

Abstract
The sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) plays a critical role in Ca2+ homeostasis via sequestration of this ion into the sarco/endoplasmic reticulum. The activity of this pump is inhibited by oxidants and impaired in ageing tissues and cardiovascular disease. We have shown previously that the myeloperoxidase- (MPO) derived oxidants HOCl and HOSCN target thiols and mediate cellular dysfunction. As SERCA contains Cys residues critical to ATPase activity, we hypothesized that HOCl and HOSCN might inhibit SERCA activity, via thiol oxidation, and increase cytosolic Ca2+ levels in human coronary artery endothelial cells (HCAEC). Exposure of sarcoplasmic reticulum vesicles to pre-formed or enzymatically-generated HOCl and HOSCN resulted in a concentration-dependent decrease in ATPase activity; this was also inhibited by the SERCA inhibitor thapsigargin. Decomposed HOSCN and incomplete MPO enzyme systems did not decrease activity. Loss of ATPase activity occurred concurrently with oxidation of SERCA Cys residues and protein modification. Exposure of HCAEC, with or without external Ca2+, to HOSCN or HOCl, resulted in a time- and concentration-dependent increase in intracellular Ca2+ under conditions that did not result in immediate loss of cell viability. Thapsigargin, but not inhibitors of plasma membrane or mitochondrial Ca2+ pumps/channels, completely attenuated the increase in intracellular Ca2+ consistent with a critical role for SERCA in maintaining endothelial cell Ca2+ homeostasis. Angiotensin II pre-treatment potentiated the effect of HOSCN at low concentrations. MPO-mediated modulation of intracellular Ca2+ levels may exacerbate endothelial dysfunction, a key early event in atherosclerosis, and be more marked in smokers due to their higher SCN− levels.
Keywords: Myeloperoxidase, calcium, oxidation, thiols, sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), hypochlorous acid, hypothiocyanous acid
Introduction
Considerable evidence has been presented in support of the hypothesis that the level of the heme enzyme myeloperoxidase (MPO), is an important risk factor for multiple inflammatory diseases including atherosclerosis, neurodegenerative conditions and some cancers (reviewed in [1-3]). This enzyme has also been linked with other human pathologies including arthritis, asthma, cystic fibrosis and kidney damage. Circulating levels of MPO have been shown to be a major risk factor for coronary artery disease, with a higher odds ratio than total cholesterol (as a ratio with high-density lipoprotein cholesterol; odds ratios of 20.4 and 4.2, respectively) in multivariable models after adjustment for other factors [4]. MPO levels can also predict health outcomes in people presenting with chest pain [5], in patients with acute coronary syndromes [6], and in patients that have suffered a myocardial infarction [7]. However, the MPO-derived species and mechanisms responsible for this elevated disease risk, and detrimental patient outcomes, remain unknown.
MPO is released by activated neutrophils, monocytes and tissue macrophages at sites of inflammation, including within atherosclerotic lesions. MPO catalyzes the reaction of hydrogen peroxide (H2O2) with halide and pseudohalide ions (Cl−, Br−, I− and SCN−) to form hypohalous acids (hypochlorous acid, HOCl, from Cl−; hypobromous acid, HOBr, from Br− and hypothiocyanous acid, HOSCN, from SCN−); these species are potent oxidants [1,2,8]. HOCl and HOSCN are the major oxidants formed by MPO with normal plasma levels of halides and SCN− (100 mM Cl−, ∼100 μM Br−, < 1 μM I−, ∼ 50 μM SCN−), as SCN− has a very high specificity constant for MPO [9]. The concentration of these oxidants formed at inflammatory sites has been estimated (using a blood concentration of neutrophils of ∼ 5 × 106 cells mL−1) as 250-425 μmoles of HOCl per hour [10,11].
These MPO-derived oxidants play a major role in killing bacteria and other invading pathogens [2,8,12,13], but excessive or misplaced generation can result in damage to both host cells (including the endothelium of the artery wall) and extracellular components. Enzymatically-active MPO protein has been shown to be present throughout the intima in all grades of human atherosclerotic lesions, and is present at particularly high levels in shoulder regions where lesion rupture is often detected [14]. The thickening of the intima in human lesions correlates with the extent of staining by a monoclonal antibody specific for HOCl-damaged proteins [15,16], and markedly elevated levels of the HOCl biomarkers, 3-chloro-tyrosine and 5-chloro-uracil, have been detected in human atherosclerotic lesions [17,18]. In contrast, the role of HOSCN in disease is less well established. It has been shown that the amount of oxidized low-density lipoproteins (LDL) present in fatty streaks in the aortae of young people correlates with serum SCN- levels [19]. Furthermore smokers, who have high SCN- levels, have larger numbers of lipid-laden macrophages than non-smokers [20].
Unlike HOCl, which has been shown to react with a wide range of biological targets (though primarily proteins) [21-23], HOSCN reacts with great specificity with thiols (both glutathione (GSH) and protein cysteine (Cys) residues) and selenium-containing species [24,25]. There are therefore very major differences in the reactivity of these two oxidants and they would be expected to have different biological effects; recent studies support this conclusion [26,27]. The selectivity of HOSCN for thiols can result in significant and specific damage to cellular enzymes that contain active site Cys residues [26-28], elicits significantly greater cellular dysfunction than identical concentrations of HOCl and HOBr, with this linked to protein thiol oxidation [27], and increases the extent of thiol oxidation in plasma from smokers compared to non-smokers [29].
The sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) plays a critical role in regulating calcium homeostasis by transporting cytosolic Ca2+ into the sarco/endoplasmic reticulum, thus lowering intracellular Ca2+ levels. Through this process of Ca2+ sequestration, SERCA mediates smooth, cardiac and skeletal muscle cell relaxation, and also participates in a variety of cellular functions relevant to growth, proliferation and apoptosis [30,31]. At least ten isoforms of SERCA have been detected in mammalian tissues, encoded by three distinct genes: SERCA1, 2 and 3. SERCA1 is expressed in fast-twitch skeletal muscle and is alternatively spliced to give the adult (SERCA1a) and neonatal (SERCA1b) forms. SERCA2 encodes the cardiac and slow-twitch muscle isoform, SERCA2a, as well as the ubiquitously expressed SERCA2b. SERCA3 is expressed in a variety of tissues including non-muscle cells and has 6 isoforms, designated SERCA3a-f [32].
SERCA activity can be modulated by reversible nitric oxide (NO.)-dependent S-glutathiolation of specific Cys residues [33], a process that is impaired in ageing tissues [34] and cardiovascular disease [33]. In pathophysiological states such as atherosclerosis, where reactive species are generated in excess, critical SERCA thiols can become irreversibly oxidized, leading to a loss of enzyme function [35]. Consistent with these data, peroxynitrite [36], NO. and NO.-derived species [37] and amino acid peroxides [38] have been shown to inhibit Ca2+-ATPase activity by selectively targeting SERCA Cys residues. In the light of these data, the present study has investigated whether HOCl and HOSCN, at pathophysiologically-relevant concentrations, can modify partially-purified SERCA (possibly via targeting of cysteine residues), thereby inhibiting Ca2+-ATPase activity, and whether the reactivity of these oxidants differ as might be expected. These studies have subsequently been extended to examine whether these MPO-derived oxidants can perturb Ca2+ homeostasis in human coronary artery endothelial cells (HCAEC) through a SERCA-dependent mechanism. Such modifications may generate or exacerbate endothelial dysfunction, a key early event in atherosclerosis.
Materials and Methods
Materials
Aqueous solutions and buffers were prepared using nanopure water filtered through a four-stage Milli-Q system (Millipore, North Ryde, Australia). Lactoperoxidase (LPO, from bovine milk; Calbiochem) was quantified by absorbance at 412 nm using a molar absorption coefficient of 112,000 M−1 cm−1[39]. H2O2 (30 % v/v, Merck) was quantified by absorbance at 240 nm using a molar absorption coefficient of 39.4 M−1 cm−1 [40]. ThioGlo1 was obtained from Calbiochem. Fura-2AM and 5-iodoacetamidofluoroescein (IAF) were from Molecular Probes (Life Technologies). NaOCl and tri-sodium citrate were from BDH (Poole, UK). Acetonitrile and HCl (HPLC grade) were from Merck. Tris-glycine 4-15 % gradient gels and Precision Plus Kaleidoscope standards were from Bio-Rad. Ammonium molybdate was from M & B. NaCl and PBS were from Amresco. MesoEndo Growth Medium and HCAEC were purchased from Cell Applications. Molecular mass cut-off devices were from Nanosep (Pall Life Sciences). All other chemicals were obtained from Sigma-Aldrich.
