions(1) is considered as the first choice treatment of multiple sclerosis, a severe neurodegenerative autoimmune disease(2). Interferon beta-1a (IFNβ1a), in contrast to interferon beta-1b (IFNβ1b)(3), is glycosylated at asparagine 80(4) carrying mainly biantennary and triantennary glycan structures(5, 6). IFNβ1a has an average molecular weight of about 22.5 kDa, contains 166 amino acid residues and is produced in Chinese hamster ovarian (CHO) cells(6). As described by Karpusas et al.(7), the protein contains five alpha helices, an intramolecular disulfide bridge between cysteines in positions 31 and 141, and a free cysteine (Cys) in position 17. Like many other protein therapeutics, IFNβ is prone to aggregation, which has been correlated with enhanced immunogenicity(8). Furthermore, it has been demonstrated that the removal of protein aggregates reduces the immunogenicity of IFNβ formulations(9–11). In addition, IFNβ1b (Betaferon), which contains more aggregates than IFNβ1a products (Avonex, Rebif), has been found to be the most immunogenic IFNβ product in several preclinical and clinical studies(12, 13).
Protein aggregates induced by metal-catalyzed oxidation (MCO) have been found to be particularly immunogenic in preclinical models. For instance, Hermeling et al. showed that recombinant human interferon alpha-2b (IFNα2b) forms immunogenic aggregates upon oxidative stress induced by Cu2+/ascorbate catalyzed oxidation, using a transgenic mouse model immune tolerant to the human protein(14, 15). Similarly, IFNβ1a aggregates and monoclonal antibody aggregates, both produced via the same oxidative system, were shown to be highly immunogenic in transgenic immune tolerant mouse models(8, 16). We therefore believe that characterization of chemical modifications and cross-links caused by Cu2+/ascorbate catalyzed oxidation is of high importance, since these data may shed light on structure-immunogenicity relationships for IFNβ1a and potentially other therapeutic proteins.
The scope of the present work was to identify the chemical modifications in Cu2+/ascorbate oxidized IFNβ1a, including covalent cross-links that may be involved in aggregation. Here we report a comprehensive characterization of oxidative modifications in IFNβ1a following MCO and discuss putative mechanisms of MCO-induced IFNβ1a aggregation. Our data strongly support the formation of electrophilic oxidation products, which can be further involved in aggregate formation via 1,4- or 1,6-type addition to pre-existing protein nucleophiles, such as primary amine groups, in agreement with our recent mechanistic studies on insulin as a model protein(17).
Materials and Methods
Materials
IFNβ1a (0.26 mg/ml protein in 20 mM sodium acetate buffer, pH 4.8, containing 154 mM arginine) was kindly provided by Biogen Idec Inc. (Cambridge, MA, USA). PNGase F, L-ascorbic acid, ethylenediamine tetraacetic acid (EDTA), copper dichloride, arginine, monobasic and dibasic sodium hydrogen phosphate, ammonium bicarbonate (ABI), sodium chloride, sodium azide, SDS, dithiothreitol (DTT), iodoacetamide, glacial acetic acid, and acetonitrile were purchased from Sigma-Aldrich (St. Louis, MO, USA). MilliQ water was used for the preparation of all the formulations. All chemicals were of analytical grade and used without further purification. 4-(aminomethyl) benzenesulfonic acid (ABS) was synthesized according to a published procedure(18). Endoproteinases Glu-C and trypsin were purchased from Promega (Madison, WI, USA).
Preparation of untreated and oxidized IFNβ1a formulations
IFNβ1a was prepared and treated as reported by van Beers et al.(8). Briefly, the protein solution was dialyzed against 100 mM sodium phosphate buffer and 200 mM sodium chloride, pH 7.2 (PBS). This dialyzed solution is referred to as untreated IFNβ1a. To obtain oxidized IFNβ1a, untreated IFNβ1a, diluted to 200 µg/ml with PBS, was incubated with 4 mM ascorbic acid and 40 μM CuCl2 for 3 hours at room temperature. The oxidation reaction was stopped by adding 100 mM EDTA to a final concentration of 1 mM as previously reported(17).
ABS derivatization
Untreated and oxidized protein (0.2 mg/mL) were dialyzed against 50 mM ABI, pH 8.0, using a 3.5-kDa MWCO Slide-A-Lyzer Cassette (Asheville, NC, USA), before derivatization with ABS. To 250 μL of dialyzed samples 7.5 μL of 0.1 M sodium hydroxide were added to adjust the pH to 9.5. Then 100 mM ABS stock solution was added to a final concentration of 10 mM. Subsequently, 5 mM K3Fe(CN)6 stock solution in milliQ water was added to a final concentration of 0.5 mM. The reaction was conducted for 1 hour at room temperature, before performing SEC analysis or fluorescence measurement (as reported below in the section Size exclusion chromatography and Fluorescence spectroscopy, respectively). Controls for non-specific fluorescence included (i) oxidized and non-oxidized protein prior to derivatization and (ii) reagents alone (i.e. ABS/K3Fe(CN)6) incubated under the same conditions.
Size exclusion chromatography
To determine the aggregate content and to investigate the effect of reducing agents on oxidized and untreated IFNβ1a, SEC was performed by using a TSKgel Super SW2000 column protected by a Super SW guard column (Sigma Aldrich, St. Louis, MO, USA). An SPD-6AV UV and fluorescence detector (Shimadzu, Columbia, MD, USA) was used to record the chromatograms at a wavelength of 280 nm and at excitation/emission wavelength combination of 295/350nm, respectively. A flow rate of 0.35 ml/min was applied using a 515 HPLC pump and 717 Plus autosampler (Waters, Milford, MA, USA). The mobile phase consisted of 200 mM sodium chloride, 0.05% (w/v) sodium azide and 0.1% (w/v) SDS in 100 mM sodium phosphate, pH 7.2, which was filtered through a 0.2-μm filter prior to use.
To detect fluorescent benzoxazole products in the ABS-derivatized samples, we used an Insulin HMWP Column, 7.8 × 300 mm (Waters) connected to a HPLC system (Shimadzu, Columbia, MD, USA) coupled with a Shimadzu RF-20A fluorescence detector. Excitation and emission wavelengths were set at 360 and 490 nm, respectively. The mobile phase composition and flow rate were as previously reported(19).
Fluorescence spectroscopy
The emission spectra of the ABS-derivatized samples, diluted twofold in ABI, were measured in a 0.5-mL quartz fluorescence cuvette with an RF-5000U fluorescence spectrophotometer (Shimadzu) with excitation and emission wavelengths range set at 360 and 400–600 nm, respectively, and bandwidths set at 5 nm.
To test the potential presence of dityrosine, fluorescence of the oxidized IFNβ1a was measured in triplicate, using excitation and emission wavelengths set at 315 and 420 nm, respectively, as described previously(20).
Tryptophan (Trp) intrinsic fluorescence was measured with a TECAN infinite M1000 fluorometer (Tecan group Ltd., Männedorf, Switzerland) upon excitation at 295 nm and emission recorded from 310–500 nm with a step size of 2 nm, 50 flashes (frequency 400 Hz), a gain of 150 and a Z-position of 20.0 mm. Duplicates of 200 μL of each sample were analyzed in black polystyrene 96-well plates (Greiner Bio-One, Frickenhausen, Germany).
Circular dichroism spectroscopy
Circular dichroism (CD) spectra of untreated and oxidized IFNβ1a (0.1 mg/mL) were recorded from 190 to 250 nm, using a Jasco J-815 CD spectrometer (Jasco International, Tokyo, Japan). Analyses were performed in a 1-mm (far-UV CD) path length quartz cuvette at 25 °C using a scan rate of 100 nm/min, a response time of 2 s, and a bandwidth of 1 nm. Each spectrum was the result of an averaging of six repeated scans and background was corrected with the corresponding buffer spectrum. The CD signals were converted to mean residue ellipticity [θ]mrw, λ, using a mean residue weight (MRW, calculated as MRW = M/(N−1), where M is the average molecular weight of the untreated (glycan-free) protein (i.e. 20027 Da, based on the protein’s chemical formula C908H1408N246O252S7) and N is the number of amino acid residues in the chain).
Reduction, alkylation and digestion
Untreated and oxidized protein (0.2 mg/mL) were dialyzed against 50 mM ABI, pH 8.0, using a 3.5-kDa MWCO Slide-A-Lyzer Cassette (Asheville, NC, USA), and reduced by addition of 50 mM DTT, freshly prepared in 50 mM ABI, pH 8.0, to a final concentration of 5 mM. The samples were incubated for 45 minutes at 45 °C using a Thermo NES heating bath (Thermo Scientific, NC, USA). Subsequently, 200 mM iodoacetamide, freshly prepared in 50 mM ABI, pH 8.0, was added to a final concentration of 20 mM. The digestion was performed as follows: IFNβ1a was incubated with Glu-C endoproteinase at a IFNβ1a/Glu-C ratio of 10:1 (w/w), for 1 hour at 37 °C. Next, trypsin was added (IFNβ1a/trypsin ratio 20:1 (w/w)) and the mixture was incubated for an additional hour at the same temperature. Finally, 4 μL of a solution of PNGase F, dissolved according to the manufacturer’s protocol, was added to 76 μL of the protein mixture, which was then incubated overnight at 37 °C.
MS/MS analysis
Digested and non-digested samples were analyzed by means of an LTQ-FT hybrid linear quadrupole ion trap Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Thermo-Finnigan, Bremen, Germany)(21). The MS/MS spectra were analyzed with the web-based software MassMatrix(22–25), which was used to simulate mass spectra and the theoretical fragment tables of the b- and y-ions(26) for the native, oxidized and cross-linked peptide products. The simulated spectra were compared to the experimental MS/MS spectra in order to validate the proposed product and cross-link structures. A mass accuracy sensitive probability-based scoring algorithm for database searching of tandem mass spectrometry data by MassMatrix software was employed for peptide identification; the manual filter was set at mass/charge ratio (m/z) accuracy < 0.1 amu for both parent peptide and fragment ions.
