

Abstract
The core mechanism of intracellular vesicle fusion consists of SNAREpin zippering between vesicular and target membranes. Recent studies indicate that the same SNARE-binding protein, Complexin (CPX), can act either as a facilitator or as an inhibitor of membrane fusion, giving rise to a major controversy. Here, we employ energetic measurements using the Surface Forces Apparatus which reveal that CPX acts sequentially on assembling SNAREpins, first facilitating zippering by nearly doubling the distance at which v- and t-SNAREs can engage, and then by clamping them into a half-zippered fusion-incompetent state. Specifically, we find that the central helix of CPX allows SNAREs to form this intermediate energetic state at 9–15 nm, but not when the bilayers are closer than 9 nm. Stabilizing the activated-clamped state at separations < 9 nm requires the accessory helix of CPX, which prevents membrane-proximal assembly of SNAREpins.
During regulated exocytosis in synaptic transmission and neuro-endocrine secretion, a pool of neurotransmitter or hormone-containing vesicles accumulate at the plasma membrane awaiting the signal for rapid and synchronous release. To create this readily releasable pool, the molecular machinery of regulated exocytosis needs to bring the vesicles into an activated state which is as close to fusion as possible, all the while preventing them from fusing prematurely1–3. Calcium-dependent exocytosis is controlled by the pairing between cognate v- and t-SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) proteins, which assemble like a zipper across the membranes that are destined to fuse, thus facilitating their close apposition and subsequent merging4,5. The assembly of such membrane-bridging complexes, called SNAREpins, is further manipulated by SNARE interacting proteins to allow fusion to be clamped or triggered where and when necessary1. Complexin (CPX) and synaptotagmin (SYT) proteins are key regulators of calcium-dependent neurotransmitter release displaying an intriguing dual activatory-inhibitory role in membrane fusion2,6–9.
In vitro, CPX inhibits SNARE-mediated liposome-liposome10 or cell-cell fusion11, and this inhibition is relieved by the addition of calcium and SYT. In contrast to this clamping effect, CPX can also activate SNAREpin assembly and membrane fusion, when added to liposomes that have already been bridged by SNAREpins12,13. In vivo studies also led to seemingly contradictory data. Notably, knock out of CPX in mouse synapses decreases both spontaneous and Calcium-evoked exocytosis14, indicating a positive requirement, whereas deletion of CPX in Drosophila or in C. elegans synapses increases spontaneous release (negative role) but also decreases calcium-evoked release (positive role)3,15, thus implying a clamping function. Clamp and activator functions seem to selectively reside in the different structural domains that comprise the CPX molecule16,17,18, which consist of an N-terminal domain (activator), an accessory helical domain (inhibitor), a central helical domain (necessary as the main binding site to the SNARE complex), and a C-terminal domain (of unclear function13,16).
Altogether, these disparate observations have led to a controversy concerning CPX and its role(s) in exocytosis. The simplest explanation would be that CPX is both a clamp and an activator, acting at different stages of SNARE assembly1. Despite these recent important insights into the structure-function properties of CPXs, the physical-chemical mechanism by which one relatively small protein can perform such opposite functions, all within a few milliseconds, remains unclear.
To investigate the molecular mechanism underlying the function of CPX, we took advantage of recent advances in Surface Forces Apparatus (SFA) measurements, which have previously allowed us to measure simultaneously the distance and the interaction energy between SNARE proteins embedded into lipid bilayers, and thus to identify intermediate states along their assembly pathway19. In our SFA system, two opposing bilayers are brought into closely controlled apposition, one containing the synaptic v-SNARE, VAMP2, the other one the synaptic t-SNARE, a complex of Stx1a and SNAP25 (derived from rat and mouse). These SNAREs are anchored to the bilayers by covalent attachment at their C-termini to the lipid phosphatidylethanolamine rather than by their natural transmembrane (TM) domain. To our knowledge, this is the only experimental system that can provide direct energetic measurements of SNARE complexes assembling between bilayers, closely mimicking the geometry that occurs in cells.
Here, we introduce the soluble form of human complexin 3 (CPX) and its various functional domains into this SNARE bilayer - SFA experiment to address the following fundamental questions: (i) How does CPX affect the structural-energetic landscape of SNAREpins as they assemble across membranes? (ii) What is the role of the different functional domains of CPX in SNAREpin activation and/or clamping? To answer these questions, SNARE bilayers were approached down to molecular contact distances and then separated from each other, all the while measuring at regular intervals (30 s) both distances (± 0.1 nm) and forces (± 1 μN) of interaction. After performing several approach-separation cycles in the absence of CPX, SNARE bilayers were separated several hundreds of nanometers apart and then incubated with CPX for 2 hours prior to initiating additional approach-separation cycles. The process was repeated on the same SNARE bilayers using increasing concentrations of CPX, ranging from 0.1 to 3 μM.