Isolation of sarcoplasmic reticulum (SR) vesicles
Native SR vesicles were isolated from 6-month old Fisher 344 × Brown Norway F1 hybrid rat hind-limb skeletal muscles (fast-twitch fibers), as previously described [38].
Incubation of SR vesicles with HOSCN and HOCl
HOSCN was prepared enzymatically using bovine milk LPO, as previously described [27,41]. Briefly, 1.5-2.0 μM LPO was incubated with 7.5 mM NaSCN and 3.75 mM H2O2 (added in 5 aliquots, 60 s apart) for 15 min in 8.5 mM potassium phosphate buffer, pH 6.6. The reaction was stopped by the addition of catalase (bovine liver, 140 IU), which was removed along with the LPO by centrifugation (5 min, 11,200 g) through 10 kDa molecular mass cut-off filters. The concentration of HOSCN was immediately determined by reaction with TNB (5-thio-2-nitrobenzoic acid; prepared by alkaline hydrolysis of DTNB with NaOH), using a molar absorption coefficient of 14,150 M−1 cm−1 at 412 nm [42]. HOSCN (0-100 μM) was incubated with 0.5 mg mL−1 SR vesicles in 50 mM MOPS buffer, pH 7.0, and 10 mM MgCl2 for 2 h at 21 °C. SR vesicles were also incubated with up to 300 μM HOSCN that had been left to decompose overnight at 21 °C. HOCl was prepared by diluting a concentrated stock solution of NaOCl in water. SR vesicles (0.5 mg mL−1) were incubated with 0-100 μM HOCl in saline-sodium citrate (SSC) buffer, pH 7.0, and 10 mM MgCl2 for 1 h at 21 °C. For the time-dependent experiments, SR vesicles were treated with 50 μM HOCl in the same manner for 1-60 min. Aliquots of control- and oxidant-treated SR were subsequently taken for analysis. Oxidant-treated SR vesicles were further incubated with 2.5 mM dithiothreitol (DTT) for 30 min at 21°C to determine whether inhibition of Ca2+-ATPase activity was reversible.
Incubation of SR vesicles with MPO-derived oxidants
SR vesicles (0.5 mg mL−1) were incubated with 100 nM MPO and 50 μM H2O2 for 1 h at 21 °C with either: 10 mM MgCl2 and 1× SSC buffer, pH 7.0 (to generate HOCl only); or 10 mM MgSO4, 50 μM NaSCN and 5 mM MOPS, pH 7.0 (to generate HOSCN only); or 10 mM MgCl2, 50 μM NaSCN and 1× SSC buffer, pH 7.0 (to generate both HOCl and HOSCN). Negative controls omitting MPO, H2O2, NaSCN and Cl− individually were also included.
Sarco/endo-plasmic reticulum Ca2+-ATPase (SERCA) activity assay
Total, Ca2+-dependent, and basal SERCA ATPase activity were determined by measurement of inorganic phosphate (Pi) in the presence of the calcium ionophore, A23187. Reactions contained 50 μg mL−1 SR in 25 mM MOPS, pH 7.0, 6 μM A23187, 5 mM MgCl2, and either 1 mM EGTA or 0.1 mM CaCl2. Five mM ATP was added to start the reaction, and Pi was measured colorimetrically as described previously [43], with 10 % (v/v) Tween-20 replacing Sterox. Phosphate standards were included to quantify the Pi content of SR samples. In order to calculate Ca2+-dependent ATPase activity, the basal activity (assays containing EGTA) was subtracted from the total activity (assays containing CaCl2). The measured activity was attributed to SERCA as the Ca2+-ATPase activity was completely inhibited by the addition of the SERCA-specific inhibitor, thapsigargin (20 μM) [44].
Quantification of thiols
Prior to thiol quantification, oxidant-treated SR samples were denatured by exposure to 10 μL 10 % (w/v) sodium dodecyl sulfate (SDS) in sodium phosphate buffer, pH 7.4, for 30 min at 37 °C. SR thiol groups were quantified by labeling Cys residues with the fluorescent dye, ThioGlo (TG1), as described previously [45]. Briefly, a stock solution of 2.6 mM TG1 was prepared in acetonitrile. Immediately prior to the assay, a 1:100 dilution of the TG1 stock was made in PBS. Fifty μL control or oxidant-treated SR vesicles were added to wells of a 96 well plate, followed by 50 μL diluted TG1. The plate was incubated for 5 min in the dark at 21 °C, with the fluorescence then measured at λex 384 nm and λem 513 nm. Thiol concentrations were calculated from a GSH standard curve.
Quantification of chloramine formation
SR vesicles (0.5 mg mL−1) were incubated with 0-100 μM HOCl in SSC buffer, pH 7.0, and 10 mM MgCl2 for 15 min at 21 °C. The formation of HOCl-derived chloramines was measured using the TNB assay as described previously [45]. Briefly, aliquots of control- and oxidant-treated SR were reacted with TNB (5-thio-2-nitrobenzoic acid; prepared by alkaline hydrolysis of DTNB with NaOH), and chloramine concentration determined by measurement of absorbance at 412 nm using a molar absorption coefficient of 14,150 M−1 cm−1 [42].
Gel electrophoresis
The electrophoretic mobility of SR proteins following treatment with 0-100 μM HOCl or HOSCN (incubated for 1 or 2 h, respectively) was determined using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions. Following exposure to oxidant (as detailed above), SR samples were mixed with Tris-glycine-SDS running buffer and loaded into Tris-glycine 4-15 % SDS-PAGE gradient gels. Gels were run at 150 V for 50 min and then stained using Coomassie Blue in isopropanol and glacial acetic acid. Electrophoresis was also carried out to examine the loss of thiol groups from HOCl- and HOSCN-treated SR in the presence of the thiol indicator, IAF, as described previously [45]. Briefly, 100 μM IAF in DMSO was added to control and oxidized SR samples and electrophoresis carried out as described above. Gels were scanned for fluorescence with λex 384 nm and λem 513 nm, followed by Coomassie Blue staining to confirm molecular mass.
Cell Culture
Commercial stocks of HCAEC were thawed and transferred into T-75 flasks containing 10 mL MesoEndo Cell Growth Medium and incubated overnight at 37 °C in a 5 % O2/95 % CO2 incubator. Medium was replaced following overnight culture, and every second day thereafter. HCAEC reached 90 % confluency within approximately 7 days, at which time HCAEC were subcultured. Existing medium was removed and HCAEC washed twice with PBS before application of Trypsin/EDTA. HCAEC were incubated for 2-5 min at 37 °C in a 5 % O2/95 % CO2 incubator before the addition of 10 mL medium to inhibit tryptic activity. HCAEC were then centrifuged at 525 g for 5 min, supernatant removed and pellet re-suspended in 3-4 mL medium. One mL re-suspended HCAEC was transferred to T-75 flasks containing 9 mL medium. Fura-2AM studies were performed on HCAEC 0-1 days following subculture.
Measurement of intracellular calcium
Intracellular calcium was monitored using the fluorescent indicator Fura-2AM (1 μM, λex 340/380 nm, λem 510 nm) in a HEPES-buffered solution (HBS) containing (in mM): KCl 5.33, MgSO4 0.41, NaCl 139, Na2HPO4 5.63, glucose 5, HEPES 20, glutamine 2, and Ca(NO3)2 2.5 (adjusted to pH 7.4 with NaOH), at 37 °C as described previously [46]. Fluorescence was measured using a Hamamatsu Orca ER digital camera attached to an inverted Nikon TE2000-U microscope. Metamorph 6.3 was used to quantify the signal by manually tracing cells. An equivalent region not containing cells was used as background and was subtracted. Ratiometric 340/380 nm fluorescence was plotted relative to the pre-treatment fluorescence assigned a value of 1.0. Fluorescent ratios recorded over 3 min were averaged 7 min following addition of either HOCl alone, HOCl in the presence of 10 mM NaSCN (thus generating HOSCN) [47], or 10 mM NaSCN alone, and reported as a percentage from the baseline pre-treatment average. In some experiments cells were pre-exposed to Ang II (1 nM) for 10 min and changes in Fura 2 fluorescence were recorded. NaSCN (10 mM) was then added, followed by increasing concentrations of HOCl as described above. Cell death was assessed after addition of propidium iodide at the end of each experiment.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism 5 for Windows (GraphPad Software, La Jolla, USA), with P < 0.05 considered significant. Details of specific tests performed for each experiment are given in the Figure legends.
