

Abstract
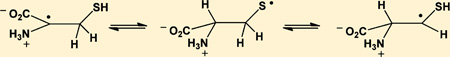
The mercapto group of cysteine (Cys) is a predominant target for oxidative modification, where one-electron oxidation leads to the formation of Cys thiyl radicals, CysS•. These Cys thiyl radicals enter 1,2- and 1,3-hydrogen transfer reactions, for which rate constants are reported in this paper. The products of these 1,2- and 1,3-hydrogen transfer reactions are carbon-centered radicals at position C3 (α-mercaptoalkyl radicals) and C2 (•Cα radicals) of Cys, respectively. Both processes can be monitored separately in Cys analogues such as cysteamine (CyaSH) and penicillamine (PenSH). At acidic pH, thiyl radicals from CyaSH permit only the 1,2-hydrogen transfer according to equilibrium 12, +H3NCH2CH2S• ⇌ +H3NCH2 •CH–SH, where rate constants for forward and reverse reaction are k12 ≈ 105 s−1 and k−12 ≈ 1.5 × 105s−1, respectively. In contrast, only the 1,3-hydrogen transfer is possible for thiyl radicals from PenSH according to equilibrium 14, (+H3N/CO2H)Cα–C(CH3)2–S• ⇌ (+H3N/CO2H)•Cα–C(CH3)2–SH, where rate constants for the forward and the reverse reaction are k14 = 8 × 104 s−1 and k−14 = 1.4 × 106 s−1. The •Cα radicals from PenSH and Cys have the additional opportunity for β-elimination of HS•/S•−, which proceeds with k39 ≈ (3 ± 1) × 104 s−1 from •Cα radicals from PenSH and k−34 ≈ 5 × 103 s−1 from •Cα radicals from Cys. The rate constants quantified for the 1,2- and 1,3-hydrogen transfer reactions can be used as a basis to calculate similar processes for Cys thiyl radicals in proteins, where hydrogen transfer reactions, followed by the addition of oxygen, may lead to the irreversible modification of target proteins.
INTRODUCTION
This work provides a kinetic analysis and equilibrium constants for hydrogen transfer reactions of cysteine (Cys) thiyl radicals, CysS•, which may play a significant role in biochemical processes of oxidative stress, but have received little attention in the field of oxidative stress biology. There is increasing evidence that thiyl radicals play an important role in enzyme-catalyzed transformations, synthetic organic chemistry, and the post-translational modification of proteins.1–4 For example, ribonucleotide reductase and pyruvate–formate lyase utilize CysS• radicals for hydrogen abstraction and addition reactions, respectively. Here, reversible hydrogen transfer reactions between radicals and C–H and S–H bonds of glycine (Gly) and Cys, respectively, provide CysS• radicals in a protein conformation-dependent pathway. The role of thiols/thiyl radicals in “polarity-reversal catalysis” has been well documented,5 for example in hydrogen abstraction reactions from alcohols and ethers,5–7 and radiation chemical experiments have provided rate constants on the order of 103–104 M−1 s−1 for the reactions of thiyl radicals with various alcohols and ethers.6,7 Subsequently, NMR experiments have provided rate constants on the order of 104 M−1 s−1 for the reactions of thiyl radicals with carbohydrates.8 Pulse radiolysis experiments have also provided rate constants on the order of 105 M−1 s−1 for the reactions of thiyl radicals with the anionic forms of Gly and alanine (Ala),9 H2N–CH(R)CO2− (R = H, CH3), i.e., charge states where the developing radical on the Cα atom experiences some capto-dative stabilization. In contrast, rate constants for the reactions of thiyl radicals with zwitterionic amino acids and N-acetyl amino acid amides are lower (on the order of 104 M−1 s−1), unless the amino acid is part of a diketopiperazine ring.10,11 Recently, pulse radiolysis experiments have provided rate constants for intramolecular hydrogen transfer reactions of CysS• radicals with Ala and Gly in model peptides, which are on the order of 104–105 s−1.12 Product studies have revealed that these reactions can lead to covalent H/D-exchange at original amino acid C–H bonds when reactions are performed in D2O.13–15 During these product studies, we obtained mass spectrometric evidence for l- to d-amino acid conversion of Ala,14 and the formation of dehydroalanine.15 A conceivable free radical mechanism of dehydroalanine formation would imply •Cα radical formation on Cys, followed by β-elimination of HS•, and H2S formation during amino acid and peptide disulfide photolysis has been recognized.16 Together with the known propensity of thiyl radicals from glutathione (GSH) to establish equilibrium with carbon-centered radicals at the γ-glutamic acid (Glu) Cα position,17,18 and possibly additional locations, including Cβ,19 these observations suggest the potential for hydrogen transfer within CysS• radicals, i.e. formal 1,2- and 1,3-hydrogen transfer reactions between the CysS• radicals and the Cα–H and the Cβ–H bond. In fact, some indirect kinetic data on a 1,2-hydrogen transfer equilibrium for thiyl radicals of β-mercaptoethanol were reported on the basis of time-resolved pulse radiolysis experiments.20 Pulse radiolysis data were also provided for intramolecular •Cα radical formation of thiyl radicals of homocysteine and Cys at alkaline pH (i.e., for the anionic forms of these amino acids).9 These •Cα radicals were kinetically monitored indirectly through their reducing propensities (reduction of methyl viologen). However, at alkaline pH there is also a formal possibility for intramolecular electron transfer from the amino group, followed by deprotonation at Cα, which would yield reducing •Cα radicals; hence, at alkaline pH the formation of reducing radicals does not require hydrogen transfer. However, by mass spectrometry we detected covalent H/D exchange at Cys, when CysS• radicals were generated in peptides13–16 and insulin21 in D2O, consistent with reversible hydrogen transfer processes. Additional support for the possibility of hydrogen abstraction from α-thio-substituted C–H bonds comes from our kinetic NMR experiments on the reaction of cysteamine thiyl radicals with the Cγ–H and Cε–H bonds of methionine (Met),11 and from electron spin resonance experiments on the photolysis of aqueous solutions of CH3SH, which resulted in the formation of •CH2SH.22 On the basis of the known photochemistry of thiols/thiolates, any formation of •CH2SH via primary photochemical processes appears unlikely, as these result in the cleavage of the C–S or S–H bond, or photoionization (of thiolate), yielding •CH3/•SH, CH3S•/•H or CH3S•/eaq −, respectively (where eaq − represent a hydrated electron). Subsequently, the reaction of •CH3, H• and eaq − with CH3SH would lead to additional CH3S• and •CH3 but not •CH2SH. Hence, the detection of •CH2SH is most likely the result of a 1,2-hydrogen transfer equilibrium between CH3S• and •CH2SH. By analogy, the formation of CH3–C•H–SH radicals during the photolysis of CH3CH2SH in a dilute glass matrix23,24 can be rationalized through a 1,2-hydrogen transfer equilibrium between CH3–C•H–SH and CH3CH2S•. ESR experiments following the generation of thiyl radicals from Cys,25 GSH,26 and penicillamine (PenSH)26 have also indicated the formation of carbon-centered radicals, and especially the presence of more than one carbon-centered radical after the oxidation of GSH26 points to the fact that the γ-Glu Cα–H bond is not the only hydrogen donor for intramolecular hydrogen transfer to the original thiyl radical. In fact, Becker et al.27 had suggested the formation of a carbon-centered radical at the Cα position of Cys upon γ-irradiation of GSH in water ice at pH 3, i.e. under conditions where the N-terminal amino group of the γ-Glu residue is protonated and would not permit the formation of a capto-datively28 stabilized radical at the Cα position of γ-Glu. All these observations lead to the notion that hydrogen transfer equilibria in thiyl radicals are possible and must be quantified in order to fully understand the reactions of thiyl radicals during biological conditions of oxidative stress. Results in support of these hydrogen transfer equilibria will be presented in this paper together with quantitative data on kinetics and equilibrium constants. By using model compounds of various structures, we are able to evaluate the kinetics for 1,2- and 1,3-hydrogen transfer reactions individually, and to provide rate constants, which are then used to quantify both reactions in thiyl radicals from Cys, together with kinetics for β-elimination of HS• from •Cα radicals of Cys and PenSH.