In the presence of CPX, the interaction energy versus distance profile of SNARE bilayers exhibited substantial changes consistent with an initial activation of cognate SNARE binding evident during the approach phase, followed by a block to completion of SNAREpin assembly evident during the separation phase of the experiment. These will be presented in the next sections.
RESULTS
CPX activates N-terminal assembly of SNAREpins
Before adding CPX (Fig. 1a and our previous work19), SNARE bilayers initially repel each other during the approach phase in a non-specific polymer-like manner between d = 20 and 10 nm (d is the separation between the opposing bilayer surfaces, which is by definition 0 when the bilayers are in contact), and with an exponential decay length of 8±2 nm. At a distance dbinding = 9±1 nm, the SNAREs start assembling, leading to a slower increase of the repulsion resembling a plateau. Then, when the bilayers are brought even closer (down to 4±1 nm), SNARE assembly is completed to the maximum extent possible (recall that the two bilayers cannot fuse due to the lipid anchors), and a stiff repulsion is encountered which persists down to dcontact = 2.5±1.5 nm; this corresponds to the compression of partially assembled (~70%) SNAREpins19.
Figure 1. CPX affects the structural-energetic landscape of SNAREpins as they assemble across membranes.

(a–c) Interaction energy versus distance profile of SNAREpins in the absence (black) or the presence (purple) of CPX at various concentrations (squares: approach; circles: separation). The interaction profiles between SNARE bilayers have been normalized to the surface density of SNAREs in the apposing bilayers, and are represented as average interaction energy (in kBT unit) per assembling SNAREpin (see Supplementary Method). At low CPX concentration (0.3 μM), the separation profile displays two adhesions. The first one (at ~ 9 nm) corresponds to the unbinding of CPX-free SNAREpins that are ~ 70% assembled (~ down to layer +6) and display a binding energy of 35 kBT19. The second one (at ~ 16 nm) corresponds to the unbinding of CPX-bound SNAREpins that are ~ 50% assembled (~ down to layer +1) and display a binding energy of 15 kBT (see text for details). (d) When more CPX is added between SNARE bilayers, the first adhesion (black) progressively decreases while the second one (purple) progressively increases (more SNAREpins are clamped by CPX). At high CPX concentration, all SNAREpins are clamped by CPX and display reduced binding energy and extent of assembly.
After incubation with CPX (Fig. 1b,c), these features are conserved qualitatively, however, all events occur at dramatically larger distances (Table 1). Notably, the distance dbinding at which SNAREpins start assembling increases up to 15±1 nm in the presence of CPX (compare distances at e ~ 10 kBT in Fig. 1a-c and see also Figs. 2 and 3). In addition, the polymer-like repulsion is of larger decay length after incubation with CPX (Table 1), indicating that either or both SNAREs extend their sequence further away from the bilayer surface. This would explain the increase in reactivity of v- with t-SNAREs at greater separations (i.e. the activation of SNAREpin assembly) when CPX is present.
Table 1.
CPX increases the interaction distances between membrane-embedded SNAREs. λpolymer is the decay length of the polymer-like repulsion measured on the SFA approaching curves19, and gives the range of the entropic repulsions between unassembled SNAREs; dcontact shows the thickness of the protein layer between two compressed bilayers; dunbinding is directly related to the extent of SNAREpin assembly. Errors are standard deviations for SFA measurements performed on 2–4 independent pairs of cognate SNARE bilayers, conducting 5–15 consecutive approach-separation cycles with these bilayers.
| λpolymer (nm) | dcontact (nm) | dunbinding (nm) | |
|---|---|---|---|
| No CPX | 7.9±1.9 | 2.3±1.4 | 9.0±2.5 |
| CPX | 11.8±1.2 | 5.8±0.6 | 15.9±2.3 |
| CPX(26-93) | 9.3±1.8 | 5.9±1.4 | 16.1±1.0 |
| CPX(58-158) | 9.6±0.9 | 4.5±0.5 | 11.0±1.8 |
| CPX(K26A) | 10.4±1.6 | 5.2±1.2 | 9.0±1.7 |
| CPX(super-clamp) | 11.9±2.1 | 5.2±0.5 | 20.4±3.2 |
Figure 2. CPX allows SNAREpins to assemble at a larger distance.