Results
Comparison of untreated and oxidized IFNβ1a by SEC and spectroscopic methods
SEC analysis was used to verify whether the aggregation profile of oxidized IFNβ1a was similar to that observed earlier(8) and to study a potential involvement of disulfide bridging in aggregate formation. Comparison of the chromatograms of untreated and oxidized IFNβ1a (Figure 1A) shows extensive aggregation of the oxidized product and both chromatograms are in close agreement with the results published by van Beers et al.(8). Furthermore, the treatment of oxidized IFNβ1a with DDT did not have a major impact on the elution behavior, indicating that the majority of the aggregates were formed by non-reducible covalent bonds. In contrast, we observed that for untreated IFNβ1a, the reduction with DDT resulted in a decrease in dimer content by approximately 60% (Figure 1A), indicating that most of the dimers in the untreated IFNβ1a were formed through disulfide bridging.
Figure 1A.
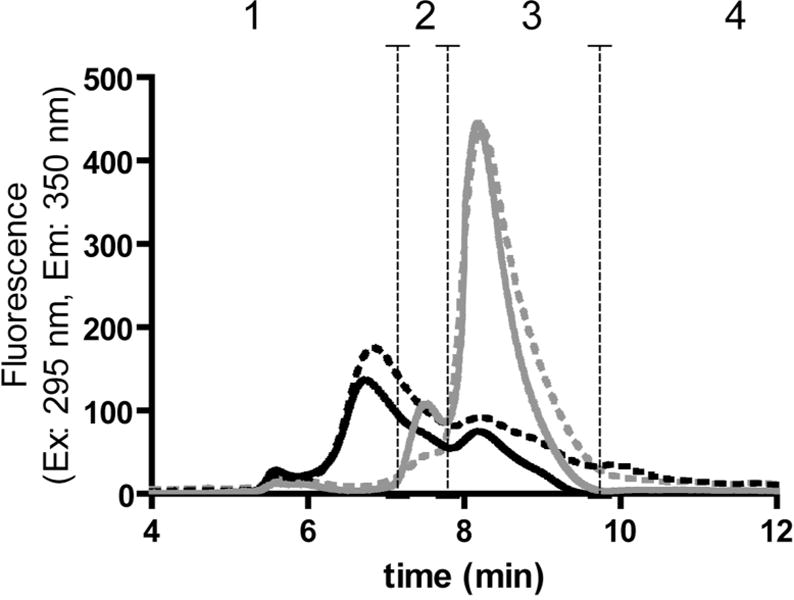
SEC (TSKgel Super SW2000 column) with intrinsic tryptophan fluorescence detection of untreated IFNβ1a before (solid grey chromatogram) and after reduction and alkylation (dashed grey) and oxidized IFNβ1a before (solid black) and after reduction and alkylation (dashed black). The vertical dashed lines show the range of each peak: (1) mainly representing oligomers, (2) dimers, (3) monomers, (4) fragments.
SEC with fluorescence detection of ABS-tagged proteins was used to study whether the monomers or aggregates contain DOPA (3,4-dihydroxyphenylalanine) and/or DOCH (2-amino-3-(3,4-dioxocyclohexa-1,5-dien-1-yl) propanoic acid), produced by oxidation of tyrosine (Tyr) and/or phenylalanine (Phe) residues and subject to fluorogenic derivatization with ABS(17) and/or 5-hydroxytryptophan, a Trp oxidation product which can also form fluorescent benzoxazole products upon ABS derivatization(27, 28). Our data show that after derivatization of the oxidized and dialyzed protein with ABS, the total SEC fluorescence peak area (i.e. the sum of all the peak areas under the curve) for the ABS-derivatized oxidized IFNβ1a is about six fold higher than that of the ABS-derivatized untreated protein (Figure 1B), indicating that MCO of IFNβ1a leads to formation of DOPA/DOCH and/or 5-hydroxytryptophan residues in the oxidized protein. ABS derivatization of non-oxidized IFNβ1a also produces a weak fluorescent signal mainly associated with protein monomer (Figure 1B), suggesting a background oxidation in the untreated protein. The ABS fluorescence of ABS derivatized oxidized and untreated IFNβ1a was also measured by steady-state fluorescence. The results show a seven fold increase in fluorescence intensity for the oxidized protein (Figure 2), which is consistent with the SEC data.
Figure 1B.
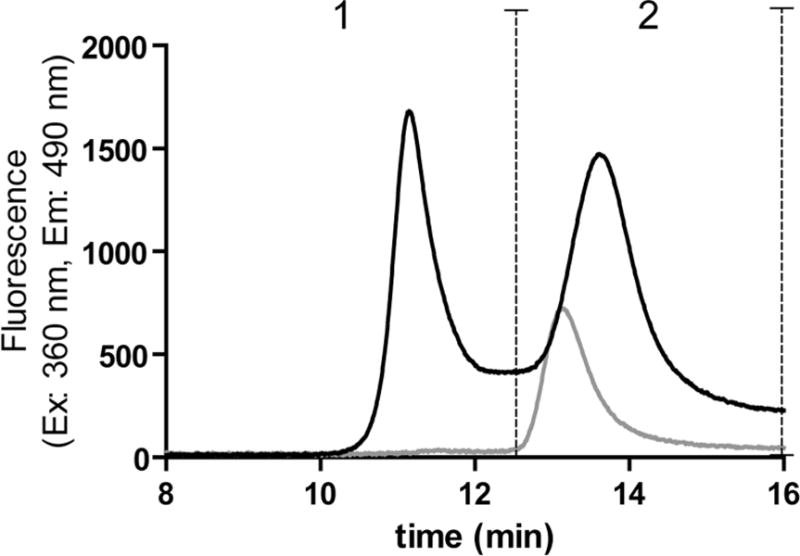
SEC (Insulin HMWP Column) with ABS fluorescence detection of ABS-derivatized untreated IFNβ1a (grey) and ABS-derivatized oxidized IFNβ1a (black). The vertical dashed lines show the range of each peak: (1) mainly representing oligomers, (2) dimers and monomers (with this column it is not possible to discriminate dimer from monomer).
Figure 2.
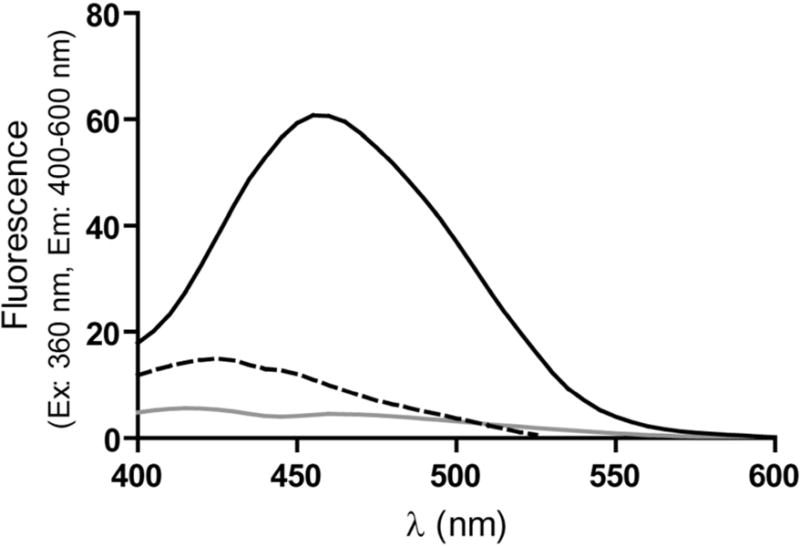
Extrinsic fluorescence emission spectra of ABS-derivatized untreated IFNβ1a (grey), ABS-derivatized oxidized IFNβ1a (black), and oxidized IFNβ1a (dashed black). Spectra represent the average of two batches.
Further structural characterization was performed by CD (Figure 3) and intrinsic steady-state fluorescence measurements (Figure 4). Likely as a result of oxidation and cross-link formation, it was found that the content of alpha helix in oxidized IFNβ1a had decreased, as indicated by the dro p in negative CD signal, with a concomitant increase in random coil structure, as indicated by the higher 208/222 nm ratio measured for the oxidized protein (1.041 ± 0.024) as compared with its native counterpart (0.729 ± 0.004)(29, 30). Moreover, a substantial decrease of Trp fluorescence was observed, pointing to chemical modification of Trp residues and/or loss of tertiary structure in general. No shift in the emission maximum was however observed.
Figure 3.
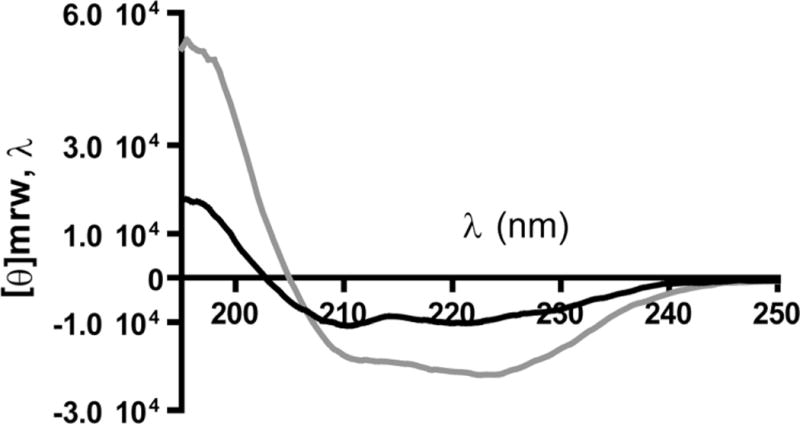
Far-UV CD spectra of untreated IFNβ1a (grey) and oxidized IFNβ1a (black). Spectra represent the average of two batches.