EXPERIMENTAL SECTION
Materials
The chemicals were provided by ABCR, (N-Ac–Dha–OMe (or: methyl 2- acetamidoacrylate), dibenzyl disulfide), Acros (NaN3), Bachem (cystine (CysSSCys), N-Ac–CysOMe), Fluka (cystamine × 2HCl, cysteamine, 3,3′-dithiodipropionic acid, penicillamine disulfide, penicillamine), Merck (H2SO4), Siegfried (KOH), Sigma-Aldrich (cysteine, methanol), TCI (2,2′-dithiodiethanol, thiocamphor), and VWR (Na2S·3H2O). Ultra pure water (18.2 MΩ) was taken from a Milli-Q system (Millipore) and t-BuOH had been recrystallized several times and checked for peroxides. The names and structues of the thiols used are shown in Table 1.
Table 1.
Names and Structures of Thiols Used
| Name | Formula |
|---|---|
| Mercaptoethanol | |
| Cysteamine (CyaSH) | |
| Cysteine (CysSH) | |
| 3-Mercaptopropionic acid | |
| Penicillamine (PenSH) | 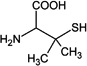 |
| Dehydoralanine (Dha) | |
| Thiocamphor | 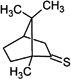 |
| Benzylthiol / Benzylmercaptane |
Pulse Radiolysis
Pulse radiolysis experiments were performed with a Febetron 705 (L-3 Communications, San Leandro, CA) 2 MeV accelerator at the ETH Zürich, as described previously.12 The irradiation of water leads to the formation of primary radicals according to reactions 1 and 2.29
| (1) |
| (2) |
Thiyl radicals were produced either by reduction of disulfides, reactions 3–6 (representatively shown for cystamine), or by oxidation of thiolates by azide, reactions 8–11 (representatively shown for cysteamine). In the reductive pathway, hydroxyl radicals are scavenged by t-BuOH, reaction 7.
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
RESULTS
A location in an amino acid is generally indicated with reference to the carbon carrying the amino and the carboxylate groups, the α-carbon atom, Cα. In the following, carbon-centered radicals at the Cα atom will be referred to as •Cα radicals. However, we will not use the term •Cβ radical for a carbon-centered radical at Cβ (the C3 position) of Cys. Instead, we use the term α-mercapto alkyl radical in order to avoid confusion with other amino acids and for comparison to non-amino-acid compounds
1. CyaSSCya and PenSSPen
1.1. Absorption Spectra
CyaSSCya (H2NCH2CH2–SS–CH2CH2NH2)
Pulse irradiation of an Ar-saturated solution containing 1 mM CyaSSCya (×2HCl), 10 mM H2SO4, and 2 M tert-butanol (t-BuOH) leads to an initial absorption spectrum (monitored 1.2 µs after the pulse) with a broad maximum at 330 nm and an unstructured UV-band with λmax < 240 nm (Figure 1A). Within the next 7 µs, the 330 nm band decays by approximately 50%. Concomitantly a new band at 280–290 nm evolves while the far-UV band (<240 nm) disappears. There is an isosbestic point at approximately 310 nm. The 330 nm band is a characteristic property of thiyl radicals generated via reactions 3–6,12,30–33 and is assigned to the cysteamine thiyl radical, CyaS•. HO• radicals are nearly quantitatively scavenged with t-BuOH (reaction 7; k7 = 6 × 108 M−1 s−1),34 preventing a significant extent of reaction of HO• radicals with CyaSSCya and/or Cl−.
Figure 1.

Cysteamine (CyaSH) and cystamine (CyaSSCya). Pulse radiolysis of Ar-saturated solutions of 1 mM CyaSSCya (2HCl), 10 mM H2SO4, and 2 M t-BuOH. Panel A: Absorption spectra at 1.2 µs (squares), 3 µs (triangles) and 8.5 µs (circles) after pulse. Dose: approximately 150 Gy. Panel B: Absorption-vs-time profiles at 280 nm (open squares) and 330 nm (gray circles). Dose: approximately 150 Gy. Panel C: Kinetic traces used to determine rate constants. Circles: experimental conditions as for panels A and B, except that data were acquired with a low dose of 31 Gy; at λ = 285 nm a fit gives kobs = 2.5 × 105 s−1. Open squares: 0.64 mM CyaSH, 0.9 M NaN3, N2O-saturated, dose = 15 Gy; at λ = 420 nm the fit gives kobs = 1 × 105 s−1. Panel D: Pulse radiolysis of N2O-saturated aqueous solution, pH ca. 3.5 (adjusted with HCl), containing 1 mM CyaSSCya (2×HCl) and 1 M t-BuOH. Dose: ca. 140 Gy.
The molar absorptivity of thiyl radicals, ε330, has recently been re-evaluated to ε330 = 150 M−1 cm−1.12 Absorption bands with maxima at 280–290 nm35–38 have been reported for α-(alkylthio)alkyl radicals (R-•CH-S-R’), and, by analogy, the current 280 nm band could be assigned to an α-mercapto alkyl radical of Cya, +H3NCH2 •CH–SH, generated according to equilibrium 12.20
| (12) |
Consistent with such an assignment, we also detected a 285 nm band after 1-electron reduction of a thioketone (thiocamphor) at acidic pH (see below), and after laser flash photolysis of 5,6-dihydroxy-1,2-dithiacyclohexane.39 Also consistent with the assignment to an α-mercapto alkyl radical, no 280–290 nm band was detected after pulse irradiation of penicillamine disulfide (PenSSPen), where the C3 hydrogens (of Cys) are substituted by methyl groups (see below).
PenSSPen (HOOC(NH2)CHC(CH3)2–SS–C(CH3)2CH(NH2)-COOH)
Pulse irradiation of an Ar-saturated solution containing 1 mM PenSSPen, 10 mM H2SO4, and 2 M t-BuOH leads to an absorption spectrum (monitored 1.2 µs after the pulse) which exhibits a maximum at 265 nm and a shoulder/band at 330 nm, which is broader than that obtained from CyaSSCya (Figure 2A). The 265 nm maximum is characteristic for amino-acid •Cα radicals of the general structure R–NH–•Cα(R′)–CO–R″.40,41 The broadening of the shoulder at 330 nm could be due to the presence of a small yield of perthiyl radicals (R–SS•, with an absorption maximum at ca. 375 nm42,43) and/or due to contributions from the •Cα radicals, which display a shoulder around 310 nm, which extends up to 400 nm40,41 Over longer times (3–9 µs after the pulse), the absorbance at λ >300 nm decays while the absorbance maximum at 265 nm persists.
Figure 2.

Penicillamine (PenSH) and penicillamine disulfide (PenSSPen). Pulse radiolysis of Ar-saturated solutions of 1 mM PenSSPen, 10 mM H2SO4, and 2 M t-BuOH. Samples were irradiated with very high doses of approximately 150 Gy. Panel A: Absorption spectra at 1.2 µs (squares), 3 µs (triangles) and 9 µs (circles) after the pulse. Panel B: Absorption-vs-time profiles at 260 nm (triangles), 280 nm (open squares) and 330 nm (diamonds). Panel C: Kinetics traces used to estimate rate constants. Gray squares: Experimental conditions like panels A and B, λ = 265 nm; the fit (full line) yields kobs = 1.5 × 106 s−1; full circles and inset, N2O-saturated solutions of 0.05 mM PenSH, 0.1 M NaN3, 10 mM KOH, dose =10 Gy, λ = 285 nm, kobs = 8 × 104 s−1.