Results
HOSCN and HOCl inhibit SERCA Ca2+-ATPase activity
Fig. 1a shows SERCA Ca2+-ATPase activity following incubation of rat skeletal muscle SR vesicles with 0-100 μM HOSCN for 2 h at 21 °C and pH 7.0, with and without subsequent incubation with 2.5 mM DTT for 30 min. Freshly prepared HOSCN significantly inhibited SERCA activity at all concentrations tested. Decomposed HOSCN at concentrations of up to 300 μM had no effect on SERCA activity (data not shown). Subsequent incubation of HOSCN-treated SR with 2.5 mM DTT for 30 min at 21 °C resulted in higher levels of SERCA activity compared to the non-DTT treated samples at 10 μM HOSCN, indicating that the oxidant-induced loss in SERCA activity was partially reversible.
Fig. 1.
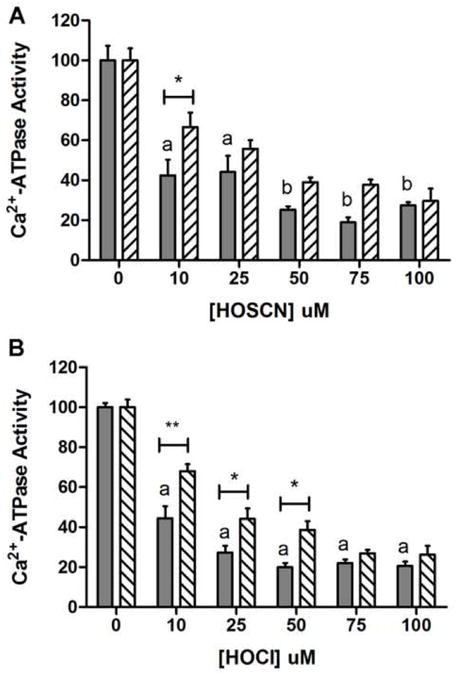
HOSCN and HOCl inhibit the Ca2+-ATPase activity in SR vesicles. (a) Ca2+-ATPase activity, attributed to SERCA, was measured after exposure to 0-100 μM freshly prepared HOSCN for 2 h (solid-filled bars), with subsequent exposure to 2.5 mM dithiothreitol (DTT) for 30 min (hatched bars) at 21 °C and pH 7.0. (b) As (a) except treatment with 0-100 μM HOCl for 1 h (solid-filled bars), and subsequent exposure to DTT (hatched bars). For both (a) and (b), data are expressed as mean + SEM of SERCA activity compared to native SR vesicles (0 μM HOCl). Letters depict statistically significant differences in activity between oxidant-treated and control vesicles by one-way ANOVA with Dunnett's post-hoc tests (a, P < 0.01; b, P < 0.001). Two-way ANOVA was carried out with Bonferroni post-hoc tests to compare data at each oxidant concentration; asterisks depict statistically significant differences between the absence and presence of DTT treatment.
Analogous experiments with HOCl also resulted in a reduction in SERCA activity at all concentrations tested (Fig. 1b). SERCA inactivation was also assessed using 50 μM HOCl over a time course between 1 and 60 min. A significant reduction in SERCA activity was observed at the first time point examined (1 min), and at all subsequent time points (data not shown), indicating that HOCl rapidly inactivates this enzyme. Subsequent incubation of the HOCl-treated SERCA with 2.5 mM DTT for 30 min at 21 °C before assessment of activity indicated that SERCA activity was partially recovered with DTT treatment, when concentrations of up to 50 μM HOCl were employed.
These data suggest that treatment of SERCA with low concentrations of oxidants (10 μM HOSCN, up to 50 μM HOCl) results in a partially reversible loss of enzyme activity, with this ascribed to the formation of reversible oxidation products, whereas high oxidant concentrations resulted in irreversible modification of the protein.
Enzymatic MPO systems inhibit SERCA Ca2+-ATPase activity
The effect of oxidants generated by enzymatic MPO/H2O2/halide/pseudohalide ions systems on SERCA activity was subsequently investigated (Fig. 2). Incubation of SR vesicles with MPO, H2O2, and either SCN− or Cl− individually, or both SCN− and Cl−, caused a significant inhibition of Ca2+-ATPase activity compared to native SERCA. Omission of individual components (i.e. MPO, H2O2 or SCN−/Cl−) from the complete reaction mixture did not result in any effect on SERCA activity.
Fig. 2.
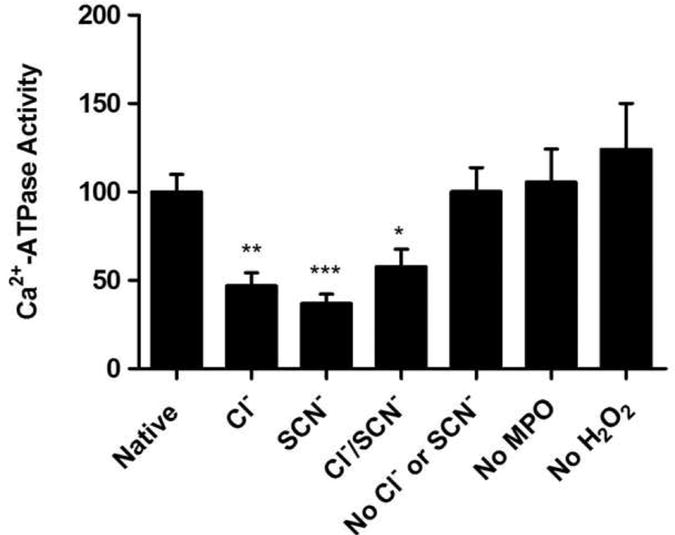
Oxidants generated by MPO inhibit SR Ca2+-ATPase activity. SR Ca2+-ATPase activity as measured after incubation with complete or incomplete MPO systems for 1 h at 21 °C and pH 7.0. The complete MPO system consisted of: 0.5 mg mL−1 SR, 100 nM MPO and 50 μM H2O2 plus either: 10 mM MgCl2 and 1× SSC buffer, pH 7.0 (to generate HOCl only); or 10 mM MgSO4, 50 μM NaSCN and 5 mM MOPS, pH 7.0 (to generate HOSCN only); or 10 mM MgCl2, 50 μM NaSCN and 1× SSC buffer, pH 7.0 (to generate both HOCl and HOSCN). Control samples containing no MPO, no H2O2, no NaSCN and no Cl− individually, were also examined. Data are expressed as mean + SEM of SERCA activity compared to native SR vesicles, with statistical analysis carried out using one-way ANOVA with Dunnett's post-hoc tests.
HOCl and HOSCN alter the electrophoretic mobility of SERCA
SDS-PAGE separation of native and oxidant-treated SERCA preparations was carried out using 4-15 % gradient gels with the protein bands visualized by Coomassie Blue staining. Following exposure of SR to increasing concentrations of HOSCN, a concentration-dependent loss of the band assigned to SERCA at ∼ 100 kDa [48] was observed, with a concomitant increase in the presence of high-molecular-mass aggregates in the wells of the gel (Fig. 3a). With HOCl, increasing concentrations of oxidant resulted in a partial loss of the protein band assigned to SERCA. In addition, high-molecular-mass aggregates were detected in the wells of the gel, but to a lesser extent than observed with HOSCN (Fig. 3b).
Fig. 3.
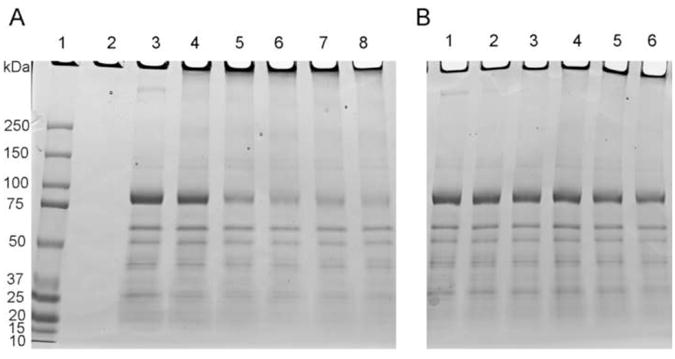
Exposure of SR vesicles to HOSCN and HOCl alters the electrophoretic mobility of SERCA protein as assessed by SDS-PAGE separation of native and oxidant-treated SR vesicles with subsequent Coomassie Blue staining. (a) Treatment of SR with 0-100 μM freshly prepared HOSCN resulted in a concentration-dependent loss of the SERCA band, and an increase in high molecular mass aggregates in the wells of the gel. Lane 1: Molecular mass markers; lane 2: negative control; lane 3: Native SR (0 μM HOSCN); Lanes 4-8: SR treated with 10, 25, 50, 75 and 100 μM HOSCN, respectively. (b) As (a) except with HOCl. Lane 1: native SR (0 μM HOCl); lanes 2-6: SR treated with 10, 25, 50, 75 and 100 μM HOCl, respectively.