Figure 4.
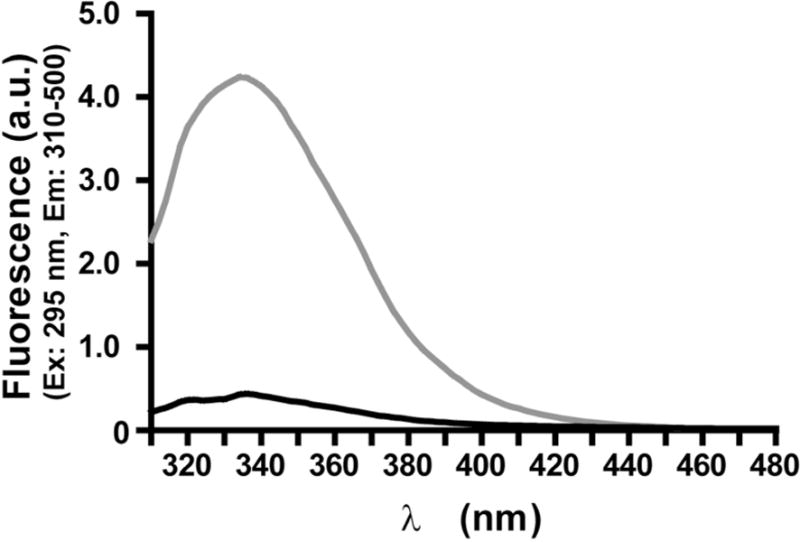
Intrinsic steady-state fluorescence of untreated IFNβ1a (grey) and oxidized IFNβ1a (black). Spectra represent the average of two batches.
Mass spectrometry analysis
Undigested IFNβ1a
The data of liquid chromatography coupled to electrospray ionization mass spectrometry (LC-ESI-MS) analysis of undigested untreated IFNβ1a (Figure 5A) are in agreement with previously reported results(31): the spectrum of the protein represents a mixture of multiple glycoforms (mainly fucosylated and sialylated protein) where the most abundant isoform contains one mono-fucosylated, biantennary structure carrying two sialic acid residues, featuring a total molecular weight of 22376 Da. The spectrum of the oxidized protein represents a single broad peak with a maximum at 22460.5 Da resulting from an overlay of multiple heterogeneously oxidized protein isoforms (Figure 5B), similar to results published for another oxidized protein (32).
Figure 5.
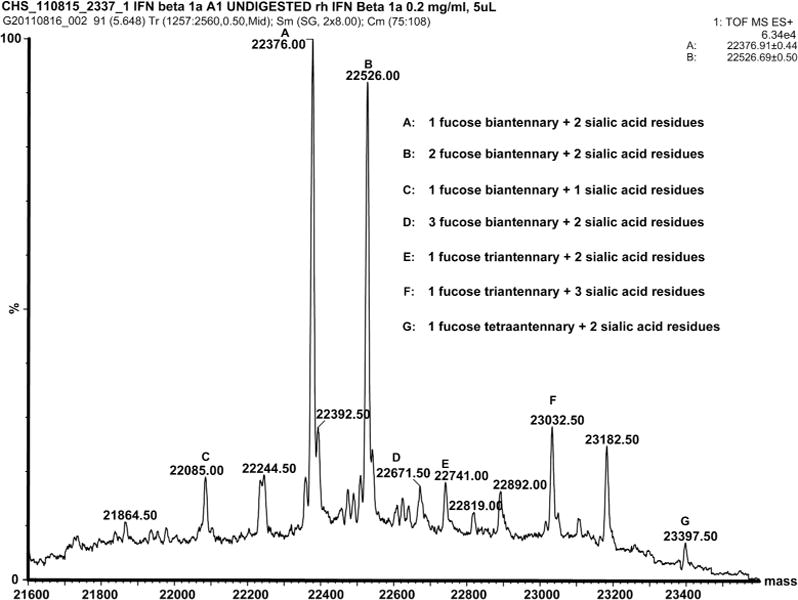
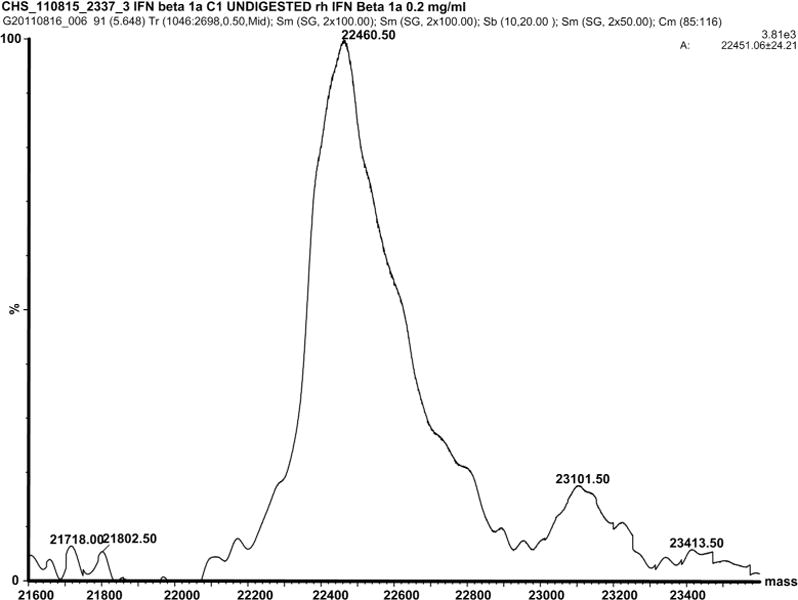
A: Deconvoluted mass spectrum of undigested untreated IFNβ1a and B: undigested oxidized IFNβ1a.
Sequence-specific analysis of IFNβ1a oxidative modifications
This section focuses on the identification of amino acid sequence-specific oxidative protein modifications through MS/MS analysis of IFNβ1a digests and the results will be presented in the following order: Methionine (Met), Phe, Histidine (His), Tyr and Trp oxidation. It is noteworthy that some peptide sequences potentially contain more than one modification, and that some MS/MS spectra may represent a mixture of peptide isoforms with the same m/z but different locations of oxidative modifications within the sequence. Table 1 lists all the chemically modified peptides detected. Although a few modifications were observed also in peptides derived from the untreated protein (Table 1, last column), most modifications were observed in peptides from oxidized IFNβ1a and involved Met, Phe, His, Tyr and Trp residues. Table 2 shows all the potential targets for MCO, including the ones found oxidized in the present study. A supplementary Table S1 shows all the potential amino acid modifications included in the settings of a database search for oxidized, reduced, alkylated and digested IFNβ1a samples. All annotated MS/MS spectra for oxidative protein modifications along with suggested peptide structures presented as inserts are shown in the supplementary Figures S1–S20. Note that in the supplementary figures, the peptide structures of the non-modified amino acid residues are displayed in a one-letter code, while the respective full structures are drawn for the suggested oxidatively modified and alkylated/deamidated residues.
Table 1.
Chemical modifications measured by LC-ESI-MS/MS in peptides from oxidized and untreated IFNβ1a obtained after reduction, alkylation and enzymic digestion.
| Chemical modifications and reference to corresponding mass spectrum | Peptide | Experimental m/z | Charge | Theoretical mass | Δm | Oxidized IFNβ1a* | Untreated IFNβ1a* |
|---|---|---|---|---|---|---|---|
| M1(O) Figure S1 |
M1SYNLLGFLQR11 | 679.35 | 2 | 1356.70 | 0.00 | + | + |
| M1(O), Y3(O) Figure S2 |
M1SYNLLGFLQR11 | 687.35 | 2 | 1372.69 | + 0.01 | + | − |
| Y3(DOCH) Figure S3 |
M1SYNLLGFLQR11 | 678.34 | 2 | 1354.68 | 0.00 | + | − |
| M1(loss CH3S) Figure S4 |
M1SY NLLGFLQR11 | 647.35 | 2 | 1292.68 | + 0.02 | + | + |
| F38(O) Figure S5(a–c) |
M36NFDIPEEIK45 | 626.30 | 2 | 1250.59 | + 0.01 | + | − |
| M36(O) Figure S6(a–c) |
M36NFDIPEEIK45 | 626.30 | 2 | 1250.59 | + 0.01 | + | + |
| M62(O) Figure S7 |
M62LQNIFAIFR71 | 634.84 | 2 | 1267.68 | 0.00 | + | − |
| M62(loss CH3S) Figure S8 |
M62LQNIFAIFR71 | 402.23 | 3 | 1203.68 | + 0.01 | + | − |
| M117(O), H121(O) Figure S9 |
L116MSSLHLK123 | 480.76 | 2 | 959.52 | 0.00 | + | − |
| F50(O), Q(deamidation in several positions) Figure S10 |
Q46LQQFQK52 | 468.76 | 2 | 935.48 | + 0.04 | + | − |
| H93(O) Figure S11 |
N86LLANVYHQINHLK99 | 846.96 | 2 | 1691.92 | 0.00 | + | − |
| H97(O) Figure S12 |
N86LLANVYHQINHLK99 | 846.96 | 2 | 1691.92 | 0.00 | + | − |
| H93(O), H97(O) Figure S13 |
N86LLANVYHQINHLK99 | 854.96 | 2 | 1707.91 | + 0.01 | + | − |
| H93(asn) Figure S14(a–b) |
N86LLANVYHQINHLK99 | 827.46 | 2 | 1652.91 | + 0.01 | + | − |
| H97(asn) Figure S15(a–b) |
N86LLANVYHQINHLK99 | 827.46 | 2 | 1652.91 | + 0.01 | + | − |
| H131(O), Y132(O) Figure S16(a–c) |
I129LHYLK134 | 401.74 | 2 | 801.48 | 0.00 | + | − |
| Y138(DOCH), H140(O) Figure S17 |
E137YSHCAWTIVR147 | 697.81 | 2 | 1393.62 | 0.00 | + | − |
| H140(O), C141(IAM)# Figure S18 |
Y138SHCAWTIVR147 | 654.81 | 2 | 1307.62 | 0.00 | + | − |
| W22(O), N25(deamidation) Figure S19 |
L20LWQLNGR27 | 508.78 | 2 | 1015.55 | + 0.01 | + | + |
| W22(N-fkyn), N25(deamidation) Figure S20 |
L20LWQLNGR27 | 516.78 | 2 | 1031.55 | + 0.01 | + | + |
+ detected; − not detected
(IAM): alkylation with iodoacetamide
Table 2.