1.2. Kinetics
CyaSSCya
At pH 2 (Figure 1B), the decay of absorptivity at 330 nm is paralleled by a concomitant increase of absorptivity at 280 nm. For quantitative kinetic measurements, experiments were repeated at lower doses (31 Gy), which yield ca. 11 µM H• atoms and 10 µM HO• radicals. The hydroxyl radicals will be scavenged by t-BuOH (reaction 7). Hydrogen atoms will react with a rate constant of 1 × 1010 M−1 s−1 with the disulfide, and, at a concentration of 1 mM disulfide, thiyl radicals will be produced with a pseudofirst order rate constant of 1 × 107 s−1 (t1/2 = 69 ns). Figure 1C (285 nm trace) shows a representative kinetic trace at 285 nm recorded at pH 2, which proceeds with significantly slower kinetics and appears, therefore, secondary to thiyl radical generation. The observed rate constant kobs(CyaSSCya, 280 nm, pH 2) = 2.5 × 105 s−1 is a function of all first-order reactions and competing radical recombination reactions. Because of the number of competing first and second order reactions, there is no analytical solution to derive kobs. The maximal influence of the competition reactions can be described by the initial rate of the fastest competing reaction, which is the thiyl radical–thiyl radical recombination; for this recombination, we approximate k ca. 1.7 × 109 M−1 s−1 by analogy to the rate constant measured for PenS•.31 Such value appears more reasonable in view of the measured recombination rate constants for other small radicals, i.e. the 2-hydroxyprop-2-yl radical, (CH3)2C•OH, and the 2-methyl-2-hydroxyprop-1-yl radical, •CH2C(CH3)2OH, where 2k = 1.6 × 109 and 1.2 × 109 M−1 s−1.44 We assume that the initially reported rate constant for the bimolecular recombination of thiyl radicals from cysteamine at acidic pH, k = 1.4 × 1010 M−1 s−1,33 is too high, likely because of the underlying fast first-order transformation according to equilibrium 12. With an initial concentration of thiyl radicals of [RS•] = 10−5 M, we calculate an initial rate of ca. 1.5 × 104 s−1. We find kobs to be more than 1 order of magnitude larger than this value, so the influence of recombination reactions on kobs is below the uncertainty of the experiment. Therefore, we can approximate kobs = k12 + k−12 = 2.5 × 105 s−1, where k12 + k−12 represents the rate constant of equilibration, which most likely proceeds via a solvent-assisted hydrogen transfer (see Discussion). We repeated this experiment with half the concentration of CyaSSCya (×2HCl) (i.e., 0.5 mM) with an entirely new optical system, involving a multipass system, allowing for 6 cm path length of the analyzing light. The kinetic fit yields kobs = 2.8 × 105 s−1, i.e., a value nearly identical to that obtained with 1 mM CyaSSCya (also the absorbance yields at 280–290 were similar to those obtained with 1 mM CyaSSCya, indicating that the ratio of [CyaSSCya]/[t-BuOH] had little influence on these yields). In a separate experiment, we generated CyaS• photochemically (266 nm laser flash photolysis) from 2 mM CyaSSCya (×2HCl) (in an N2O-saturated aqueous solution containing 10 mM H2SO4 and 2 M t-BuOH), and monitored the first-order build up at 285 nm (kobs ca. 3.5 × 105 s−1).70 Hence, a variation of the CyaSSCya concentration by a factor of 4 resulted in values of kobs between 2.5 × 105 s−1 and ca. 3.5 × 105 s−1, strongly indicating that the observed reaction does not depend on the concentration of the disulfide.
The rate constant k12 was independently confirmed by an experiment at pH 11 (Figure 1C; 420 nm trace). Here, we pulse-irradiated N2O-saturated solutions containing 0.64 mM CyaS− and 0.1 M NaN3. Under these conditions, the reaction of HO•/O•− with N3− yields N3• radicals (reaction 8), which are in equilibrium with •N6− (equilibrium 9),9 and rapidly oxidize CyaS− to CyaS• (reaction 10), which exists in equilibrium 11 with the dimeric radical anion, CyaSSCya•−.32 CyaS• radicals will undergo the 1,2-H-transfer (reaction 12); however, based on the pKa of the cysteamine mercapto group, pKa = 8.3,45 at pH 11, the ensuing α-(mercapto)alkyl radical is expected to deprotonate (reaction 13) so that the 1,2-Htransfer will be quasi irreversible due to the low propensity of carbon-centered radicals to react with a deprotonated mercapto group.46
| (13) |
For the disappearance of the absorption at 420 nm we measure kobs(420 nm, pH 11) = (6 ± 3) × 104 s−1. Importantly, the rate of formation of CyaS• through reaction 10 is computed to k10[CyaS−] = 6 × 105 s−1. Hence, for the determination of kobs in Figure 1C (420 nm trace) we utilized the absorption vs time trace only for times at >10 µs after the pulse. The concentration of CyaSSCya•− is proportional to the concentration of thiyl radicals (equilibrium 11). Equilibrium constants K11 for the pH-dependent formation of CyaSSCya•− are known where K11 ≈ 1000 M−1 at pH 11,45 so that k12 can be derived as k12= kobs (1 + 1/K11[CyaS−]) ≈ 105 s−1.
In a control experiment, we ensured that the spectral changes observed in Figures 1A–1C are not caused by the reaction of tert-butyl radicals, •CH2C(CH3)2OH, with CyaSSCya. An N2O-saturated aqueous solution, pH 3.5, containing 1 mM CyaSSCya and 1 M t-BuOH was pulse-irradiated and the absorption vs time traces recorded at 280 and 330 nm are displayed in Figure 1D. Under these experimental conditions, ca. 92% of the hydrated electrons react with N2O according to reaction 2, generating HO• radicals which subsequently react with t-BuOH. Hence, the initial yield of HO• radicals is ca. twice as high as compared to the experiments performed under Ar-saturation. Figure 1D shows that we do not observe any absorption increase at 280 nm over 10 µs, confirming that tert-butyl radicals in the presence of CyaSSCya are not responsible for the spectral changes recorded in Figures 1A–C. The fast initial absorption decays, observed at both 280 and 330 nm over ca. 2 µs in Figure 1D, are likely due to the finite lifetime of intermediary [CyaSSCya]•− generated through reaction 3 (ca. 5% of the hydrated electrons will react with CyaSSCya under the experimental conditions). This was independently confirmed through an absorption vs time trace recorded at 420 nm (not shown).
PenSSPen
At pH 2, the equilibration between PenS• and the •Cα radicals (equilibrium 14) is completed within 2 µs (Figure 2B; pulse irradiation of an Ar-saturated solution containing 1 mM PenSSPen, 10 mM H2SO4, and 2 M t-BuOH). Hence, even kinetic data recorded with high doses can be used to derive kobs(PenSSPen, pH 2, 265 nm) = k14 + k−14 = 1.5 × 106 s−1 (Figure 2C, open squares).
| (14) |
Because of the high molar absoptivity of •Cα radicals we can also monitor k14 through the alkaline reduction of PenSSPen. Here, the product •Cα radical cannot be detected at its absorption maximum at 265 nm because of the interference of thiolate; instead 285 nm was chosen for detection (Figure 2C, filled circles). A build-up with a rate constant of k14 = 8 × 104 s−1 was derived. An additional experiment (Figure 2C, insert) analogous to that described above for Cya was performed with 0.1 mM PenS− in an Ar-saturated solution containing 10 mM KOH and 100 mM NaN3, generating PenS• and [PenSSPen]•− according to reactions 15 and 16.
| (15) |
| (16) |
The insert in Figure 2C representatively displays data recorded at λ = 330 nm. However, the absorption decays via apparent first-order kinetics with kobs = (3 ± 1) × 104 s−1 at all observed wavelengths, 285, 330, and 420 nm. This observation is consistent with a 1,3-H-transfer of the zwitterionic PenS• radical (reaction 17), followed by a fragmentation process and not a radical–radical recombination, which will be discussed below. Analogous to the 1,2-H-transfer at alkaline pH observed for CyaS•, we expect that the 1,3-H-transfer in PenS• is followed by a deprotonation of the mercapto group at alkaline pH (equilibrium 18).
| (17) |
| (18) |
2. CysSSCys
At pH 2, the reduction of CysSSCys with hydrogen atoms leads to an absorption spectrum with a maximum at 260 nm and a shoulder at 330 nm at 1.5 µs after the pulse (Figure 3A), consistent with an initial production of CysS• radicals that are in a fast equilibrium with •Cα radicals (equilibrium 19), analogous to the reactions detected for PenSSPen. Also here, the equilibrium is located on the side of thiyl radicals. The •Cα radicals can only be detected because their molar absorptivity is approximately 2 orders of magnitude larger than that of the thiyl radicals. At longer times, 9 µs after the pulse, the characteristic absorption of •Cα radicals has disappeared, while the absorption at 330 nm is reduced to approximately half its original value and a new band at 280– 290 nm can be observed (Figures 3A and 3B). This is consistent with the delayed formation of considerable amounts of α-mercaptoalkyl radicals (equilibrium 20), analogous to the reactions described for CyaSSCya (see above).
| (19) |
| (20) |
Figure 3.

Cysteine (CysSH) and cystine (CysSSCys). Panels A and B: Pulse radiolysis of Ar-saturated solutions of 1 mM CysSSCys, 10 mM H2SO4, and 2 M t-BuOH. Samples were irradiated with very high doses of approximately 150 Gy. Panel A: Spectra are shown for 1.5 µs (squares), 3 µs (triangles) and 9 µs (circles) after pulse. The bands at 330 nm (thiyl radicals) and 265 nm (•Cα radicals) decay quickly while a band at 280 nm (α-mercaptoalkyl radicals) forms. Panel B: Absorption-vs-time profiles at 280 nm (open squares) and 330 nm (filled squares); dose: approximately 150 Gy. Panel C: Pulse radiolysis of Ar-saturated solutions of 1 mM N-Ac–Cys–OMe, 10 mM KOH, 0.1 M t-BuOH; dose =21 Gy. The thiyl radical concentration is monitored via the equilibrium to the disulfide radical anion at 420 nm; the solid line represents a first-order fit, which yields kobs = 1 × 105 s−1. Inset: decay of the carbon-centered radicals monitored at λ = 330 nm; dose =25 Gy.