SERCA thiol groups are oxidized on treatment with HOSCN and HOCl
The potential loss of Cys residues in SR vesicles on oxidant exposure was examined using the ThioGlo assay. Incubation of SR with 0-100 μM HOSCN for 2 h at 21 °C and pH 7.0 followed by SDS denaturation for 30 min at 37 °C resulted in a significant loss of thiol groups for ≥ 25 μM HOSCN, when compared to control samples (Fig. 4a). Similarly, exposure of SR to 0-100 μM HOCl for 1 h at 21 °C and pH 7.0, with subsequent SDS denaturation for 30 min at 37 °C, produced significant SR thiol loss at concentrations of 25 μM HOCl and above (Fig. 4b). For HOSCN, the loss of SR thiol groups occurred in a concentration-dependent manner similar to the observed inhibition of Ca2+-ATPase activity, suggesting HOSCN is targeting the key SERCA thiol groups critical for enzyme activity. In contrast, the loss of free thiol groups in HOCl-treated SR vesicles was preceded by significant inhibition of SERCA activity.
Fig. 4.
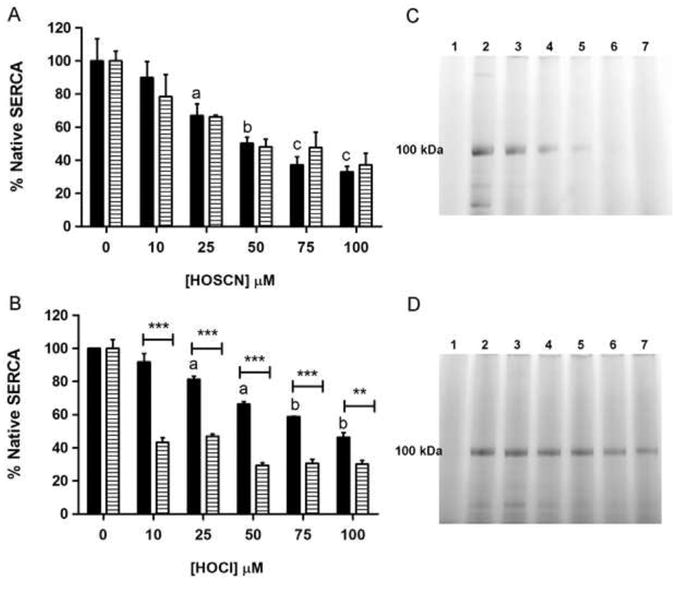
Exposure of SR preparations to HOSCN and HOCl results in oxidation of thiol groups as assessed using the ThioGlo assay. (a) SR were treated with 0-100 μM HOSCN for 2 h at 21 °C and pH 7.0, followed by SDS denaturation for 30 min at 37 °C. Black bars indicate concentration of free thiol groups, and hatched bars the corresponding Ca2+-ATPase activity. (b) As (a) except with 0-100 μM HOCl for 1 h. Bars depict mean + SEM of thiol groups or SERCA activity. In both (a) and (b) oxidant-induced thiol loss relative control SR was assessed by one-way ANOVA with Dunnett's post-hoc tests, with letters depicting statistically significant difference versus the untreated control (a, P < 0.05; b, P < 0.01; c, P < 0.001). Two-way ANOVA with Bonferroni post-hoc tests was used to compare thiol loss and SERCA activity data at each oxidant concentration: asterisks denote statistically-significant differences between these two parameters at the P < 0.05 level. (c) and (d), SDS-PAGE separation of native and oxidant-treated SR labeled with the fluorescent thiol tag, IAF; (c) treatment with HOSCN, (d) treatment with HOCl. For both gels: lane 1 is a negative control; lane 2, native SR (0 μM oxidant); lanes 3-7: SR treated with 10, 25, 50, 75 and 100 μM oxidant, respectively. The band at ∼ 100 kDa is assigned to SERCA.
To determine whether Cys residues present on SERCA were being modified by HOSCN and HOCl (as opposed to those on other proteins which might be present in the partially-purified preparations), control and oxidized SERCA preparations were treated, post-oxidant exposure, with the fluorescent thiol-derivatization agent, IAF (100 μM), and separated by SDS-PAGE (see Materials and Methods). Incubation with 0-100 μM HOSCN for 2 h resulted in a loss of the IAF-reactive groups present on the protein band assigned to SERCA, indicating that HOSCN oxidizes SERCA Cys residues in a concentration-dependent manner. Almost complete loss of the Cys residues was detected with HOSCN concentrations ≥ 50 μM (Fig. 4c). Similar experiments with SR and 0-100 μM HOCl for 1 h resulted in a partial loss of SERCA thiol groups (Fig. 4d), which correlated with the loss of SR thiols quantified by the ThioGlo assay. These results support the hypothesis that SERCA Cys residues are oxidized by MPO-derived oxidants, with this occurring with greater selectivity with HOSCN compared to HOCl.
HOCl-mediated formation of chloramines on SR vesicles
The potential oxidation of other targets on SR vesicles by HOCl was examined by assay of chloramine (RNHCl formed by reaction of HOCl with amine groups) formation on SR vesicles, by use of the TNB assay, following exposure to 0-100 μM HOCl. A dose-dependent increase in chloramines was detected upon treatment of SR vesicles with HOCl, which was statistically significant at 75 μM or higher HOCl concentrations (Fig. 5). At 100 μM HOCl, ∼13 μM chloramines were detected, suggesting that a significant proportion of the HOCl is reacting with targets apart from Cys residues on the SR proteins. These data are likely to underestimate attack by HOCl at non-Cys targets as reaction with methionine (Met) and cystine residues cannot be quantified using this method, despite these being highly reactive targets for this oxidant [22]. HOSCN does not react at a significant rate with these other amino acids, and does not generate chloramines [24].
Fig. 5.

Quantification of chloramine formation on SR vesicle proteins on exposure to HOCl. SR vesicles were exposed to 0-100 μM HOCl for 15 min at 21 °C and pH 7.0, and chloramines subsequently quantified by reaction with TNB at 412 nm. Bars depict mean + SEM of chloramine concentration, with statistical analysis carried out using one-way ANOVA with Dunnett's post-hoc tests compared to native SR.
Exposure of HCAEC to HOCl and HOSCN increases intracellular Ca2+ in human coronary artery endothelial cells (HCAEC)
In the light of the above data that indicate that the MPO enzymatic system and preformed oxidants can oxidize Cys residues and attenuate SERCA activity, subsequent experiments were carried out to determine whether similar oxidant exposure increased intracellular Ca2+ levels in HCAEC as a result of the potential inhibition of SERCA activity, a key Ca2+ sequestrating system in such cells.
Changes in Fura-2AM 340/380 nm ratiometric fluorescence in HCAEC, assigned to an increased level of cytosolic Ca2+, were detected on exposure of the cells to increasing concentrations of HOCl. Treatment with HOCl alone resulted in a concentration-dependent increase in Fura-2AM fluorescence in the absence of cell death, as assessed by the subsequent addition of propidium iodide (Fig. 6a). Similar results were recorded in the absence of extracellular Ca2+, suggesting the source of the higher cytosolic Ca2+ concentrations was intracellular (Fig. 6b). To further explore this, HCAEC were exposed to 4 μM nisoldipine, a blocker of L-type Ca2+ channels, or 4 μM Ru360, an inhibitor of mitochondrial calcium uniporter. Neither nisoldipine nor Ru360 attenuated the increase in Fura-2AM fluorescence after exposure to HOCl (Fig. 7a,b). However, exposure of HCAEC to 3 μM thapsigargin, an agent that blocks Ca2+ re-uptake into internal stores (including via SERCA), significantly attenuated the increase in Fura-2AM fluorescence in response to HOCl (Fig. 7c). Similar results were recorded in the absence of extracellular Ca2+ (Fig. 7d).
Fig. 6.