Oxidation prone amino acid residues and oxidized amino acid detected in peptides derived from oxidized IFNβ1a.1
| Methionine | Phenylalanine | Tyrosine | Tryptophan | Histidine |
|---|---|---|---|---|
|
| ||||
| 1 | 8 | 3 | 22 | 93* |
| 36 | 15 | 30 | 79# | 97* |
| 62 | 38 | 60 | 143# | 121* |
| 117 | 50 | 92 | 131 | |
| 67 | 125 | 140 | ||
| 70# | 126 | |||
| 111 | 132 | |||
| 154# | 138 | |||
| 156 | 155 | |||
| 163 | ||||
Bold numbers: oxidized residues; bold underlined numbers: oxidized residues involved in H-bond critical for the correct folding of IFNβ1a; underlined numbers: residues involved in H-bond critical for the correct folding of IFNβ1a (Karpusas et al.7); plain italic numbers: non-oxidized residues.
Hydrophobic residues that stabilize the core of the molecule.
Residues that coordinate a zinc ion, responsible for dimer formation.
Methionine oxidation
Met1 has been found susceptible to oxidation already by other authors(7, 31). The b2+ ion in Figure S1, where the peptide M1SYNLLGFLQR11 is displayed, shows the incorporation of one oxygen atom (+ 16 Da) into the sequence Met1Ser2: here, Ser is less prone to oxidation than Met, hence the target of oxidation is likely Met1. Figure S2 shows the same peptide with two oxygen atoms incorporated (+ 32 Da), which may represent a mixture of two sequences where either Met1 is oxidized to its sulfone, or DOPA (formed from Tyr3) and Met sulfoxide are present together. This is illustrated in the inserts to Figure S2, showing two potential b2+ ions, one with a mass increase of + 16 Da (mono oxidized, indicated as Met ox) and another with the mass increase of + 32 Da (di-oxidized, indicated as Met (ox)2). The second insert shows two fragments with m/z 1123.57 and 1139.25, which may represent the y9+ ions for the unmodified and oxidized form of Tyr3; the ratio of these peaks suggests that the peptide containing oxidized Met1 and DOPA in position 3 is far less abundant than the unmodified peptide (a fraction of less than 10%). However, this value does not necessarily reflect a lower rate of DOPA formation compared to Met sulfone; rather, it may indicate a higher reactivity of DOPA, resulting in DOCH and further cross-linked products. A spectrum of the doubly charged peptide ion with m/z 678.34 in Figure S3 shows such an example where Tyr is oxidized to DOCH based on a mass increase of 14 Da, which corresponds to the incorporation of one oxygen atom and loss of two hydrogen atoms, albeit at very low abundance of both y9+ and b3+−H2O sequence-indicating ions. We were also able to detect a characteristic loss of methane sulfenic acid (CH3SOH, −64 Da, compared with the mono oxidized sequence) from the N-terminus oxidized Met (Figure S4), which was suggested earlier as diagnostic for identifying peptides containing oxidized Met(26, 33).
Met36 is located in a solvent exposed domain of IFNβ1a(7, 31). Figure S5a and S5b represent the MS/MS spectra matching the sequence M36NFDIPEEIK45, in which either Met36 or Phe38 is oxidized. These two products are not resolved by our HPLC method and the respective spectra overlay, as evidenced by the presence of two potential fragment ions for y8+ at m/z 990.48 and 1006.44 (Figure S5b) for oxidized Met36 and oxidized Phe38, respectively. As additional evidence for oxidized Phe38, the ion at m/z 228.18 can be considered, which most likely represents the b2+−H2O ion (indicated as F ox) from the parent peptide with oxidized Phe38 (Figure S5c). Though the fraction of the peptide with oxidized Phe38 is rather small (ca. 3–5% based on the relative abundance of fragment ion y8+ in Figure S5b, indicated as F ox), it is noteworthy that in the control spectra (non-oxidized IFNβ1a) both fragments at 1006.44 and at 228.18 were not observed (Figure S6a–S6c). Altogether, these data suggest that untreated IFNβ1a already contains some oxidized Met36 residues, whereas oxidized Phe38 is only present in oxidized IFNβ1a.
Met62 is a buried residue(7, 31). It was resistant to oxidation by hydrogen peroxide(31), however, in conditions of MCO, we detected MS/MS spectra for the sequence M62LQNIFAIFR71 containing the oxidized Met62 (Figure S7). Again, the spectrum for the loss of methane sulfenic acid in position 62, indicative of Met62 oxidation, was observed (Figure S8).
Met117 is a solvent exposed residue(7, 31) and is most susceptible to oxidation (under mild oxidative conditions via hydrogen peroxide), as compared to Met1, Met36 and Met62(31). Figure S9 represents a spectrum for the sequence L116MSSLHLK123, where besides Met117, His121 is oxidized to 2-oxo-His. The distinct b2+ and y3+ ions indicate oxidation of Met117 and His121, respectively.
Phenylalanine oxidation
The fragment containing oxidized Phe38 (M36NF38DIPEEIK45) has been described above in the section Methionine oxidation.
Phe50 is adjacent to several Gln residues, which may undergo deamidation, during digestion or MCO. Figure S10 presents a typical MS/MS spectrum indicating the oxidation of Phe50, in addition to deamidation of one of the Gln residues. Formation of pyroglutamic acid, which should be attributed only to the cyclization of N-terminal Gln in the peptide (− 18 Da) during the LC-MS analysis, was also detected.
Histidine and tyrosine oxidation
His oxidation to 2-oxo-His is well documented and has been measured in proteins such as insulin(34–36). Stadtman et al. showed that His oxidation also can yield asparagine as a potential product(37).
The sequence N86LLANVYHQINHLK99 obtained after digestion with a combination of trypsin and endoproteinase Glu-C, contains two His residues in positions 93 and 97 and one Tyr residue in position 92. Several observed combinations of His93 or His97 mono oxidation (+ 16 Da on His), oxidation of both His residues (+ 32 Da), and formation of Asn in positions 93 and 97 are shown in Figures S11, S12, S13, S14(a–b), S15(a–b), where the respective modifications are proven by b7+, b8+, and b12+ ions, as well as y3+, y7+, y8+, and y12++ ions. In Figure S15 are shown only the ions which differ from Figure S14.
His121 and Tyr3 oxidation was already demonstrated for the fragments L116MSSLHLK123 and M1SYNLLGFLQR11, respectively, as discussed above.
Sequence I129LHYLK134 contains two potential sites for oxidation: His131 and Tyr132. The ions highlighted in Figure S16a fit to oxidation of either His131 or Tyr132, whereas in Figure S16b, based on multiple fragments for the y4+ ion, oxidation of both amino acid residues is suggested. The ions y4+−H2O and b4+ in Figure S16b, at m/z 574.26 and m/z 559.29, respectively, support the simultaneous oxidation of His and Tyr. Also, the region of the spectrum between 200 and 400 m/z contains two peaks identified as b3+ in which His (Figure S16b, H ox, m/z 380.28) or Tyr (Figure S16c, Y ox, m/z 364.11) are oxidized. Furthermore, in Figure S16c is depicted the ion y3++ supporting Tyr oxidation. Altogether, it seems that three different peptides containing either oxidized His131, oxidized Tyr132, or both amino acids oxidized, were detected simultaneously.
In the sequence E137YSHCAWTIVR147, containing a missed cleavage site and 2 additional oxygen atoms, there are 3 potential oxidation sites: Tyr138, His140, and Trp143. In the spectrum shown in Figure S17 the y5+−H2O ion at m/z 656.96 rules out Trp oxidation, y8++ at m/z 501.36, in the insert, supports His oxidation, and b3+ and b4++ are in accordance with Tyr oxidation to DOCH. Further evidence for His140 oxidation is shown in Figure S18, where Cys is derivatized with iodoacetamide, and N-terminal Glu (E) is cleaved-off by Glu-C.
Tryptophan oxidation
Trp22 oxidation was detected in both the oxidized and the control samples, suggesting a spontaneous oxidation of this residue during protein storage or handling. Spectra presented in Figures S19 and S20 show the formation of the major oxidative products, hydroxyl Trp (probably hydroxylated at position 5), and N-formyl kynurenine(38, 39), respectively, as evidenced through the presence of the sequence-indicative ions y3+−y7+ and some b+ ions, particularly b3+. In addition to Trp oxidation, Asn25 deamidation was detected in peptide L20LWQLNGR27 from both oxidized and control IFNβ1a, which could be expected given the highly deamidation prone N25G sequence(40).