We performed an additional experiment analogous to that for PenSSPen under alkaline conditions: an Ar-saturated solution of 1 mM N-Ac–Cys–OMe (disulfide), 100 mM t-BuOH in 10 mM KOH was pulse irradiated with doses of about 20 Gy (Figure 3C). At 420 nm, a monotonic decay was observed, which was further analyzed. At 330 nm, we detect a second-order decay process with 2k/ε ≈ 1 × 105 Mcm−1s−1 (Figure 3C, insert). If this absorption were caused by a •Cα radical (ε330 ≈ 3000 M−1 cm−1)12,41 then we would derive 2k ≈ 3 × 108 M−1 s−1, a reasonable value for the recombination of C-centered radicals in amino acids. If, instead, a first order mechanism is assumed a first-order reaction rate constant of <1 × 103 s−1 would result.
3. Assignment of the Absorption Band of α-Mercaptoalkyl Radicals
To confirm the position of the absorption maximum assigned to α-mercaptoalkyl radicals, we selected various independent experimental entries for their generation.
One-Electron Reduction of a Thioketone (Thiocamphor, 1,7,7-Trimethylbicyclo[2.2.1]heptan-2-thione)
The pulse irradiation of 1.3 mM thiocamphor in a 65:35 (v/v) mixture of water and 2-propanol, containing 20 mM H2SO4, led to the transient spectrum shown in Figure 4 at 2 µs after the pulse. Under these conditions, the radiolysis results only in reducing species (H• and CH3C•(OH)CH3). The spectrum shows an absorption maximum at 285 nm, which is assigned to the α-mercaptoalkyl radical derived from the reduction of thiocamphor (reactions 21–25). Theoretical studies support the addition of hydrogen atoms to the sulfur moiety of thioketones.47 We note that the reduction by CH3C•(OH)CH3 likely proceeds via an intermediary adduct analogous to the addition of a series of alkyl radicals to thioketones;48 in case of thiocamphor such an adduct represents a thiohemiacetal, which likely decomposes into acetone and thioketone radical anion, which protonates (reactions 24 and 25). The 285 nm maximum observed after one-electron reduction of the thioketone is consistent with the assignment of the analogous 280 nm bands from Cya and Cys to α-mercaptoalkyl radicals.
| (21) |
| (22) |
| (23) |
| (24) |
| (25) |
Figure 4.

Absorption spectrum recorded at 2 µs after pulse irradiation of an Ar-saturated solution of 1.3 mM thiocamphor in 2-propanol/water (35:65; v/v), containing 20 mM H2SO4. The molar absorptivity was calculated based on a radiation chemical yield of G = 6.3.
One-Electron Reduction of 3,3′-Dithiodipropionic acid (HOOCCH2CH2–S–S–CH2CH2COOH)
Pulse irradiation of an Ar-saturated solution containing 1 mM 3,3′-dithiodipropionic acid, 2 M t-BuOH, and 10 mM KOH leads to an optical spectrum at 1 µs after the pulse (Figure 5), which displays a broad structure-less absorbance in the far UV region (<250 nm), typical for carboxyethyl radicals,49 and a narrow peak with λmax = 285 nm with a half width, at half-maximum, of 10 nm, similar to the absorption band obtained from Cya and Cys (which is absent for PenSH). The broad absorbance at λ > 300 nm can be due to small yields of perthiyl radicals and/or residual thiyl radicals. The α-mercaptoalkyl radical would be generated through a 1,2-H-transfer analogous to the reactions proposed for thiyl radicals from Cya and Cys.
Figure 5.

Absorption spectra recorded at 1, 2, and 8 µs after pulse irradiation of an Ar-saturated aqueous solution containing 1.8 mM 3,3′-dithiodipropionic acid, 1 M t-BuOH, and 10 mM KOH; dose ≈150 Gy.
One-Electron Reduction of 2,2′-dithiodiethanol (HOCH2CH2-S–S–CH2CH2OH)
Pulse irradiation of an Ar-saturated solution containing 2,2′-dithiodiethanol, 1 M BuOH, and 10 mM H2SO4 leads to optical absorbance spectra, which show a time-dependent decrease at 330 nm, indicative for a reaction of thiyl radicals, paralleled by a time-dependent increase of a narrow band with λmax = 285 nm (Figure 6). Importantly, no absorption maximum at λ < 285 is detected. Qualitatively, these data are consistent with the formation of α-mercaptoalkyl radicals; however, due to the rather small absorbance yields (close to the detection limit) the spectral shape will not be discussed in more detail.
Figure 6.

Pulse irradiation of Ar-saturated aqueous solutions containing 1 mM 2,2′-dithiodiethanol, 10 mM H2SO4, and 1 M t-BuOH. Panel A: Absorption spectra after 3, 5, and 10 µs after the pulse; dose =130 Gy. Panel B: Absorption-vs-time trace recorded at 285 nm; dose: 52 Gy.
One-Electron Reduction of Dibenzyl Disulfide (Ph–CH2–S–S–CH2–Ph)
Pulse irradiation of 2.8 × 10−4 M dibenzyl disulfide in 80% methanol and 20% water (v/v) saturated with Ar and containing 10−2 MKOH leads to the transient spectra shown in Figure 7. Under such conditions the primary species will be solvated electrons and hydroxymethyl radicals according to reactions 1, 26 and 27 (based on k26 = 2.6 × 106 M−1 s−1,34 ca. 90% of H• are expected to react with CH3OH).
| (26) |
| (27) |
Figure 7.

Pulse irradiation of Ar-saturated solution of 0.29 mM dibenzyl disulfide in 80% 2-propanol/20% H2O (v/v), containing 10 mM KOH. Panel A: Absorption spectra at 5 and 30 µs after the pulse (dose ≈25 Gy), corrected by subtraction of the absorbance spectra (normalized to the yield of primary radicals) generated in a control experiment displayed in panel C. Panel B: Absorption spectra at 5 and 30 µs after the pulse (uncorrected); dose ≈25 Gy. Panel C: Control experiment: pulse irradiation of an N2O-saturated solution of 10 mM KOH in 80% 2-propanol/20% H2O (v/v).
The reduction of dibenzyl disulfide by solvated electrons will occur nearly exclusively at the disulfide group because the reaction of electrons with disulfides is generally 3 orders of magnitude faster than that with an aromatic reference compound such as toluene.34 The disulfide radical anions will readily fragment to a thiolate and a thiyl radical. Any interconversion between thiyl and α-mercaptoalkylradicals should be quasi irreversible because of the ensuing deprotonation of the mercapto group, which will prevent the reverse reaction.46 Our product spectra do not show the typical characteristics expected for a thiyl radical, which has only a weak absorption band with ε330 ~ 150 M−1 cm−1.12 On the basis of G(eaq−) = 2.7,29 this would yield a maximal expected absorption at 330 nm of 4.2 × 10−5 absorption units (AU)/Gy, which amounts to only 4% of the experimentally measured absorbance of 1.0 × 10−3 AU/Gy (at 5 µs after the pulse). In addition thiyl radicals only display weak absorbances at 260 nm, where in our experimental spectrum we see a strong maximum. We detect no perthiyl radicals (λmax(R–SS•) ≈ 375 nm)42,43 or unsubstituted benzyl radicals (absorption bands at 258, 307, and 318 nm)50 which could theoretically form via reductive cleavage of the carbon–sulfur bond (reactions 28 and 29).
| (28) |
| (29) |
Clearly, the bands at 260 and 330 nm are caused by the reaction of hydrated electrons with Ph–CH2–SS–CH2–Ph: these bands are not present in a control experiment, when hydrated electrons are removed through reaction with N2O (reaction 2), yielding hydroxyl radicals which convert methanol to hydroxymethyl radicals/radical anions (reaction 27). On the basis of these control experiments we expect no significant reduction of Ph–CH2–SS–CH2–Ph by hydroxymethylradical anions (•CH2O−) over the time scale of our experiment, consistent with the comparably lower reactivity of •CH2O− with disulfide bonds (cf. k = 2.4 × 108 M−1 s−1 for the reduction of 3,4-dihydroxy-1,2-dithiacyclohexane, oxidized dithiothreitol, at 25 °C).51
The prominent spectral features shown in the difference spectrum (Figure 7A) must be related to the initial formation of thiyl radicals, and point to a rather stable product radical (no decay over 100 µs). Our control experiments in the presence of N2O exclude hydroxymethyl radicals as a source of this species. Therefore, we rationalize the transient spectrum by formation of the deprotonated α-mercaptobenzyl radical through a 1,2-Htransfer within the original thiyl radical, followed by deprotonation (reactions 30–32).
| (30) |
| (31) |
| (32) |
This assignment is supported by the well-known spectral features of benzyl and substituted benzyl radicals. Unsubstituted benzyl radicals exhibit a prominent absorption band at around 260 nm and weaker bands between 305 and 320 nm.50 The 305–320 bands are known to be red-shifted in benzyl radicals, which contain electron-donating substituents in the α-position, such as, for example, to λmax =340 nm in α-aminobenzyl radicals.52 The 330 nm band recorded after pulse-irradiation of Ph–CH2–SS–CH2–Ph is consistent with such an expected red shift, i.e., from 305 to 318 to 330 nm, for a benzyl radical, which contains an additional electron-donating substituent, i.e., Ph–•CH–SH/Ph–•CH–S−.