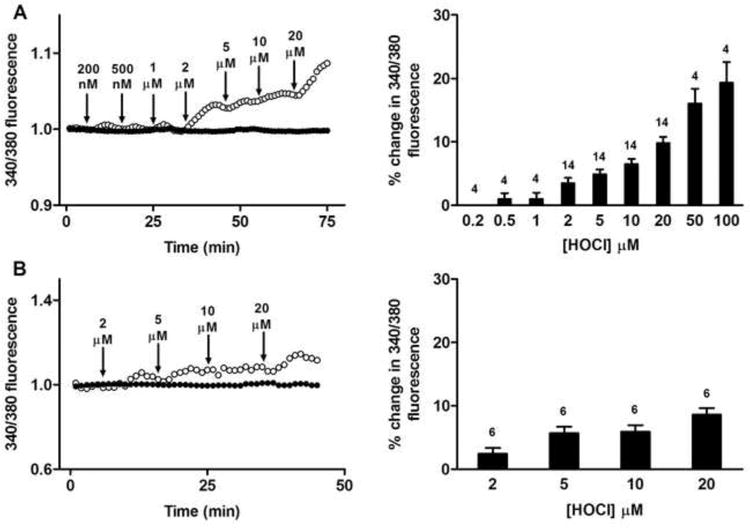
Exposure of human coronary artery endothelial cells (HCAEC) to HOCl increases intracellular calcium levels as detected by Fura-2AM fluorescence. (A) Left panel: Fura-2AM 340/380 nm fluorescence (340/380 fluorescence) recorded from HCAEC before and after exposure to increasing concentrations of HOCl (open circles), and untreated (control) HCAEC (filled circles). Arrows indicate sequential HOCl additions at the indicated concentrations. Right panel: quantification of the changes from multiple cells with increasing HOCl concentrations with data expressed as mean + SEM of % changes in 340/380 fluorescence. Numbers above bars represent n (cell number). (B) As (A), except with cells present in Ca2+-free media.
Fig. 7.
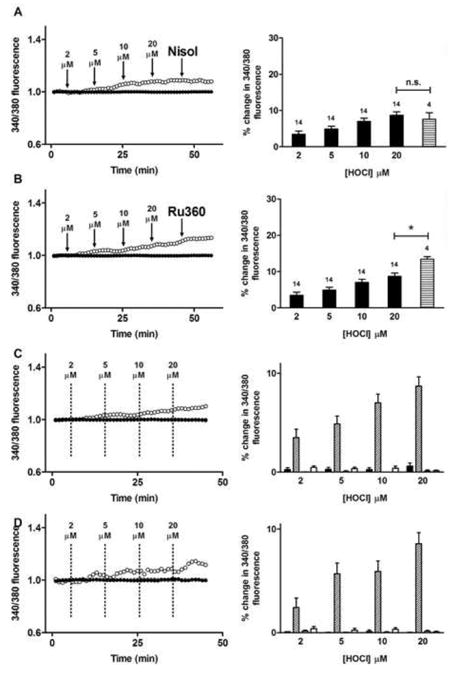
Internal calcium stores contribute to the increase in intracellular calcium observed in HCAEC after exposure to HOCl. (A) Left panel: Fura-2AM 340/380 nm fluorescence recorded from HCAEC before and after exposure to increasing concentrations of HOCl (open circles) followed by 4 μM nisoldipine (Nisol) as indicated, and untreated (control) HCAEC (filled circles). Arrows indicate time points of sequential HOCl additions at the stated concentrations and addition of Nisol. Right panel: quantification of the changes from multiple cells with increasing HOCl concentrations (black bars) and nisoldipine (horizontal striped bar). Numbers above bars represent n (cell number). (B) As (A) except with HOCl and 4 μM Ru360 (horizontal striped bar); * P < 0.05. (C) Fura-2AM 340/380 nm fluorescence recorded from HCAEC before and after exposure to increasing concentrations of HOCl (open circles), increasing concentrations of HOCl after pretreatment with 3 μM thapsigargin for 10 min (cross symbols), and untreated (control) HCAEC (filled circles). Dashed lines indicate time points of sequential HOCl additions at the stated concentrations. Right panel: quantification of changes with increasing concentrations of HOCl. Black bars, untreated cells (n = 8); diagonally hatched bars, HOCl treated cells (n = 14); grey bars, thapsigargin-treated cells in absence of HOCl (n = 4); white bars, thapsigargin-treated cells in presence of HOCl (n = 12). (D) As panel (C), except cells were present in media that did not contain Ca2+. Black bars, untreated cells (n = 6); diagonally hatched bars, HOCl-treated cells (n = 6); grey bars, thapsigargin-treated cells in absence of HOCl (n = 8); white bars, thapsigargin-treated cells in presence of HOCl (n = 16). Data in right panels are expressed as mean + SEM of % changes in 340/380 fluorescence.
The effect of HOSCN on intracellular Ca2+ was also examined. Treatment of HCAEC with HOSCN resulted in a concentration-dependent increase in Fura-2AM fluorescence in the absence of cell death (Fig. 8a). Exposure to nisoldipine or Ru360 did not alter the increase in Fura-2AM fluorescence (Fig. 8b and c), however, thapsigargin significantly attenuated the increase in Fura-2AM fluorescence in response to HOSCN (Fig. 8d). Pre-treatment of the HCAEC with Ang II (1 nM), which activates the NADPH oxidase complex of endothelial cells as well as multiple other pathways (e.g. [49]), significantly enhanced the increase in intracellular Ca2+ induced by low levels of HOSCN (2 μM), but this stimulatory action was lost at higher doses of HOSCN (Fig. 9).
Fig. 8.
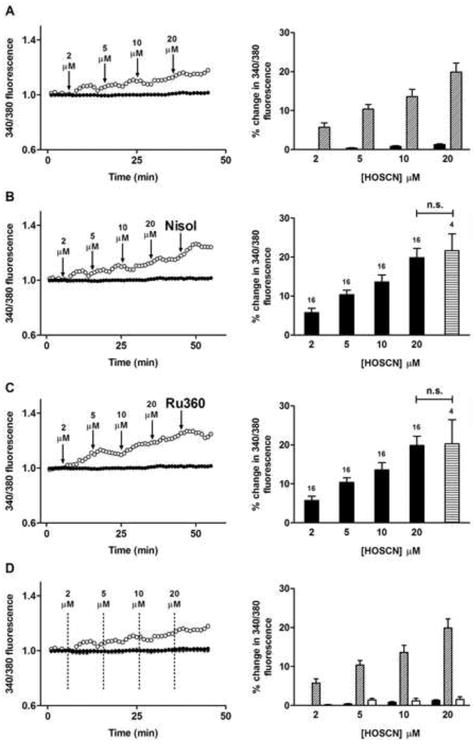
Exposure of HCAEC to HOSCN increases intracellular calcium. (A) Left panel: Fura-2AM 340/380 nm fluorescence (340/380 fluorescence) recorded from HCAEC before and after exposure to increasing concentrations of HOCl in the presence of 10 mM NaSCN to generate HOSCN (open circles), and HCAEC exposed to 10 mM NaSCN only (filled circles). Arrows indicate time points of sequential HOCl treatments at the stated concentrations. Right panel: quantification of the changes from multiple cells exposed to increasing concentrations of HOSCN. Black bars, cells exposed to 10 mM NaSCN only (n = 16); white bars, cells exposed to HOSCN generated from HOCl in the presence of 10 mM NaSCN (n = 16). (B) Left panel: as (A) except with 4 μM nisoldipine (Nisol) in the presence of 10 mM NaSCN, with arrows indicating time points of HOSCN treatment and Nisol addition. Right panel: quantification of the changes from multiple cells exposed to increasing concentrations of HOSCN and nisoldopine (horizontal striped bar). Numbers above bars represent n (cell number). (C) As (A) and (B), except with 4 μM Ru360 (horizontal striped bar). (D) Fura-2AM 340/380 nm fluorescence recorded from HCAEC before and after exposure to increasing concentrations of HOSCN (open circles, generated from 10 mM NaSCN plus HOCl), increasing concentrations of HOSCN (cross symbols) after pretreatment with 3 μM thapsigargin for 10 min, and HCAEC treated with 10 mM NaSCN alone (filled circles). Dashed lines indicate time points of sequential HOSCN additions at the stated concentrations. Right panel: quantification of changes with increasing concentrations of HOSCN. Black bars, cells exposed to 10 mM NaSCN alone (n = 16); diagonally hatched bars, cells exposed to HOSCN generated from HOCl in the presence of 10 mM NaSCN (n = 16); white bars, cells pre-treated with 3 μM thapsigargin for 10 min exposed to HOSCN generated from HOCl in the presence of 10 mM NaSCN (n = 12). Data in all right panels are expressed as mean + SEM of % changes in 340/380 fluorescence.