Cross-links
This section focuses on MS/MS identification of covalent cross-links formed through 1,4- and/or 1,6-type addition to the electrophilic product of Tyr oxidation, i.e. DOCH, resulting from MCO of IFNβ1a. As we have only MS/MS but no NMR data of our reaction products, we have to assume that both reactions are possible. However, the structures displayed in Figures 6–9 representatively show only the products of 1,4-addition. Table 3 lists all the cross-linked peptides detected by MS/MS and Figures 6–9 represent the corresponding MS/MS spectra along with the peptide cross-links. Data will be presented starting from the first chemically modified electrophilic amino acid measured, i.e. DOCH in the first observed position (Tyr3) of IFNβ1a. Data reported were obtained from IFNβ1a that was oxidized, reduced, alkylated and digested.
Figure 6A.
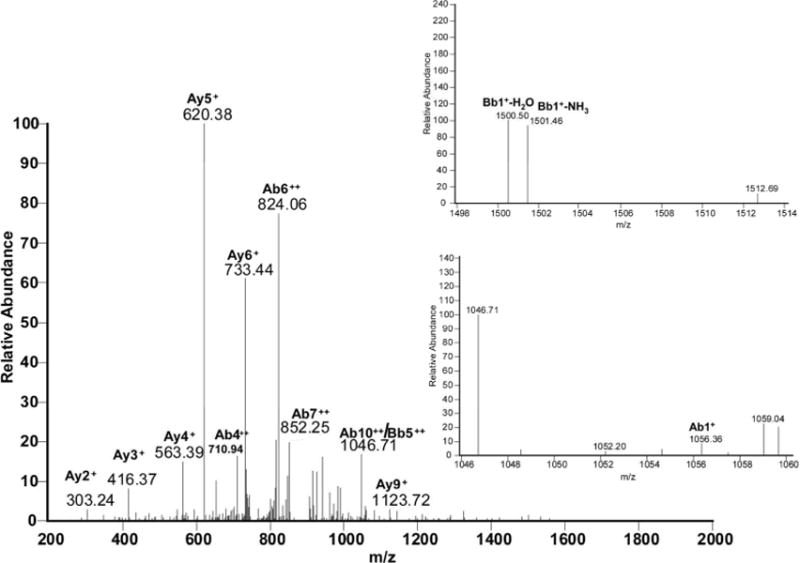
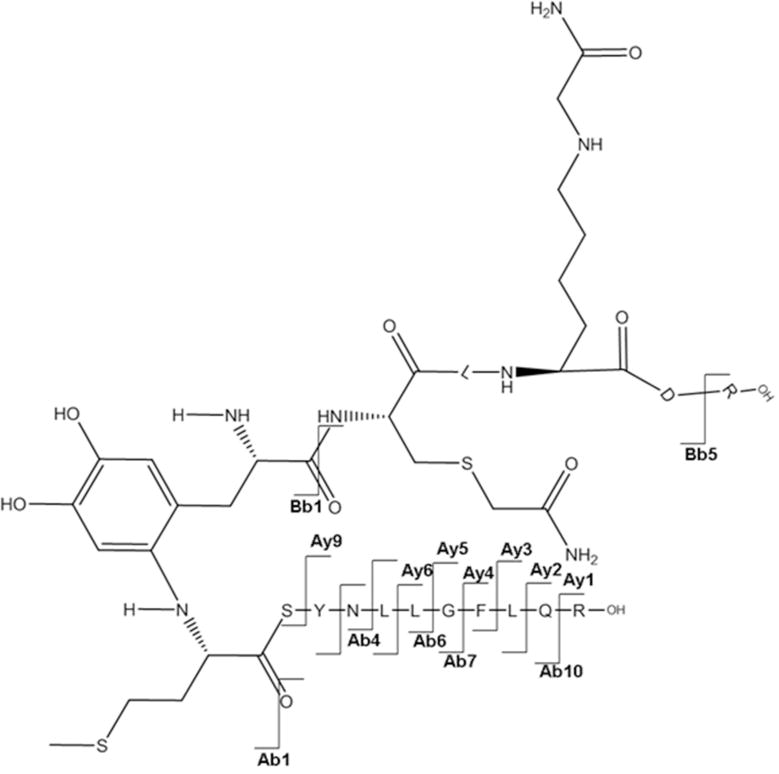
a: MS/MS spectrum for the sequence M1SYNLLGFLQR11 cross-linked through Met1 to the sequence Y30C(IAM)LK(IAM)DR35. b: Suggested chemical structure of the cross-linked sequence (theoretical mass 2265.11, experimental m/z 756.02, [M+H]3+, Δm from parent peptide − 0.05).
Table 3.
Cross-links measured by LC-ESI-MS/MS in peptides derived from oxidized IFNβ1a after reduction, alkylation and enzymic digestion (in blue nucleophilic amino acids, in red amino acids oxidized to DOCH).
| Cross-link* | Experimental m/z | Charge | Theoretical mass | Δm |
|---|---|---|---|---|
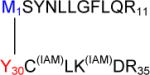 Figure 6A |
756.02 | 3 | 2265.11 | −0.05 |
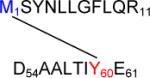 Figure 6B |
756.03 | 3 | 2265.11 | −0.02 |
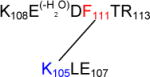 Figure 6C |
598.3 | 2 | 1194.60 | +0.00 |
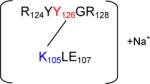 Figure 6D |
569.79 | 2 | 1137.56 | +0.02 |
(IAM): alkylation with iodoacetamide; (−H2O): loss of 1 water molecule.
Met1 – Tyr30
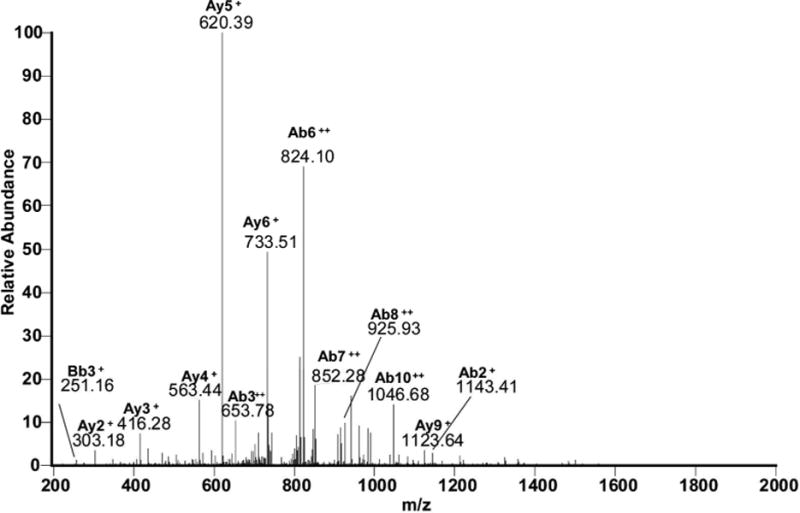
The free N-terminus of Met1 is a good nucleophile for 1,4- or 1,6-type addition under our applied conditions (pH 7.2), where the N-terminal amino group is in part deprotonated. Figure 6A, panel a, shows the MS/MS spectrum corresponding with the cross-link between Met1 and Tyr30 in the respective tryptic peptides, as illustrated by the scheme in Figure 6A, panel b. The series of y-ions in the peptide A (where the prefix A indicates the ions that belong to the peptide M1SYNLLGFLQR11) of the cross-link (Ay2+ to Ay9+) together with Ab1+ in the lower insert (zoomed region m/z 1046 –1060) demonstrate that the peptide A is linked to peptide B through the Met1 amino group, whereas Bb1+−H2O and Bb1+−NH3 ions (zoomed m/z region 1498–1514) indicate the involvement of Tyr30 of the peptide B in the cross-link formation. Note that in the sequence Y30CLKDR35 both Cys and Lysine (Lys) residues are in alkylated form (displayed by the mass of the parent peptide and fragment ions Bb1+ and Bb5++). Furthermore, we noticed that both Met1 and Tyr3 in the cross-linked peptide are not oxidized, suggesting that the cross-link may protect these residues from oxidation.
Met1 – Tyr60
Figure 6B, panel a, reports the MS/MS spectrum for the cross-link involving Met1 and Tyr60. The structure provided in panel b, where Met1 is covalently bound to Tyr60, was drawn for the position 6 of the aromatic ring of DOPA (or DOCH), which is sterically less hindered than the positions 1 and 2. In the peptide M1SYNLLGFLQR11, an additional mass shift of + 16 should be attributed to Tyr3 oxidation to DOPA, as suggested by the presence of ion Ab3++, ruling out the oxidation of another sensitive residue, Phe8. The ion Ab2+ supports the structure shown in Figure 6B, panel b. In addition, fragments matching ions Ay9+ and Bb3+ disprove the involvement of sequences D54AA56 and Y3NLLGFLQR11 in the cross-link formation.
Figure 6B.
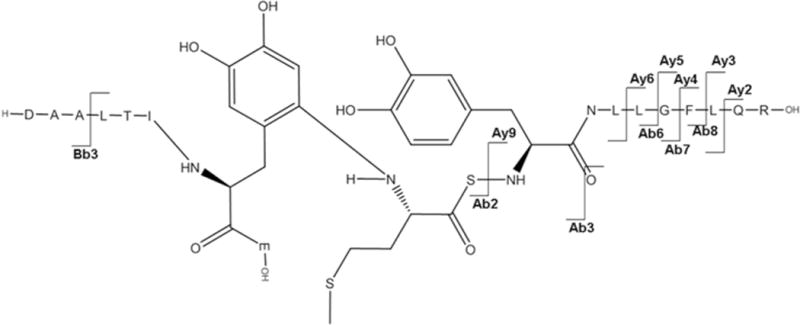
a: MS/MS spectrum of sequence M1SYNLLGFLQR11 cross-linked through Met1 to the sequence D54AALTIY60E61. b: Suggested chemical structure of the cross-linked sequence (theoretical mass 2265.11, experimental m/z 756.03, [M+H]3+, Δm from parent peptide − 0.02).