So far our data are consistent with (i) the generation of α-mercaptoalkylradicals from thiyl radicals of the general structure R–CH2S•, (ii) •Cα radicals from thiyl radicals of amino acids, and (iii) for Cys at least two equilibria which are rationalized by 1,2- and 1,3-hydrogen transfer reactions, that is, equilibria between thiyl radicals and •Cα and α-mercaptoalkyl radicals. For •Cα radicals, the evaluation of reaction kinetics must take into account the possibility of β-elimination of HS•, yielding dehydroalanine (Dha). Such reaction would display first-order kinetics. For thiyl radicals from PenSH, we observed a first-order decay at longer time scales at all wavelengths, consistent with the possibility of β-elimination. We could not unambiguously isolate such a first-order decay for radicals derived from Cys, suggesting that the β-elimination of a Cys •Cα radical is less efficient. In the following, we examined the kinetics of such β-elimination reactions in amino acids.
4. β-Elimination from •Cα Radicals
Reaction of N-Acetyl-dehydroalanine-amide (N–Ac–Dha amide) with S•−
Pulse irradiation of N2O-saturated solutions containing 0.18 mM Na2S, 0.85 mM N-Ac–Dha amide (Ac–NH–C(=CH2)– CONH2), 10 mM NaN3 and 10 mM KOH (pH 12) leads to the transient spectra displayed in Figure 8A, recorded between 1 and 8 µs after the pulse. At pH 12, sulfide exists in equilibrium between HS− and S2− (pKa ca. 1253). The N3• radicals can enter two different reaction pathways: one is the oxidation of HS−/S2− to S•− (reaction 33) (at pH 11, we expect exclusively the formation of S•−54), which can add to N-Ac– Dha amide, generating •Cα radicals of N-acetyl cysteine (thiolate) amide (reaction 34), consistent with the spectrum displayed in Figure 8. The rate constant for the formation of •Cα radicals, k34 = 6 × 108 M−1 s−1, was determined at 270 nm (Figure 8B, crosses)
| (33) |
| (34) |
.
Figure 8.

Reversible addition of S•− to Dha. Panel A: Raw difference spectra recorded after pulse irradiation of N2O-saturated solutions of 0.85 mM N-Ac–Dha–OMe, 0.18 mM Na2S, 10 mM KOH, and 10 mM NaN3, dose ≈30 Gy. Panel B: Absorption-vs-time traces for experiment and controls. Crosses: 0.85 mM N-Ac–Dha–OMe, 0.18 mM Na2S, 10 mM KOH and 10 mM NaN3, λ = 270 nm; dose =33 Gy. Gray diamonds: 1 mM N-Ac–Dha–OMe + 100 mM NaN3, 1 mM KOH, λ = 275 nm; dose =28 Gy. Black circles: 10 mM NaN3, 30 Gy, λ = 275 nm. Open triangles: 10 mM NaN3, 1 mM Na2S, λ = 275 nm; dose =33 Gy. Panel C: solution as in panel A, λ = 330 nm, dose = 34 Gy.
The other process is the, potentially reversible, addition of •N3 to N-Ac–Dha amide (reaction 35), for which a representative trace is shown in Figure 8B (open diamonds).
| (35) |
The net result of reaction 35 would be a delayed production of S•− and N-actetyl cysteine (thiolate) amide •Cα radicals. Importantly, the •Cα radicals are rather stable and decay via a second order process, indicating that the reverse β-elimination of S•− must be negligible under our experimental conditions. At 330 nm (Figure 8C), we derive 2k/ε ≈ 1 × 105 M−1 cm−1 s−1, the same value as found for the decay of the absorption of radicals of Cys under the same conditions (see above).
One-Electron Reduction of N-Ac–Cys Methyl Ester
In a separate experiment, we pulse-irradiated an Ar-saturated solution containing 1 mM N-Ac–Cys–OMe disulfide, 0.1 M t-BuOH, 10 mM KOH at low doses (25 Gy) and monitored the absorbance at 260 nm, i.e. the •Cα radicals, CH3CO– NH–•Cα(CH2SH/S−)CO2CH3, generated through intramolecular 1,3-hydrogen transfer from the initial thiyl radicals (Figure 9, middle trace). For comparison, Figure 9 (higher trace) also contains the absorption vs time trace obtained after the addition of HS/S•− to N-Ac–Dha amide. Clearly, both absorption vs time traces show some distinct differences, and the bottom trace in Figure 9 represents the difference between these traces, which can be fitted to first order kinetics, representing the β-elimination of HS/S•− from CH3CO– NH–•C(CH2SH/S−)CO2CH3 (see Discussion).
Figure 9.

Comparison of the decay kinetics of Cys-•Cα radicals between two complementary experiments in order to quantify the kinetics of β-elimination of HS•/S•− from Cys-•Cα radicals. Pulse-irradiation of (black squares) Ar-saturated solutions containing 1 mM N-Ac–Cys–OMe disulfide, 100 mM t-BuOH, and 10 mM KOH (dose =25 Gy), producing 8.5 µM CysS•, and (gray circles) N2O-saturated solutions containing 0.85 mM N-Ac–Dha–OMe, 0.18 mM Na2S, 10 mM NaN3 and 10 mM KOH (applied dose =11 Gy), producing 6.9 µM S•−. To match the inititial absorptivities, the Cys trace was scaled by a factor of 1.8. Then the difference of the two traces (open trangles) was analyzed with a first-order fit, assumed to represent the β-elimination of HS•/S•− from Cys–•Cα radicals.
DISCUSSION
The one-electron oxidation of Cys generates Cys thiyl radicals. In the area of oxidative stress biology, such thiyl radicals have commonly been considered as source of disulfides, sulfenic, sulfinic, and sulfonic acid. However, additional, more deleterious reaction pathways of Cys thiyl radicals may need to be considered such as the intermediary formation of carboncentered radicals at both the C2 (Cα) and C3 position of Cys. Earlier ESR experiments have identified at least two distinct carbon-centered radicals originating from the oxidation of GSH, and also carbon-centered radicals originating from the oxidation of penicillamine26 and Cys.25 Both, Cα- and additional carbon-centered radicals were recently reported after pulse irradiation of GSH and GSSG, which leads to the initial formation of thiyl radicals.19 However, it remained unclear where these additional carbon-centered radical(s) were located. The pulse radiolysis data presented here are consistent with these earlier experiments, are able to localize the positions of the generated carbon-centered radicals, and provide rate and equilibrium constants for reversible hydrogen transfer reactions in thiyl radicals from various precursors.