Fig. 9.
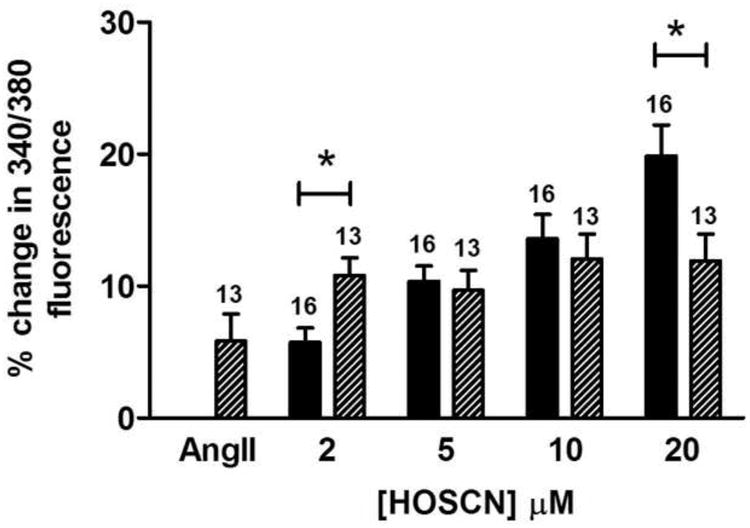
Effect of Ang II on HOSCN mediated changes in intracellular calcium. Fura 2 fluorescence was recorded in cells before and after exposure to Ang II (1 nM) for 10 min to determine baseline changes induced by Ang II. HCAECs were then treated with increasing concentrations of HOSCN. Dashed bars are treatment with Ang II alone (bar labeled AngII) or Ang II plus the stated concentration of HOSCN. Black bars are control cells (no Ang II) treated with the stated levels of HOSCN. The number of experiments are indicated above each bar. * indicates P < 0.05 for the Ang II pre-treatment with HOSCN, versus HOSCN alone.
Discussion
Substantial evidence links elevated levels of MPO, and the oxidants that it generates, with multiple inflammatory diseases including atherosclerosis, neurodegenerative conditions, arthritis, asthma, cystic fibrosis, kidney damage and some cancers (reviewed in [1-3]). This association is believed to arise from the MPO-mediated generation of hypochlorous acid (HOCl, from Cl−); hypobromous acid (HOBr, from Br−) and hypothiocyanous acid (HOSCN, from SCN−) by H2O2, and subsequent reaction with cellular targets. The proportion of each of these species is determined, at least in part, by the relative concentrations of the parent halide/pseudohalide ions in biological fluids, with the yield of HOSCN higher in subjects who smoke, or consume certain foodstuffs [29].
Previous studies have indicated that both HOCl and HOSCN rapidly oxidize thiol groups present on both low-molecular-mass molecules (e.g. the key intracellular antioxidant and co-factor, GSH) and on proteins (Cys residues), and that damage to proteins predominates over other targets in most situations [23]. Whilst HOSCN has been shown to react in a selective manner with thiol (RSH) [24,25,50] and selenol (RSeH) groups [25], HOCl reacts rapidly with multiple other residues including disulphides (cystine), methionine (Met) and amine-containing (lysine and histidine) residues [21,22].
Maintenance of intracellular Ca2+ levels is critical to cell function and survival, with failure to maintain Ca2+ concentrations within particular limits associated with aberrant cell signaling, cellular dysfunction and apoptosis/necrosis [51,52]. SERCA plays a critical role in regulating Ca2+ homeostasis, by sequestering cytosolic Ca2+ released from cellular organelles (e.g. mitochondria or the endoplasmic reticulum), or entering the cell from external pools. This enzyme pumps Ca2+ into the sarco/endoplasmic reticulum at the expense of ATP, thus lowering intracellular Ca2+ levels. Through this process of Ca2+ sequestration, SERCA mediates smooth, cardiac and skeletal muscle cell relaxation, and also participates in a variety of cellular functions relevant to growth, proliferation and apoptosis [30,31]. Previous studies have shown that SERCA activity can be modulated by the reversible oxidation of specific Cys residues (e.g. NO. -dependent S-glutathionylation) [33]; this is impaired in ageing tissues [34] and cardiovascular disease [33]. When reactive species are generated at higher levels, SERCA Cys residues can become irreversibly oxidized, with a consequent loss of function [35]. Putative biological oxidants such as peroxynitrite [36], NO.-derived species [37] and amino acid peroxides [38] have been shown to induce irreversible inactivation by selective targeting of critical Cys residues on SERCA.
In the current study, both pre-formed HOCl and HOSCN (at pathophysiologically-relevant concentrations) and an active MPO enzyme system have been shown to inhibit the Ca2+-ATPase activity of semi-purified SERCA. This loss of activity could be partially restored by subsequent incubation of the oxidant-treated samples with DTT, consistent with reversible oxidation of at least some of the affected targets. Significant loss of activity occurred even at relatively low (10 μM) concentrations of HOCl and HOSCN, while decomposed HOSCN did not induce any significant loss of activity. Furthermore, a fast reaction rate of HOCl was observed, with no time-dependent changes in activity detected after 60 s of incubation with the oxidant. The rapid inhibition of activity with HOCl is consistent with previous rate constant data for the reactions of this species [21,22]. Examination of analogous samples using SDS-PAGE resulted in a marked loss in staining of the protein band assigned to SERCA with increasing HOSCN concentrations, but much less marked loss in the case of HOCl. In the former case, the loss of parent protein was associated with an increased level of high-molecular-mass materials that remained in the protein wells; this is consistent with the presence of very large oxidant-induced aggregates. No additional discrete bands were detected with either oxidant, suggesting that fragmentation or specific dimer formation is not occurring to a significant extent. The less dramatic changes observed with HOCl are consistent with reaction of this oxidant at other sites (cf. the chloramine data) as well as with critical Cys residues; this conclusion is supported by the less pronounced loss of Cys residues detected in the thiol tagging experiments using IAF (cf. Fig. 4).
The effect of HOCl on SERCA activity has been investigated previously and it has been concluded that this oxidant can modulate SERCA activity [53-56]. However, the buffers used in these studies (e.g. HEPES) contained amine groups, which react readily with HOCl to form chloramines [21], particularly at the high concentrations of HOCl used (up to 3 mM) relative to the SERCA protein concentration (0.1 − 1 mg mL−1). The presence of this alternative target for the oxidant, and potential secondary reactions of the chloramines (which retain some of the oxidizing power of HOCl [57]) complicates the analysis of this previous data and may account for the discrepancies between these sets of data. In the present study, saline-sodium citrate buffer, pH 7.0, which does not contain any amine groups, was employed to avoid such confounding factors, thereby enabling the effects of HOCl per se to be examined.
Inhibition of SERCA activity was also detected with a complete MPO/H2O2/halide/pseudohalide system. SERCA activity was impaired by 50% with the MPO/H2O2/Cl− system relative to untreated samples, and 65% with a MPO/H2O2/SCN− system. The presence of both Cl− and SCN− in the enzyme system also resulted in marked activity loss. In contrast, the absence of these ions (i.e. treatment with MPO/H2O2 alone) did not result in a significant loss of enzyme activity, eliminating MPO peroxidase activity (i.e. one-electron oxidation reactions; see [2]) as a source of this damage.
The potential role of oxidation of thiol groups (i.e. Cys residues) on SERCA in the observed loss of activity was examined by quantification of total reduced thiols in the preparations, and by specific IAF labeling of reduced thiols on the protein band ascribed to SERCA on protein gels. As a result of oxidant exposure, the total levels of thiols in the preparations (which may include some contributions from other proteins also present in the isolated SR, evident as weak additional protein bands in Fig. 3), decreased in a concentration-dependent manner. The extent of thiol loss at low oxidant concentrations was more marked with HOSCN than HOCl; this is in accord with recent studies on the selectivity of HOSCN for thiols [24,29], and the reaction of HOCl at other sites, as discussed above.