Lys105 – Phe111
Panel a of Figure 6C displays the MS/MS spectrum consistent with a cross-link between Lys105 and Phe111. It must be noticed that the mass for this cross-link is 18 Da lower than the mass that would be expected. This is due to loss of water on Glu109 in the sequence K108EDFTR113, which can arise from the cyclization with its N-terminal amino group or with the more nucleophilic amino group of Lys1, which would lead to the formation of a 1,4 diazocine-like structure as shown by Koriatopoulou et al.(41). The presence of an intense fragment ion Bb1++ in particular shows that the sequence K108EDFTR113 is covalently attached to Lys105, as illustrated in Figure 6C, panel b. Furthermore, the fragment ion By2+ proves that neither Leu106 nor Glu107 of the sequence K105LE107 are involved in the cross-linked structure.
Figure 6C.
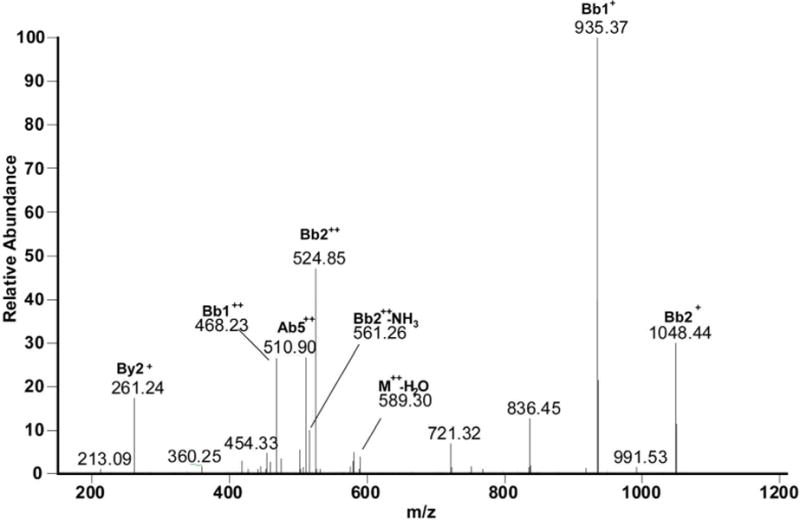
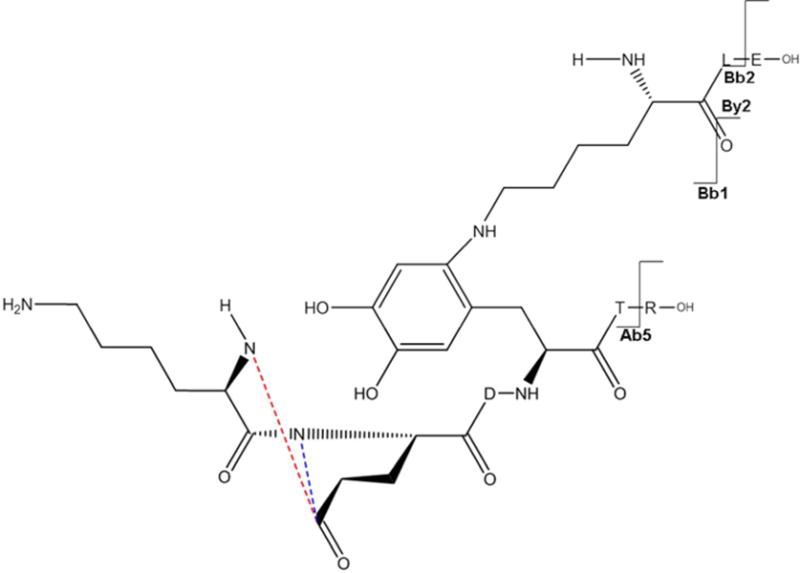
a: MS/MS spectrum of sequence K105LE107 cross-linked through Lys105 to the sequence K108E(−H2O)DF111TR113. b: Suggested chemical structure of the cross-linked sequence (theoretical mass 1194.60, experimental m/z 598.30, [M+H]2+, Δm from parent peptide +0.00).
Lys105 – Tyr126
The cross-linked sequence R124YYGR128 with K105LE107 through Lys105 and Tyr126 is presented in Figure 6D. Here there are two adjacent Tyr residues (in R124YYGR128) which can be involved in the cross-link. The Ay3++ and Ay4++ related ions however suggest that Tyr126 (and not Tyr125) serves as electron acceptor in the cross-link. [Bb1+ Na]+ and [Bb2 + Na]+ fragments prove the involvement of Lys105 in the peptide cross-linking. The sodiation of the respective fragment ions (except for By2+) is due to complex formation between the C-terminal carboxyl of Arg128 and residual sodium ions present in samples, likely due to incomplete removal of sodium phosphate buffer during protein dialysis after MCO.
Figure 6D.
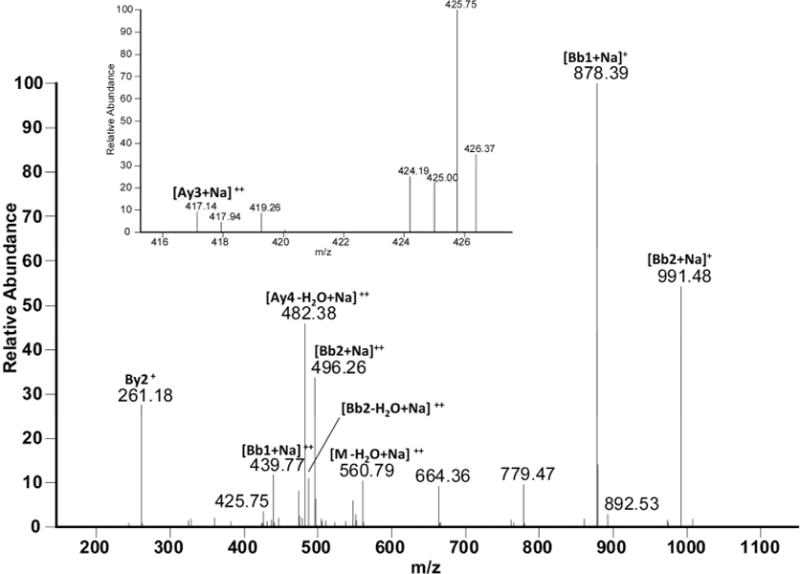
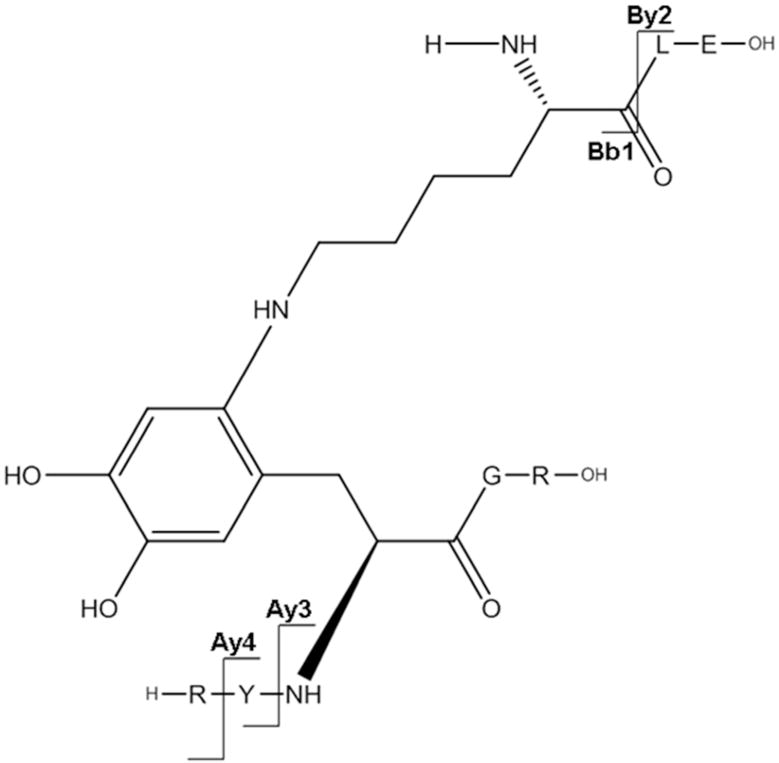
a: MS/MS spectrum of sodiated sequence K105LE107 linked through Lys105 to the sequence R124YY126GR128 (the inset show the MS/MS spectrum of figure 11, panel A zoomed in the m/z range 415–428. b: Suggested chemical structure of the cross-linked sequence (theoretical mass 1137.56, experimental m/z 569.79, [M+H]2+, Δm from native parent peptide + 0.02).
Discussion
Oxidative chemical modification of amino acid residues may alter the secondary and tertiary protein structure, favoring interaction between protein surfaces and subsequently leading to non-covalent aggregation. Indeed, MCO (using the oxidative system presented in this paper) of proteins, such as growth hormone(42), IFNα2b(14), IFNβ1a(8), IgG1(16), IgG2(43), insulin and PEGylated insulin(19), relaxin(44), recombinant SHa (29–231) prion protein(45), and superoxide dismutase(46), have been shown to lead to extensive aggregation. Moreover, protein aggregates generated by MCO have been shown to be particularly immunogenic. For instance, Hermeling et al.(14, 15), using a transgenic immune tolerant mouse model, discovered that aggregated IFNα2b, oxidized via metal catalysis was more immunogenic than IFNα2b aggregates that were produced otherwise. More recently, van Beers and colleagues hypothesized that a particular combination of oxidation and covalent aggregation could be responsible for the immune response against IFNβ1a exposed to MCO, though no particular mechanisms for chemical cross-linking were demonstrated(8). Similarly, MCO of an IgG1 was found to produce immunogenic aggregates(16).