Cysteamine: The 1,2-Hydrogen Transfer
A distinct feature of the one-electron reduction of CyaSSCya is the initial formation of CyaS•, indicated by a 330 nm band, followed by the reversible conversion of CyaS• into a transient species absorbing with λmax ≈ 285 nm. By analogy to α-(alkylthio)alkyl radicals,35–38 and based on complementary data from the reduction of a thioketone, this 285 nm transient is assigned to the α-mercaptoalkyl radical formed according to equilibrium 12. Under the assumption that equilibrium 12 is established at 8.5 µs after the pulse, the bleaching at 330 nm between 1.2 and 8.5 µs approximately matches the absorption increase at 285 nm. This is also reflected in the absorption vs time traces in Figure 1B, and would suggest that the molar absorptivities of +H3NCH2CH2S• and +H3NCH2 •CH–SH are comparable within 1 order of magnitude. The absorption vs time trace at 330 nm shows a biphasic decay where the initial 10 µs represent the equilibration process (equilibrium 12), followed by a significantly slower radical–radical reaction. During equilibration, the 330 nm species decays by ca. 50%, suggesting that K12 ca. 1. Independently, K12 can be derived from kinetic analysis of the absorption vs time traces displayed in Figure 1C, which were recorded at low doses to minimize the extent of second order reactions. Here, the build-up at 285 nm at pH 2 yields kobs = k12 + k−12 = 2.5 × 105 s−1 while the decay at 420 nm at pH 12 yields k12 ≈ 105 s−1. Therefore, k−12 = kobs – k12 ≈ 1.5 × 105 s−1, and K12 = k12/k−12 = 0.67. With an equilibrium constant K12 ca. 1, any one-electron oxidation of thiols would yield significant concentrations of α-mercaptoalkyl radicals. We note that theoretical calculations for thiyl radicals from 2-mercaptoethanol predict that equilibrium 36 should be located nearly quantitatively on the left-hand side, i.e., on the side of the thiyl radical.55 Analogous calculations for the interconversion of CH3S• and •CH2SH gave comparable results,56 and the C–H and S–H bond dissociation energies of CH3SH were determined to 387 kJ/mol57 and 379 kJ/mol,58 respectively.
| (36) |
Such a situation is clearly not consistent with our experimental observations: (i) for CyaS• we detect a clear conversion of a species absorbing at 330 nm (assigned to +H3NCH2CH2S•) into a species absorbing at 285 nm (assigned to +H3NCH2 •CH–SH). The only other possibility for a carbon-centered radical of cysteamine would be the α-amino substituted radical, H2N–•CHCH2–SH, but such radicals display structure-less absorption spectra with λmax < 250 nm.36,59 Moreover, at pH 2 the formation of an α-amino substituted radical would be disfavored through the protonation of the amino group. (ii) The 285 nm band is clearly present also when thiyl radicals are generated from Cys (Figure 3A), 3-mercaptopropionate (through reduction of 3,3′-dithiodipropionic acid; Figure 5), and from 2-mercaptoethanol (reduction of 2,2′-dithiodiethanol; Figure 6). However, the 285 nm absorbance is absent when thiyl radicals are generated from penicillamine, a β,β′-dimethyl-substituted Cys derivative.
Our experimental data on the conversion of +H3NCH2CH2S• into +H3NCH2 •CH–SH during equilibration (equilibrium 12) suggest that ε(+H3NCH2 •CH–SH)285 ≈ ε(+H3NCH2CH2S•)330 ≈ 150 M−1 cm−1. This value for +H3NCH2 •CH–SH is a factor of 24 lower than that measured for α-mercaptoalkyl radicals from thiocamphor (and a factor of ca. 20 lower than the absorptivity of α-(alkylthio)alkyl radicals obtained via deprotonation of thioether radical cations35–38). This discrepancy could indicate an experimental overestimation of K12: taking ε(α-mercaptoalkyl radical)285 = 3700 M−1 cm−1 from the reduction of thiocamphor (Figure 4), one would calculate that K12 ca. 1/24 = 0.04. However, such a value for K12 would imply at least one additional pathway for the decomposition of +H3NCH2CH2S• yielding a product without any significant absorbance between 240 and 400 nm. To our knowledge there exists no report of an α–β cleavage of aliphatic thiyl radicals analogous to fragmentations reported for alkoxyl radicals (reaction 37).
| (37) |
.
In the hypothetical case of +H3NCH2CH2S• such a fragmentation would generate thioformaldehyde and an α- amino radical (after deprotonation), which should display a pronounced absorbance at λ < 250 nm.36,59 Such an absorption is not detected at the time when equilibrium 12 is established (Figure 1A).
Calculated activation energies for 1,2-hydrogen shifts are generally high.60 In fact, the 1,2-hydrogen shift within alkoxyl radicals (reaction 38) requires solvent participation.61 Analogous solvent effects may contribute to 1,2-hydrogen transfer reactions of thiyl radicals.
| (38) |
Importantly, when neat CH3SH was photolyzed in the frozen state at 77 K only CH3S• radicals were detected by ESR spectroscopy. However, when a saturated solution of CH3SH in water was frozen and photolyzed at 77 K, ESR analysis revealed the formation of both CH3S• and •CH2SH (and •CH3).22 The formation of •CH2SH is not easily rationalized by a primary photoprocess, and, at that time, was ascribed to the reaction of a carbon-centered radical with CH3SH. However, pulse radiolysis studies have shown that carbon-centered radicals would selectively react with the mercapto group of thiols,46 and we take the formation of •CH2SH during the photolysis of CH3SH in water as additional evidence for a (potentially solvent-catalyzed) 1,2-hydrogen transfer in the primary radical CH3S•.
Ab initio calculations by Rauk and co-workers provide a value for the C3–H bond dissociation energy of Cys of 378.3 kJ/ mol.62 This value is close to the bond dissociation energy of the C2–H bond of propan-2-ol, 381 kJ/mol,58 a substrate for which intermolecular hydrogen transfer to thiyl radicals has been observed and which exhibits a rate constant of k ~ 104 M−1 s−1.7 Similarly, the rate constants for hydrogen abstraction at the γC–H and εC–H bonds of Met is 104 M−1 s−1.11 With an experimental concentration of 1 mM CyaSSCya, and under the assumption of a rate constant of also 104 M−1 s−1 for an intermolecular hydrogen transfer between CyaS• and CyaSSCya, we derive a reaction rate of 10 s− 1. This is 4 orders of magnitude slower than the measured value, kobs > 105 s−1. We conclude that we do not observe an intermolecular hydrogen transfer during our pulse radiolysis experiment.
Penicillamine: the 1,3-Hydrogen Transfer
For penicillamine thiyl radicals, PenS•, there exists the possibility of a 1,3-hydrogen transfer, followed by β-elimination of HS•/S•− (reaction 39). We obtained k14 = 8 × 104 s−1 and k14 + k−14 = 1.5 × 106 s−1, i.e., k−14 = (1.5–0.08) × 106 s−1 ≈ 1.4 × 106 s−1 and K14 = k14/k−14 ≈ 0.06. The 1,3-hydrogen transfer generates a carbon-centered radical,•Cα, consistent with the detection of carbon-centered radicals by ESR, when penicillamine was subjected to oxidation.26
| (39) |
The unimolecular decay of •Cα radicals from penicillamine on longer time scales is consistent with equilibrium 39, for which kobs = k39 + k−39[(+H3N/CO2H)C=C(CH3)2] = (3 ± 1) × 104 s−1 (Figure 2C). At the low doses (10 Gy) used for the kinetics in Figure 2C, the final concentration of (+H3N/CO2H)Cα=C(CH3)2 is on the order of [(+H3N/CO2H)Cα=C(CH3)2]final ≈ 5.7 × 10−6 M. Approximating k−39 ≤ 109 M−1 s−1 (by analogy to the addition of thiyl radicals to substituted ethylenes59), we estimate k−39[(+H3N/CO2H)Cα=C-(CH3)2]final ≤ 6000. Hence, the upper limit for k−39[(+H3N/CO2H)Cα=C(CH3)2]final is on the order of the uncertainty of our measurement, so that in a first approximation kobs = k39 + k−39 ≈ k39 = (3 ± 1) × 104 s−1. We conclude that the β-elimination from penicillamine •Cα radicals proceeds with k39 ≈ (3 ± 1) × 104 s−1, significantly slower compared to the elimination from thiyl radical adducts to styrene.63