The present study subsequently examined whether these MPO-derived oxidants can perturb Ca2+ homeostasis in HCAEC through a SERCA-dependent mechanism. Such modifications may generate or exacerbate endothelial dysfunction, a key early event in atherosclerosis. Exposure of isolated cells, with or without external Ca2+, to either HOSCN or HOCl resulted in concentration-dependent increase in intracellular Ca2+ levels under conditions that did not result in an immediate loss of cell viability. The magnitude of the elevation in Ca2+ levels was dependent on the oxidant employed, with HOSCN (generated by reaction of HOCl with excess SCN− [47]) giving rise to a greater increase than the corresponding concentration of HOCl. SCN− alone had no effect on intracellular Ca2+ levels.
Preliminary studies employing acute Ang II pre-treatment, which would be expected to enhance NADPH oxidase activity and thereby increase O2−• and H2O2 levels [49], enhanced the rise in intracellular Ca2+ induced by low (but not high) levels of HOSCN. This additional rise is tentatively ascribed to the additional effects of O2−• and H2O2 on intracellular targets; whether this results in additional SERCA inhibition (e.g. induced by H2O2) or via effects on other processes (e.g. kinase and phosphatase activity, effects on cell signaling) remains to be determined.
The absence of extracellular Ca2+ did not result in any significant differences in the nature or the extent of cytosolic Ca2+ accumulation, indicating that an influx of extracellular Ca2+ is not responsible for the observed effects. Similarly, the presence of either nisoldipine (a blocker of L-type Ca2+ channels) or Ru360 (an inhibitor of mitochondrial calcium uniporter) had no significant effect on the intracellular Ca2+ increases, consistent with neither influx of Ca2+ via L-type channels, or Ca2+ release from mitochondria being major pathways. In contrast, thapsigargin, a non-competitive inhibitor of SERCA, completely attenuated the Ca2+ accumulation induced by both HOCl and HOSCN. As thapsigargin is a re-uptake inhibitor of cytosolic Ca2+ in the sarco/endoplasmic reticulum, rather than a release inhibitor, these data suggest that Ca2+ release from one or more (unidentified) intracellular stores must be occurring, and that the effect of the oxidants is on the removal of accumulated cytosolic Ca2+; this is consistent with the loss of SERCA activity seen in the experiments with semi-purified SERCA. The source of the cytosolic Ca2+ has not been identified, and is the subject of on-going studies, though it should be noted that previous studies have provided evidence for oxidant-mediated (H2O2 and HOCl) release of Ca2+ from internal stores [53,54].
Previous studies have demonstrated that the accumulation of cytosolic Ca2+ has multiple downstream consequences, including the perturbation of critical cellular signaling pathways, modulation of the activity of key enzymes, and when the extent of Ca2+ accumulation is large, the induction of apoptosis [51,52]. It has also been reported that exposure of cells to HOSCN and HOCl can induce both apoptosis and necrosis, and that apoptosis is a key process with HOSCN, at least with some cell types [27]. The data reported here may provide a rationale for these observations, though this may be cell-type dependent [58]. Furthermore, as many enzymes are Ca2+-dependent, the perturbations in Ca2+ levels induced by these oxidants may contribute to wider cellular dysfunction, a key and early process in many inflammatory diseases, including atherosclerosis [59].
Acknowledgments
HMV is recipient of a Biomedical Postgraduate Research Scholarship from NHMRC and National Heart Foundation of Australia. LCH is Australian Research Council Future Fellow and Honorary NHMRC Senior Research Fellow. Financial support of this work by the Australian Research Council (through the Centre of Excellence and Discovery programs, CE0561607 and DP0988311), the National Heart Foundation and the National Institutes of Health (P01AG12993) (VS, CS) is gratefully acknowledged.
Abbreviations
- Ang II
angiotensin II
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- DTT
dithiothreitol
- GSH
glutathione
- HCAEC
human coronary artery endothelial cells
- HOCl
the mixture of hypochlorous acid and its anion present at pH 7.4
- HOSCN
the mixture of hypothiocyanous acid and its anion present at pH 7.4
- IAF
5-iodoacetamidofluoroescein
- MPO
myeloperoxidase
- PBS
phosphate-buffered saline
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- SCN−
thiocyanate ions
- SR
sarco/endoplasmic reticulum
- SSC buffer
saline-sodium citrate buffer
- TNB
5-thio-2-nitrobenzoic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies MJ. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J Clin Biochem Nutr. 2011;48:8–19. doi: 10.3164/jcbn.11-006FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies MJ, Hawkins CL, Pattison DI, Rees MD. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 3.van der Veen BS, de Winther MP, Heeringa P. Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal. 2009;11:2899–2937. doi: 10.1089/ars.2009.2538. [DOI] [PubMed] [Google Scholar]
- 4.Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 5.Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, Hazen SL. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 6.Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, Simoons ML, Hamm CW. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 7.Mocatta TJ, Pilbrow AP, Cameron VA, Senthilmohan R, Frampton CM, Richards AM, Winterbourn CC. Plasma concentrations of myeloperoxidase predict mortality after myocardial infarction. J Am Coll Cardiol. 2007;49:1993–2000. doi: 10.1016/j.jacc.2007.02.040. [DOI] [PubMed] [Google Scholar]
- 8.Kettle AJ, Winterbourn CC. Myeloperoxidase: a key regulator of neutrophil oxidant production. Redox Rep. 1997;3:3–15. doi: 10.1080/13510002.1997.11747085. [DOI] [PubMed] [Google Scholar]
- 9.Van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kettle AJ, Winterbourn CC. Assays for the chlorination activity of myeloperoxidase. Meth Enzymol. 1994;233:502–512. doi: 10.1016/s0076-6879(94)33056-5. [DOI] [PubMed] [Google Scholar]
- 11.Weiss SJ, Klein R, Slivka A, Wei M. Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. J Clin Invest. 1982;70:598–607. doi: 10.1172/JCI110652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiss SJ, LoBuglio AF. Phagocyte-generated oxygen metabolites and cellular injury. Lab Invest. 1982;47:5–18. [PubMed] [Google Scholar]
- 13.Thomas EL. Myeloperoxidase, hydrogen peroxide, chloride antimicrobial system: nitrogen-chlorine derivatives of bacterial components in bactericidal action against Escherichia coli. Infect Immun. 1979;23:522–531. doi: 10.1128/iai.23.2.522-531.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94:437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hazell LJ, Arnold L, Flowers D, Waeg G, Malle E, Stocker R. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J Clin Invest. 1996;97:1535–1544. doi: 10.1172/JCI118576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hazell LJ, Baernthaler G, Stocker R. Correlation between intima-to-media ratio, apolipoprotein B-100, myeloperoxidase, and hypochlorite-oxidized proteins in human atherosclerosis. Free Radic Biol Med. 2001;31:1254–1262. doi: 10.1016/s0891-5849(01)00717-1. [DOI] [PubMed] [Google Scholar]
- 17.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalysed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeshita J, Byun J, Nhan TQ, Pritchard DK, Pennathur S, Schwartz SM, Chait A, Heinecke JW. Myeloperoxidase generates 5-chlorouracil in human atherosclerotics tissue: A potential pathway for somatic mutagenesis by macrophages. J Biol Chem. 2005;281:3096–3104. doi: 10.1074/jbc.M509236200. [DOI] [PubMed] [Google Scholar]
- 19.Scanlon CEO, Berger B, Malcom G, Wissler RW. Evidence for more extensive deposits of epitopes of oxidized low density lipoproteins in aortas of young people with elevated serum thiocyanate levels. Atherosclerosis. 1996;121:23–33. doi: 10.1016/0021-9150(95)05672-6. [DOI] [PubMed] [Google Scholar]
- 20.Botti TP, Amin H, Hiltscher L, Wissler RW. A comparison of the quantitation of macrophage foam cell populations and the extent of apolipoprotein E deposition in developing atherosclerotic lesions in young people: high and low serum thiocyanate groups as an indication of smoking. Atherosclerosis. 1996;124:191–202. doi: 10.1016/0021-9150(96)05825-x. [DOI] [PubMed] [Google Scholar]