Here, performing an extensive mass spectrometric characterization of IFNβ1a oxidized under conditions identical to those in the above examples (i.e., Cu2+/ascorbate catalysis), we describe chemical modifications of multiple residues that can affect the structure of IFNβ1a. Results of the current study, in particular, reveal that the oxidative modification of Phe and Tyr results in an electron acceptor structure, namely DOCH, which may be involved in IFNβ1a cross-linking through a 1,4- or 1,6-type addition mechanism of primary amines to DOCH. The results reported in this work are in agreement with the data we recently published using insulin as a model protein(17). However, since Met and Trp are not present in insulin, we did not know whether these are involved in covalent cross-linking of other proteins such as IFNβ1a. Although we detected oxidation of 4 Met residues and 1 Trp residue in oxidized IFNβ1a, we found no evidence of Met or Trp being involved in covalent cross-links. These data are in contrast with the common opinion that Trp oxidation could lead to new carbonyl groups and subsequent cross-linking via Schiff base formation(47). However, the imine structure generated through Schiff base formation is reversible unless it is reduced with sodium borohydride or cyanoborohydride to form a secondary amine group(48), which may explain why Trp oxidation did not seem to be involved in aggregation of IFNβ1a.
Our data on ABS derivatization of the control samples (Figure 1B) support the hypothesis that DOCH is an intermediate in the covalent cross-links of IFNβ1a aggregates induced by MCO. SEC analysis under reducing conditions did not point to disulfide-mediated covalent cross-links. Furthermore, fluorescence spectroscopy data and MS/MS analysis helped to exclude other potential mechanisms of aggregation, such as dityrosine formation. This suggests that the reaction between two tyrosyl radicals is not favorable under our experimental conditions(49).
In addition to the putative cross-links via a 1,4- or 1,6-type addition mechanism, extensive oxidative modifications of numerous amino acid residues were induced during MCO: Met in positions 1, 36, 62 and 117 were found to be oxidized to sulfoxides. Met1 was also oxidized to a higher oxidation state to form Met sulfone. His oxidation to 2-oxo-His, as observed before in insulin and other proteins exposed to similar oxidative conditions(50), targeted IFNβ1a residues in positions 93, 97, 121, 131 and 140. Moreover, oxidative modification to Asn was detected for His93 and His97.
Unfortunately, we failed to detect reliable spectra for ABS-derivatized peptides derived from oxidized IFNβ1a (unpublished data). This could be attributed to either relatively low abundance of such modifications or further chemical reactions, which may occur during the analysis, thus interfering with the detection based on a mass increase of 179, 196 and 366 Da(27).
Table 2 demonstrates that not all of the potential targets for MCO were found oxidized. Most of the unmodified amino acids, such as Phe70, Trp79 and Tyr125, are in fact residues which are responsible for stabilizing the hydrophobic core of the molecule through a broad network of hydrogen bonds(7). This could protect them from MCO and chemical cross-linking. A similar explanation could be given for the relatively low number of detected cross-linked peptides, which is far below the theoretically possible number of cross-links.
Besides the chemical modifications discussed above, based on far-UV CD measurements we noticed that MCO leads to changes in IFNβ1a’s secondary structure resulting in the conversion of alpha helices to disordered structures(29, 51). Interestingly, this was also seen upon MCO of IFNα2b (14), but not of IgG1, which has only minor alpha helical content and for which its betasheet content seemed to be largely maintained after MCO(16). Nevertheless, MCO of IgG1 did result in immunogenic aggregates(16), comparable to the observations with IFNα2b and IFNβ1a. This suggests that the change in secondary structure in oxidized IFNα2b and IFNβ1a is not a major factor contributing to the immunogenicity of these products. Rather, the combination of covalent aggregation and chemical modifications, which are probably very similar across the different proteins, likely contribute to the immunogenicity of proteins exposed to MCO. The structural changes observed in the present study, i.e. oxidation, conformational changes and aggregation, are comparable to the ones previously measured in IFNα2b, although at that time a detailed MS/MS analysis was not performed and only Met oxidation was investigated(14). However, recently DOPA formation in IFNα2b was measured by our group (unpublished data), and, considering that both interferons share similar structural features, we suggest that the same type of chemical modifications and cross-links could be expected in IFNα2b.
Conclusions
In this work we mapped the modifications of the primary structure in oxidized and immunogenic aggregates of IFNβ1a, focusing on the amino acids potentially involved in covalent cross-linking. Several oxidative modifications were identified, especially in oxidation prone solvent-exposed amino acids. The primary role of DOPA and DOCH, i.e. Tyr and Phe oxidation products, as electron acceptors for 1,4- or 1,6-type addition, has been confirmed. These results are in agreement with the mechanistic studies previously performed on insulin, and potentially might be extended to other proteins exposed to similar oxidative conditions.
Supplementary Material
Acknowledgments
The authors thank Dr. Nadya Galeva for performing MS measurements, The University of Kansas and the National Institutes of Health (PO1AG12993) for financial support, and Leiden University Fund/Slingelands (LUF, 1111/19-4-11\O, SI) and Nederlandse Stichting voor Farmacologische Wetenschappen (NSFW) for travel grants to Riccardo Torosantucci.
References
- 1.Kaynor C, Xin M, Wakefield J, Barsoum J, Qin XQ. Direct evidence that IFN-beta functions as a tumor-suppressor protein. J Interferon Cytokine Res. 2002;22:1089–1098. doi: 10.1089/10799900260442511. [DOI] [PubMed] [Google Scholar]
- 2.Calabresi PA. Diagnosis and management of multiple sclerosis. Am Fam Physician. 2004;70:1935–1944. [PubMed] [Google Scholar]
- 3.Runkel L, Meier W, Pepinsky RB, Karpusas M, Whitty A, Kimball K, Brickelmaier M, Muldowney C, Jones W, Goelz SE. Structural and functional differences between glycosylated and non-glycosylated forms of human interferon-beta (IFN-beta) Pharm Res. 1998;15:641–649. doi: 10.1023/a:1011974512425. [DOI] [PubMed] [Google Scholar]
- 4.Nelissen I, Martens E, Van Den Steen PE, Proost P, Ronsse I, Opdenakker G. Gelatinase B/matrix metalloproteinase-9 cleaves interferon-beta and is a target for immunotherapy. Brain. 2003;126:1371–1381. doi: 10.1093/brain/awg129. [DOI] [PubMed] [Google Scholar]
- 5.Kagawa Y, Takasaki S, Utsumi J, Hosoi K, Shimizu H, Kochibe N, Kobata A. Comparative-Study of the Asparagine-Linked Sugar Chains of Natural Human Interferon-Beta-1 and Recombinant Human Interferon-Beta-1 Produced by 3 Different Mammalian-Cells. J Biol Chem. 1988;263:17508–17515. [PubMed] [Google Scholar]
- 6.Conradt HS, Egge H, Peter-Katalinic J, Reiser W, Siklosi T, Schaper K. Structure of the carbohydrate moiety of human interferon-beta secreted by a recombinant Chinese hamster ovary cell line. J Biol Chem. 1987;262:14600–14605. [PubMed] [Google Scholar]
- 7.Karpusas M, Nolte M, Benton CB, Meier W, Lipscomb WN, Goelz S. The crystal structure of human interferon beta at 2.2-angstrom resolution. Proc Natl Acad Sci U S A. 1997;94:11813–11818. doi: 10.1073/pnas.94.22.11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Beers MM, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W. Oxidized and aggregated recombinant human interferon beta is immunogenic in human interferon beta transgenic mice. Pharm Res. 2011;28:2393–2402. doi: 10.1007/s11095-011-0451-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Beers MM, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W. Aggregated recombinant human interferon Beta induces antibodies but no memory in immune-tolerant transgenic mice. Pharm Res. 2010;27:1812–1824. doi: 10.1007/s11095-010-0172-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rifkin RA, Maggio ET, Dike S, Kerr DA, Levy M. n-Dodecyl-beta-d-Maltoside Inhibits Aggregation of Human Interferon-beta-1b and Reduces Its Immunogenicity. J Neuroimmune Pharmacol. 2011;6:158–162. doi: 10.1007/s11481-010-9226-7. [DOI] [PubMed] [Google Scholar]
- 11.Seefeldt MB, Rosendahl MS, Cleland JL, Hesterberg LK. Application of high hydrostatic pressure to dissociate aggregates and refold proteins. Curr Pharm Biotechnol. 2009;10:447–455. doi: 10.2174/138920109788488914. [DOI] [PubMed] [Google Scholar]