Cysteine: 1,2- and 1,3-Hydrogen Transfer
In a first approximation, the reactions of CysS• are represented by the sum of the reactions of PenS• and CyaS•: In CyaS•, at acidic pH, the 1,3-H-transfer is inhibited by (i) the electron-withdrawing R–NH3+ group and (ii) the lack of a carboxylic acid moiety necessary for captodative stabilization, whereas in PenS•, the 1,2-H-transfer is suppressed by the substitution of the C3–H bonds by C3–CH3 groups. For CysS•, at pH 2, we expect •Cα –radical formation to be possible as for PenS•; however, CysS• does not possess the C3 methyl groups so that the 1,3-H-transfer may be conformationally less restricted. Figure 3A shows, indeed, a fast initial formation of •Cα-radicals, which subsequently disappear, coupled to the equilibrium with α-mercaptoalkyl-radicals. The rates and equilibria derived from CyaS• and PenS• appear to add up to represent the situation for CysS•: the adjustment of the 1,3-H-transfer equilibrium is 1 order of magnitude faster than that of the 1,2-H-transfer (k14 + k−14 ≈ 1.5 × 106 s−1 vs k12 + k−12 ≈ 2.5 × 105 s−1); but the corresponding equilibrium constant is 1 order of magnitude lower (k12/k−12 ≈ 0.67 vs k14/k−14 ≈ 0.06). However, calculated bond dissociation energies for Cα–H (348 kJ/mol) and S–H (370 kJ/mol) bonds of Cys64 imply that the •Cα radical should be lower in energy than the thiyl radical. For such 1,3-hydrogen transfer in a thiyl radical of N-formylcysteinamide, an enthalpy of activation of 115 kJ/mol has been calculated.62 However, earlier pulse radiolysis studies by Zhao et al.9 in alkaline solution, indirectly monitoring the formation of reducing αC• radicals, revealed a rate constant for a formal 1,3-hydrogen transfer on the order of 104 s−1, consistent with our experimental data. The C3–H bond of Cys is predicted to have a bond energy of 378.3 kJ/mol (at 298 K),62 which implies that K12 is on the order of 10−3. We are not able to resolve the discrepancy between these predictions and our experimentally derived numbers. An interesting aspect would be the pKa(S–H) of the α-mercaptoalkyl radical: in α-hydroxy-substituted carbon-centered radicals the pKa(O–H) is lowered by ca. 4–5 units compared to the native alcohols.65,66 If such pKa-shifts would operate in α-mercaptoalkyl radicals, too, they could have important effects on the stability of the respective radical species. We note that in a recent mass spectrometric experiment thiyl radicals from N-Ac–Cys were generated in the gas phase; these thiyl radicals equilibrate with •Cα radicals (ca. 70:30), via 1,3-H-shift despite a calculated activation energy of 117 kJ/mol.67 More important is the observation, that the generation of thiyl radicals in Cys-containing model peptides in D2O causes H/D exchange via the general reactions 40 and 41 on otherwise inert C–H bonds, for example at those of Cys itself.13–16
| (40) |
| (41) |
Cysteine: Estimation of the Rate Constant for β-Elimination from the •Cα Radical
In an attempt to estimate the rate constant for β-elimination of HS•/S•− from Cys •Cα radicals, we compared the decay kinetics in the cases where such radicals were produced (a) by one-electron reduction of CysSSCys under alkaline conditions (1 mM N-Ac–Cys–OMe disulfide, 100 mM t-BuOH, 10 mM KOH, Ar-saturated solution, 25 Gy) and (b) by addition of S•− radicals to Dha when Dha is in large excess (0.85 mM N-Ac–Dha–OMe, 0.18 mM Na2S, 10 mM KOH, N2O-saturated solution, 11 Gy). In case (b), if S•− is eliminated from Cys •Cα radicals, such radicals will immediately react with the large excess of Dha to reform Cys •Cα, so that the equilibrium lies on the side of the Cys •Cα radical. On the other hand, in case a, when initially no excess of Dha exists, β-elimination is practically irreversible. However, during the course of the reaction Dha accumulates and with increasing Dha concentration the result will more and more resemble case b. We chose experiments where the initial concentration of sulfur-centered radicals were comparable (8.5 µM for case a vs 6.9 µM for case b). If CysS• would mainly produce •Cα radicals, we would expect a similar initial absorbance, a behavior we do not observe experimentally (Figure 9). Instead, the initial absorbance in case a amounts only to 55% of the initial absorbance in case b. This is consistent with the rate constants derived for the 1,3-H-transfer in PenS• (k14 ≈ 8 × 104 s−1) and for the 1,2-H-transfer in CyaS• (k12 ≈ 105 s−1), suggesting that approximately equal amounts of 1,2- and 1,3-H-transfer will occur from CysS• if the reverse reaction is suppressed by the high pH, a prediction which is nicely confirmed by these experiments. In further support, the rate constant of the build-up kinetics in case (a) would represent kobs = k19 + k20 ≈ 1.6 × 105 s−1 ≈ k13 + k14. Spectrophotometrically, however, only the •Cα radicals are dominant, because their molar absorptivity is 2 orders of magnitude larger than the one of α-mercaptoalkyl radicals. For the display of the data we scaled the traces to match the initial absorbances (Figure 9). We then calculated the difference between the two traces and fitted a first-order decay to the resulting absorbance vs time profile. The derived rate constant is assigned tentatively to reaction -34, with k−34 ≈ 5 × 103 s−1. Importantly, these kinetic data are supported by product studies, where thiyl radical formation in Cys-containing model peptides led to the formation of Dha.15
Relevance to Protein Oxidation
The 1,2- and 1,3-hydrogen transfer equilibria of Cys thiyl radicals are of significance for oxidation mechanisms of proteins. Through the mechanisms delineated in this paper, a Cys thiyl radical can convert into a carbon-centered radical, which may further react with oxygen. For example, the 1,3-hydrogen transfer of CysS• yields the •Cα radical, which can add oxygen to give a peroxyl radical. The extent to which such a hydrogen transfer does occur in a protein certainly depends on secondary structure, as DFT calculations predict that αC–H bond energies are controlled by secondary structure.64 Another difference to our model studies is the fact that in proteins the N-terminal amino group and C-terminal carboxylate group of Cys will be part of amide linkages, which can affect the stability of a αC• radical. Nevertheless, our product studies have demonstrated that reversible hydrogen transfer reactions are possible for thiyl radicals of Cys residues embedded in polypeptide structures, i.e. we have demonstrated covalent H/D exchange for Cys in a series of modelf peptides and insulin.13–16
Ultimately, in a protein, peroxyl radicals at the Cα position of an amino acid induce cleavage between the respective amino acid and the amino acid in position n − 1.68 Additionally, •Cα radicals can be the source of l/d racemization even if the carbon centered radical is “repaired” by an antioxidant before peroxidation can occur. Hence, the thiyl radical can induce irreversible protein damage. More importantly, any radical and/ or oxidant which can generate a thiyl radical can induce irreversible protein damage through the ensuing hydrogentransfer reactions.69 Such mechanisms may become important especially for radicals of low reactivity, which will not react with any amino acid in a protein except Cys. In vivo, such reactions may lead to protein inactivation and accelerated protein turnover. In vitro, the oxidation of protein pharmaceuticals may lead to pharmaceuticals with lower potency, altered half-life, or even immunogenic potential.