- 21.Pattison DI, Davies MJ. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem Res Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 22.Pattison DI, Davies MJ. Reactions of myeloperoxidase-derived oxidants with biological substrates: gaining insight into human inflammatory diseases. Curr Med Chem. 2006;13:3271–3290. doi: 10.2174/092986706778773095. [DOI] [PubMed] [Google Scholar]
- 23.Pattison DI, Hawkins CL, Davies MJ. What are the plasma targets of the oxidant hypochlorous acid? A kinetic modeling approach. Chem Res Toxicol. 2009;22:807–817. doi: 10.1021/tx800372d. [DOI] [PubMed] [Google Scholar]
- 24.Skaff O, Pattison DI, Davies MJ. Hypothiocyanous acid reactivity with low- molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem J. 2009;422:111–117. doi: 10.1042/BJ20090276. [DOI] [PubMed] [Google Scholar]
- 25.Skaff O, Pattison DI, Morgan PE, Bachana R, Jain VK, Priyadarsini KI, Davies MJ. Selenium-containing amino acids are major targets for myeloperoxidase- derived hypothiocyanous acid: determination of absolute rate constants and implications for biological damage. Biochem J. 2012 doi: 10.1042/BJ20101762. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lane AE, Tan JT, Hawkins CL, Heather AK, Davies MJ. The myeloperoxidase-derived oxidant HOSCN inhibits protein tyrosine phosphatases and modulates cell signalling via the mitogen-activated protein kinase (MAPK) pathway in macrophages. Biochem J. 2010;430:161–169. doi: 10.1042/BJ20100082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd MM, Van Reyk DM, Davies MJ, Hawkins CL. Hypothiocyanous acid is a more potent inducer of apoptosis and protein thiol depletion in murine macrophage cells than hypochlorous acid or hypobromous acid. Biochem J. 2008;414:271–280. doi: 10.1042/BJ20080468. [DOI] [PubMed] [Google Scholar]
- 28.Hawkins CL. The role of hypothiocyanous acid (HOSCN) in biological systems. Free Radic Res. 2009;43:1147–1158. doi: 10.3109/10715760903214462. [DOI] [PubMed] [Google Scholar]
- 29.Morgan PE, Pattison DI, Talib J, Summers FA, Harmer JA, Celermajer DS, Hawkins CL, Davies MJ. High plasma thiocyanate levels in smokers are a key determinant of thiol oxidation induced by myeloperoxidase. Free Radic Biol Med. 2011;51:1815–1822. doi: 10.1016/j.freeradbiomed.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 30.Lipskaia L, Hulot JS, Lompré AM. Role of sarco/endoplasmic reticulum calcium content and calcium ATPase activity in the control of cell growth and proliferation. Pflugers Archive - Eur J Physiol. 2009;457:673–685. doi: 10.1007/s00424-007-0428-7. [DOI] [PubMed] [Google Scholar]
- 31.Tong XY, Evangelista A, Cohen RA. Targeting the redox regulation of SERCA in vascular physiology and disease. Curr Opin Pharmacol. 2010;10:133–138. doi: 10.1016/j.coph.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Periasamy M, Kalyanasundraram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35:430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 33.Adachi T, Weisbrod RM, Pimentel DR, Yang J, Sharov VS, Schöneich C, Cohen RA. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 34.Sharov VS, Dremina ES, Galeva NA, Williams TD, Schöneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxdation during biological aging. Biochem J. 2006;394:605–615. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen RA, Adachi T. Nitric-oxide-induced vasodilatation: regulation by physiologic S-glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc Med. 2006;16:109–114. doi: 10.1016/j.tcm.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Viner RI, Hühmer AFR, Bigelow DJ, Schöneich C. The oxidative inactivation of sarcoplasmic reticulum Ca2+-ATPase by peroxynitrite. Free Radic Res. 1996;24:243–259. doi: 10.3109/10715769609088022. [DOI] [PubMed] [Google Scholar]
- 37.Viner RI, Williams TD, Schöneich C. Nitric oxide-dependent modification of the sarcoplasmic reticulum Ca-ATPase: localisation of cysteine target sites. Free Radic Biol Med. 2000;29:489–496. doi: 10.1016/s0891-5849(00)00325-7. [DOI] [PubMed] [Google Scholar]
- 38.Dremina ES, Sharov VS, Davies MJ, Schöneich C. Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem Res Toxicol. 2007;20:1462–1469. doi: 10.1021/tx700108w. [DOI] [PubMed] [Google Scholar]
- 39.Furtmüller PG, Jantschko W, Regelsberger G, Jakopitsch C, Arnhold J, Obinger C. Reaction of lactoperoxidase compound I with halides and thiocyanate. Biochemistry. 2002;41:11895–11900. doi: 10.1021/bi026326x. [DOI] [PubMed] [Google Scholar]
- 40.Nelson DP, Kiesow LA. Enthalpy of decomposition of hydrogen peroxide by catalase at 25 °C (with molar extinction coefficients of H2O2 in the UV) Anal Biochem. 1972;49:474–478. doi: 10.1016/0003-2697(72)90451-4. [DOI] [PubMed] [Google Scholar]
- 41.Thomas EL. Lactoperoxidase-catalyzed oxidation of thiocyanate: equilibria between oxidized forms of thiocyanate? Biochemistry. 1981;20:3273–3280. doi: 10.1021/bi00514a045. [DOI] [PubMed] [Google Scholar]
- 42.Eyer P, Worek F, Kiderlen D, Sinko G, Stuglin A, Simeon-Rudolf V, Reinerc E. Molar absorption coefficients for the reduced Ellman reagent: reassessment. Anal Biochem. 2003;312:224–227. doi: 10.1016/s0003-2697(02)00506-7. [DOI] [PubMed] [Google Scholar]
- 43.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 44.Xu C, Ma H, Inesi G, Al-Shawi MK, Toyoshima C. Specific structural requirements for the inhibitory effect of thapsigargin on the Ca2+-ATPase SERCA. J Biol Chem. 2004;279:17973–17979. doi: 10.1074/jbc.M313263200. [DOI] [PubMed] [Google Scholar]
- 45.Hawkins CL, Morgan PE, Davies MJ. Quantification of protein modification by oxidants. Free Radic Biol Med. 2009;46:965–988. doi: 10.1016/j.freeradbiomed.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Viola HM, Arthur PG, Hool LC. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ Res. 2007;100:1036–1044. doi: 10.1161/01.RES.0000263010.19273.48. [DOI] [PubMed] [Google Scholar]
- 47.Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–15977. doi: 10.1021/ja0438361. [DOI] [PubMed] [Google Scholar]
- 48.Louis CF, Irving I. Protein components of sarcoplasmic reticulum membranes from different animal species. Biochim Biophys Acta. 1974;365:193–202. doi: 10.1016/0005-2795(74)90263-3. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen Dinh Cat A, Touyz RM. Cell signaling of angiotensin II on vascular tone: novel mechanisms. Curr Hypertens Rep. 2011;13:122–128. doi: 10.1007/s11906-011-0187-x. [DOI] [PubMed] [Google Scholar]
- 50.Nagy P, Jameson GN, Winterbourn CC. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem Res Toxicol. 2009;22:1833–1840. doi: 10.1021/tx900249d. [DOI] [PubMed] [Google Scholar]
- 51.Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1–21. doi: 10.1146/annurev.physiol.67.040103.152647. [DOI] [PubMed] [Google Scholar]
- 52.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calciumapoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 53.Favero TG, Webb J, Papiez M, Fisher E, Trippichio RJ, Broide M, Abramson JJ. Hypochlorous acid modifies calcium release channel function from skeletal muscle sarcoplasmic reticulum. J Appl Physiol. 2003;94:1387–1394. doi: 10.1152/japplphysiol.00645.2002. [DOI] [PubMed] [Google Scholar]
- 54.Favero TG, Colter D, Hooper PF, Abramson JJ. Hypochlorous acid inhibits Ca2+-ATPase from skeletal muscle sarcoplasmic reticulum. J Appl Physiol. 1998;84:425–430. doi: 10.1152/jappl.1998.84.2.425. [DOI] [PubMed] [Google Scholar]
- 55.Strosova M, Karlovska J, Spickett CM, Grune T, Orszagova Z, Horakova L. Oxidative injury induced by hypochlorous acid to Ca-ATPase from sarcoplasmic reticulum of skeletal muscle and protective effect of trolox. Gen Physiol Biophys. 2009;28:195–209. doi: 10.4149/gpb_2009_02_195. [DOI] [PubMed] [Google Scholar]
- 56.Strosova M, Skuciová M, Horakova L. Oxidative damage to Ca2+-ATPase sarcoplasmic reticulum by HOCl and protective effect of some antioxidants. BioFactors. 2005;24:111–116. doi: 10.1002/biof.5520240113. [DOI] [PubMed] [Google Scholar]
- 57.Peskin AV, Winterbourn CC. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic Biol Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 58.Bozonet SM, Scott-Thomas AP, Nagy P, Vissers MC. Hypothiocyanous acid is a potent inhibitor of apoptosis and caspase 3 activation in endothelial cells. Free Radic Biol Med. 2010;49:1054–1063. doi: 10.1016/j.freeradbiomed.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 59.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]