- 12.Bertolotto A, Malucchi S, Sala A, Orefice G, Carrieri PB, Capobianco M, Milano E, Melis F, Giordana MT. Differential effects of three interferon betas on neutralising antibodies in patients with multiple sclerosis: a follow up study in an independent laboratory. J Neurol Neurosurg Psychiatry. 2002;73:148–153. doi: 10.1136/jnnp.73.2.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scagnolari C, Bellomi F, Turriziani O, Bagnato F, Tomassini V, Lavolpe V, Ruggieri M, Bruschi F, Meucci G, Dicuonzo G, Antonelli G. Neutralizing and binding antibodies to IFN-beta: relative frequency in relapsing-remitting multiple sclerosis patients treated with different IFN-beta preparations. J Interferon Cytokine Res. 2002;22:207–213. doi: 10.1089/107999002753536176. [DOI] [PubMed] [Google Scholar]
- 14.Hermeling S, Aranha L, Damen JMA, Slijper M, Schellekens H, Crommelin DJA, Jiskoot W. Structural characterization and immunogenicity in wild-type and immune tolerant mice of degraded recombinant human interferon alpha2b. Pharm Res. 2005;22:1997–2006. doi: 10.1007/s11095-005-8177-9. [DOI] [PubMed] [Google Scholar]
- 15.Hermeling S, Schellekens H, Maas C, Gebbink MF, Crommelin DJ, Jiskoot W. Antibody response to aggregated human interferon alpha2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci. 2006;95:1084–1096. doi: 10.1002/jps.20599. [DOI] [PubMed] [Google Scholar]
- 16.Filipe V, Jiskoot W, Basmeleh AH, Halim A, Schellekens H. Immunogenicity of different stressed IgG monoclonal antibody formulations in immune tolerant transgenic mice. MAbs. 2012;4:740–752. doi: 10.4161/mabs.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torosantucci R, Mozziconacci O, Sharov V, Schöneich C, Jiskoot W. Chemical Modifications in Aggregates of Recombinant Human Insulin Induced by Metal-Catalyzed Oxidation: Covalent Cross-Linking via Michael Addition to Tyrosine Oxidation Products. Pharm Res. 2012;29:2276–2293. doi: 10.1007/s11095-012-0755-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q, Olah ME, van Galen PJ, et al. Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torosantucci R, Kukrer B, Mero A, Van Winsen M, Tantipolphan R, Jiskoot W. Plain and mono-pegylated recombinant human insulin exhibit similar stress-induced aggregation profiles. J Pharm Sci. 2011;100:2574–2585. doi: 10.1002/jps.22523. [DOI] [PubMed] [Google Scholar]
- 20.Giulivi C, Davies KJ. Dityrosine: a marker for oxidatively modified proteins and selective proteolysis. Methods Enzymol. 1994;233:363–371. doi: 10.1016/s0076-6879(94)33042-5. [DOI] [PubMed] [Google Scholar]
- 21.Ikehata K, Duzhak TG, Galeva NA, Ji T, Koen YM, Hanzlik RP. Protein targets of reactive metabolites of thiobenzamide in rat liver in vivo. Chem Res Toxicol. 2008;21:1432–1442. doi: 10.1021/tx800093k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu H, Freitas MA. A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu H, Freitas MA. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 2009;9:1548–1555. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu H, Yang L, Freitas MA. A robust linear regression based algorithm for automated evaluation of peptide identifications from shotgun proteomics by use of reversed-phase liquid chromatography retention time. BMC Bioinformatics. 2008;9:347. doi: 10.1186/1471-2105-9-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu H, Zhang L, Freitas MA. Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J Proteome Res. 2008;7:138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 27.Sharov VS, Dremina ES, Galeva NA, Gerstenecker GS, Li X, Dobrowsky RT, Stobaugh JF, Schöneich C. Fluorogenic Tagging of Peptide and Protein 3-Nitrotyrosine with 4-(Aminomethyl)-benzenesulfonic Acid for Quantitative Analysis of Protein Tyrosine Nitration. Chromatographia. 2010;71:37–53. doi: 10.1365/s10337-009-1409-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharov VS, Dremina ES, Pennington J, Killmer J, Asmus C, Thorson M, Hong SJ, Li X, Stobaugh JF, Schöneich C. Selective fluorogenic derivatization of 3-nitrotyrosine and 3,4-dihydroxyphenylalanine in peptides: a method designed for quantitative proteomic analysis. Methods Enzymol. 2008;441:19–32. doi: 10.1016/S0076-6879(08)01202-0. [DOI] [PubMed] [Google Scholar]
- 29.Bobst CE, Abzalimov RR, Houde D, Kloczewiak M, Mhatre R, Berkowitz SA, Kaltashov IA. Detection and characterization of altered conformations of protein pharmaceuticals using complementary mass spectrometry-based approaches. Anal Chem. 2008;80:7473–7481. doi: 10.1021/ac801214x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 31.Orru S, Amoresano A, Siciliano R, Napoleoni R, Finocchiaro O, Datola A, De Luca E, Sirna A, Pucci P. Structural analysis of modified forms of recombinant IFN-beta produced under stress-simulating conditions. Biol Chem. 2000;381:7–17. doi: 10.1515/BC.2000.002. [DOI] [PubMed] [Google Scholar]
- 32.Sharov VS, Galeva NA, Dremina ES, Williams TD, Schöneich C. Inactivation of rabbit muscle glycogen phosphorylase b by peroxynitrite revisited: does the nitration of Tyr613 in the allosteric inhibition site control enzymatic function? Arch. Biochem Biophys. 2009;484:155–166. doi: 10.1016/j.abb.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Hair RAJ, Reid GE. Neighboring group versus cis-elimination mechanisms for side chain loss from protonated methionine, methionine sulfoxide and their peptides. Eur Mass Spectrom. 1999;5:325–334. [Google Scholar]
- 34.Cheng RZ, Kawakishi S. Site-specific oxidation of histidine residues in glycated insulin mediated by Cu2+ Eur J Biochem. 1994;223:759–764. doi: 10.1111/j.1432-1033.1994.tb19050.x. [DOI] [PubMed] [Google Scholar]
- 35.Sadineni V, Schöneich C. Selective oxidation of Zn2+ – Insulin catalyzed by Cu2+ J Pharm Sci. 2007;96:1844–1847. doi: 10.1002/jps.20834. [DOI] [PubMed] [Google Scholar]
- 36.Hovorka SW, Biesiada H, Williams TD, Huhmer A, Schöneich C. High sensitivity of Zn-2+ insulin to metal-catalyzed oxidation: Detection of 2-oxo-histidine by tandem mass spectrometry. Pharm Res. 2002;19:530–537. doi: 10.1023/a:1015164200431. [DOI] [PubMed] [Google Scholar]
- 37.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 38.Anderson LB, Maderia M, Ouellette AJ, Putnam-Evans C, Higgins L, Krick T, MacCoss MJ, Lim H, Yates JR, 3rd, Barry BA. Posttranslational modifications in the CP43 subunit of photosystem II. Proc Natl Acad Sci U S A. 2002;99:14676–14681. doi: 10.1073/pnas.232591599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor SW, Fahy E, Murray J, Capaldi RA, Ghosh SS. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. J Biol Chem. 2003;278:19587–19590. doi: 10.1074/jbc.C300135200. [DOI] [PubMed] [Google Scholar]
- 40.Palmisano G, Melo-Braga MN, Engholm-Keller K, Parker BL, Larsen MR. Chemical Deamidation: A Common Pitfall in Large-Scale N-Linked Glycoproteomic Mass Spectrometry-Based Analyses. J Proteome Res. 2012;11:1949–1957. doi: 10.1021/pr2011268. [DOI] [PubMed] [Google Scholar]
- 41.Koriatopoulou K, Karousis N, Varvounis G. Novel synthesis of the pyrrolo[2,1-c][1,4]benzodiazocine ring system via a Dieckmann condensation. Tetrahedron. 2008;64:10009–10013. [Google Scholar]
- 42.Hovorka SW, Hong JY, Cleland JL, Schöneich C. Metal-catalyzed oxidation of human growth hormone: Modulation by solvent-induced changes of protein conformation. J Pharm Sci. 2001;90:58–69. doi: 10.1002/1520-6017(200101)90:1<58::aid-jps7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 43.Luo QZ, Joubert MK, Stevenson R, Ketchem RR, Narhi LO, Wypych J. Chemical Modifications in Therapeutic Protein Aggregates Generated under Different Stress Conditions. J Biol Chem. 2011;286:25134–25144. doi: 10.1074/jbc.M110.160440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S, Nguyen TH, Schöneich C, Borchardt RT. Aggregation and precipitation of human relaxin induced by metal-catalyzed oxidation. Biochemistry. 1995;34:5762–5772. doi: 10.1021/bi00017a008. [DOI] [PubMed] [Google Scholar]
- 45.Requena JR, Groth D, Legname G, Stadtman ER, Prusiner SB, Levine RL. Copper-catalyzed oxidation of the recombinant SHa(29–231) prion protein. Proc Natl Acad Sci U S A. 2001;98:7170–7175. doi: 10.1073/pnas.121190898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- 47.Rauniyar N, Prokai L. Detection and identification of 4-hydroxy-2-nonenal Schiff-base adducts along with products of Michael addition using data-dependent neutral loss-driven MS3 acquisition: method evaluation through an in vitro study on cytochrome c oxidase modifications. Proteomics. 2009;9:5188–5193. doi: 10.1002/pmic.200900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dohno C, Okamoto A, Saito I. Stable, specific, and reversible base pairing via Schiff base. J Am Chem Soc. 2005;127:16681–16684. doi: 10.1021/ja054618q. [DOI] [PubMed] [Google Scholar]
- 49.Kato Y, Kitamoto N, Kawai Y, Osawa T. The hydrogen peroxide/copper ion system, but not other metal-catalyzed oxidation systems, produces protein-bound dityrosine. Free Radic Biol Med. 2001;31:624–632. doi: 10.1016/s0891-5849(01)00623-2. [DOI] [PubMed] [Google Scholar]
- 50.Schöneich C. Mechanisms of metal-catalyzed oxidation of histidine to 2-oxo-histidine in peptides and proteins. J Pharm Biomed Anal. 2000;21:1093–1097. doi: 10.1016/s0731-7085(99)00182-x. [DOI] [PubMed] [Google Scholar]
- 51.Greenfield N, Fasman GD. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry. 1969;8:4108–4116. doi: 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.