ACKNOWLEDGMENTS
Financial support by the NIH (PO1AG12993) and the ETH Zürich is gratefully acknowledged.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Stubbe J, van der Donk A. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 2.Himo F. J. Phys. Chem. B. 2002;106:7688–7692. [Google Scholar]
- 3.Himo F, Siegbahn PEM. Chem. Rev. 2003;103:2421–2456. doi: 10.1021/cr020436s. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Gallo AA, Florián J, Liu Y, Mora S, Xu W. J. Phys. Chem. B. 2010;114:5497–5502. doi: 10.1021/jp910349q. [DOI] [PubMed] [Google Scholar]
- 5.Roberts BP. J. Chem. Soc. Perkin Trans. 1996;2:2719–2725. [Google Scholar]
- 6.Akhlaq MS, Schuchmann HP, von Sonntag C. Int. J. Radiat. Biol. 1987;51:91–102. doi: 10.1080/09553008714550531. [DOI] [PubMed] [Google Scholar]
- 7.Schöneich Ch, Asmus KD, Bonifačić M. J. Chem. Soc. Faraday Trans. 1995;91:1923–1930. [Google Scholar]
- 8.Pogocki D, Schöneich Ch. Free Radical Biol. Med. 2001;31:98–107. doi: 10.1016/s0891-5849(01)00559-7. [DOI] [PubMed] [Google Scholar]
- 9.Zhao R, Lind J, Merényi G, Eriksen TE. J. Am. Chem. Soc. 1994;116:12010–12015. [Google Scholar]
- 10.Nauser T, Schöneich Ch. J. Am. Chem. Soc. 2003;125:2042–2043. doi: 10.1021/ja0293599. [DOI] [PubMed] [Google Scholar]
- 11.Nauser T, Pelling J, Schöneich Ch. Chem. Res. Toxicol. 2004;17:1323–1328. doi: 10.1021/tx049856y. [DOI] [PubMed] [Google Scholar]
- 12.Nauser T, Casi G, Koppenol WH, Schöneich Ch. J. Phys. Chem. B. 2008;112:15034–15044. doi: 10.1021/jp805133u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mozziconacci O, Sharov V, Williams TD, Kerwin B, Schöneich Ch. J. Phys. Chem. B. 2008;112:9250–9257. doi: 10.1021/jp801753d. [DOI] [PubMed] [Google Scholar]
- 14.Mozziconacci O, Kerwin BA, Schöneich Ch. J. Phys. Chem. B. 2010;114:6751–6762. doi: 10.1021/jp101508b. [DOI] [PubMed] [Google Scholar]
- 15.Mozziconacci O, Kerwin BA, Schöneich Ch. J. Phys. Chem. B. 2011;115:12287–12305. doi: 10.1021/jp2070453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mozziconacci O, Kerwin BA, Schöneich Ch. J. Phys. Chem. B. 2010;114:3668–3688. doi: 10.1021/jp910789x. [DOI] [PubMed] [Google Scholar]
- 17.Grierson L, Hildenbrand K, Bothe E. Int. J. Radiat. Biol. 1992;62:265–277. doi: 10.1080/09553009214552111. [DOI] [PubMed] [Google Scholar]
- 18.Zhao R, Lind J, Merényi G, Eriksen TE. J. Chem. Soc. Perkin Trans. 1997;2:569–574. [Google Scholar]
- 19.Hofstetter D, Nauser T, Koppenol WH. Chem. Res. Toxicol. 2010;23:1596–1600. doi: 10.1021/tx100185k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Zhang N, Schuchmann HP, von Sonntag C. J. Phys. Chem. 1994;98:6541–6547. [Google Scholar]
- 21.Mozziconacci O, Williams TD, Kerwin BA, Schöneich Ch. J. Phys. Chem. B. 2008;112:15921–15932. doi: 10.1021/jp8066519. [DOI] [PubMed] [Google Scholar]
- 22.Volman DH, Wolstenholme J, Hadley SG. J. Phys. Chem. 1967;71:1708–1803. doi: 10.1021/j100865a037. [DOI] [PubMed] [Google Scholar]
- 23.Skelton J, Adam FC. Can. J. Chem. 1971;49:3536–3543. [Google Scholar]
- 24.Elliott AJ, Adam FC. Can. J. Chem. 1974;52:102–110. [Google Scholar]
- 25.Neta P, Fessenden RW. J. Phys. Chem. 1971;75:2277–2283. doi: 10.1021/j100676a003. [DOI] [PubMed] [Google Scholar]
- 26.Karoui H, Hogg N, Fréjaville C, Tordo P, Kalyanaraman B. J. Biol. Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 27.Becker D, Swarts S, Champagne M, Sevilla MD. Int. J. Radiat. Biol. 1988;53:767–786. doi: 10.1080/09553008814551121. [DOI] [PubMed] [Google Scholar]
- 28.Sustmann R, Korth HG. Adv. Phys. Org. Chem. 1990;26:131–178. [Google Scholar]
- 29.Von Sonntag C. The Chemical Basis of Radiation Biology. London: Taylor & Francis; 1987. [Google Scholar]
- 30.Purdie JW. J. Chem. Soc. Chem. Commun. 1971:1163–1165. [Google Scholar]
- 31.Purdie JW, Gillis HA, Klassen NV. Can. J. Chem. 1973;51:3132–3142. [Google Scholar]
- 32.Simic M, Hoffman MZ. J. Am. Chem. Soc. 1970;92:6096–6098. [Google Scholar]
- 33.Hoffman MZ, Hayon E. J. Am. Chem. Soc. 1972;94:7950–7957. [Google Scholar]
- 34.Buxton GV, Greenstock CL, Helman WP, Ross AB. J. Phys. Chem. Ref. Data. 1988;17:513–886. [Google Scholar]
- 35.Bonifačić M, Möckel H, Bahnemann D, Asmus KD. J. Chem. Soc. Perkin Trans. 1975;2:675–685. [Google Scholar]
- 36.Hiller KO, Masloch B, Göbl M, Asmus KD. J. Am. Chem. Soc. 1981;103:2734–2743. [Google Scholar]
- 37.Schöneich Ch, Aced A, Asmus KD. J. Am. Chem. Soc. 1993;115:11376–11383. [Google Scholar]
- 38.Schöneich Ch, Bobrowski K. J. Am. Chem. Soc. 1993;115:6538–6547. [Google Scholar]
- 39.Barrón LB, Waterman KC, Filipiak P, Hug GL, Nauser T, Schöneich Ch. J. Phys. Chem. A. 2004;108:2247–2255. [Google Scholar]
- 40.Hayon E, Simic M. J. Am. Chem. Soc. 1971;93:6781–6786. doi: 10.1021/ja00754a013. [DOI] [PubMed] [Google Scholar]
- 41.Rao PS, Hayon E. J. Phys. Chem. 1975;79:109–115. [Google Scholar]
- 42.Wu Z, Back TG, Ahmad R, Yamdagni R, Armstrong DA. J. Phys. Chem. 1982;86:4417–4422. [Google Scholar]
- 43.Everett SA, Schöneich Ch, Stewart JH, Asmus KD. J. Phys. Chem. 1992;96:306–314. [Google Scholar]
- 44.Mezyk SP, Madden KP. J. Phys. Chem. 1999;103:235–242. [Google Scholar]
- 45.Mezyk SP, Armstrong DA. J. Chem. Soc. Perkin Trans. 1999;2:1411–1419. [Google Scholar]
- 46.Von Sonntag C. In: Sulfur-Centered Reactive Intermediates in Chemistry and Biology. Chatgilialoglu C, Asmus K-D, editors. New York: Plenum Press; 1990. pp. 359–366. [Google Scholar]
- 47.Macrae RM, Carmichael IJ. Phys. Chem. A. 2001;105:3641–3651. [Google Scholar]
- 48.Scaiano JC, Ingold KU. J. Am. Chem. Soc. 1976;98:4727–4732. [Google Scholar]
- 49.Neta P, Simic M, Hayon E. J. Phys. Chem. 1969;73:4207–4213. [Google Scholar]
- 50.Christensen HC, Sehested K, Hart EJ. J. Phys. Chem. 1973;77:983–987. [Google Scholar]
- 51.Akhlaq MS, Murthy CP, Steenken S, von Sonntag C. J. Phys. Chem. 1989;93:4331–4334. [Google Scholar]
- 52.Wang Y, Lucia LA, Schanze KS. J. Phys. Chem. 1995;99:1961–1968. [Google Scholar]
- 53.Handbook of Chemistry and Physics. 66th ed. Boca Raton, FL: CRC Press; 1986. [Google Scholar]
- 54.Das TN, Huie RE, Neta P, Padmaja S. J. Phys. Chem. 1999;103:5221–5226. [Google Scholar]
- 55.Naumov S, von Sonntag C. J. Phys. Org. Chem. 2005;18:586–594. [Google Scholar]
- 56.Fossey J, Sorba J. J. Mol. Struct. (THEOCHEM) 1989;186:305–319. [Google Scholar]
- 57.Ruscic B, Berkowitz J. J. Chem. Phys. 1992;97:1818–1823. [Google Scholar]
- 58.McMillen DF, Golden DM. Annu. Rev. Phys. Chem. 1982;33:493–532. [Google Scholar]
- 59.Hiller KO, Asmus KD. J. Phys. Chem. 1983;87:3682–3688. [Google Scholar]
- 60.Viskolcz B, Lendvay G, Körtvælyesi T, Seres L. J. Am. Chem. Soc. 1996;118:3006–3009. [Google Scholar]
- 61.Konya KG, Paul T, Lin S, Lusztyk J, Ingold KU. J. Am. Chem. Soc. 2000;122:7518–7527. [Google Scholar]
- 62.Rauk A, Yu D, Armstrong DA. J. Am. Chem. Soc. 1998;120:8848–8855. [Google Scholar]
- 63.Ito O. Res. Chem. Intermed. 1995;21:69–93. [Google Scholar]
- 64.Rauk A, Yu D, Armstrong DA. J. Am. Chem. Soc. 1997;119:208–217. [Google Scholar]
- 65.Asmus KD, Henglein A, Wigger A, Beck G. Ber. Bunsen-Ges. Phys. Chem. 1966;70:756. [Google Scholar]
- 66.Laroff GP, Fessenden RW. J. Phys. Chem. 1973;77:1283–1288. [Google Scholar]
- 67.Osburn S, Berden G, Oomens J, O”Hair RAJ, Ryzhof V. J. Am. Soc. Mass Spectrom. 2011;22:1794–1803. doi: 10.1007/s13361-011-0198-5. [DOI] [PubMed] [Google Scholar]
- 68.Davies MJ. Biochim. Biophys. Acta. 2005;1703:93–109. doi: 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 69.Schöneich Ch. Biochem. Soc. Trans. 2011;39:1254–1259. doi: 10.1042/BST0391254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nauser T, Koppenol WH, Schöneich Ch. Unpublished results [Google Scholar]