SNARE bilayers were approached down to a specific minimal interbilayer distance (dmin) and kept in contact for at least 30 min (which is the time necessary to reach an optimal adhesion19) before being separated (squares: approach; circles: separation). In the absence of CPX (red and black profiles), if dmin > ~ 9 nm, SNAREs do not assemble during the approach phase and no adhesion is observed upon separation; the red curve shows one example with dmin ~ 11 nm. In the presence of CPX (green profile), SNAREs form stable membrane-bridging complexes as soon as dmin < ~ 15 nm; the green curve gives one example where dmin ~ 14 nm. CPX thus allows the SNAREs to find each other and to assemble at a larger distance.
Figure 3. CPX activates and clamps SNAREpin assembly.
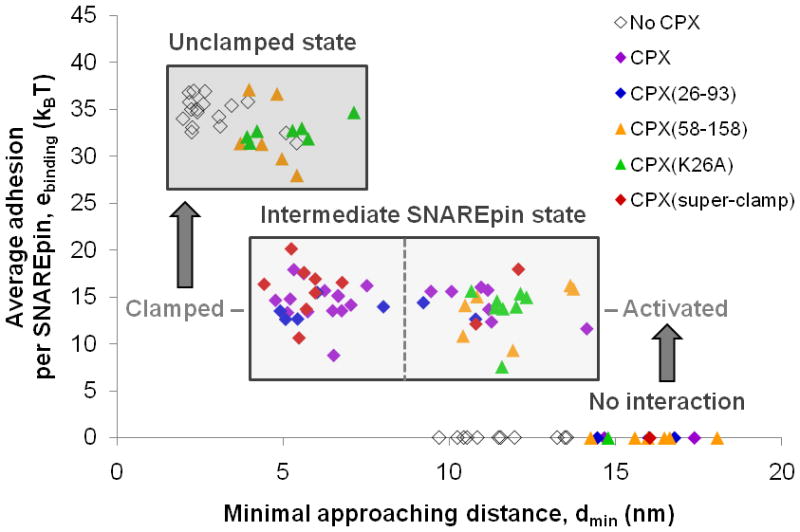
Experiments similar to those presented in Fig. 2 were repeated with many different dmin in the presence of various CPX variants (each data point corresponds to a single approach-separation cycle). Such experiments allow one to determine exactly at which distance SNAREpins start assembling, and to measure the energy of assembly at a given distance. All data points were obtained with 1 μM of CPX because, at this concentration, >80% of the SNAREpins are bound to CPX (affinity better than 0.2 μM; see Fig. 4). In the absence of CPX, SNAREs do not begin to assemble until the bilayers are within ~ 9 nm, and they zipper into highly energetic (~ 35 kBT) SNAREpins. CPX allows the SNAREpins to zipper into an intermediate (~ 15 kBT) energetic state when the bilayers are as far as ~ 15 nm apart, suggesting an activation of SNAREpin assembly by CPX. The energy of this intermediate state is unchanged over a wide range of separations (dmin ~ 5–15 nm), suggesting a clamping effect of CPX on SNAREpin assembly. Activation of SNAREpin assembly is observed with all CPX variants tested, and is thus controlled by the central helix of CPX. When the interbilayer distance becomes smaller than 9 nm, only mutants having the native or strengthened accessory helix can hold the intermediate (~15 kBT) state in place. Mutants with deleted or weakened accessory helix cannot prevent C-terminal assembly of SNAREs, which leads to the highly energetic (~35 kBT) SNAREpin state19.
To further test this conclusion, we performed experiments during which SNARE bilayers were approached down to a specific minimal distance (dmin, which is systematically varied from one experiment to the next) at which they were allowed to react for at least 30 min (which is sufficient for maximum adhesion to occur19). Then, the bilayers were separated and the energy required to do this was determined. If there was no assembly at dmin, then there will be no binding energy to overcome during separation; but if assembly did occur at dmin, then force will be needed to separate the bilayers away from dmin, the amount of which measures the average adhesion (binding) energy per SNAREpin (Figs. 2 and 3).
In the absence of CPX, there is no adhesion until dmin is less than 9±1 nm, confirming that isolated SNAREpins can only start assembling when bilayers are 9±1 nm apart or closer. CPX increases dmin to 15±1 nm, which means that SNAREs can now assemble when the bilayers are 50% further away, providing an explanation of why CPX activates SNARE assembly.
CPX clamps C-terminal assembly of SNAREpins
In the absence of CPX and upon separation, SNARE bilayers remain bound through membrane-bridging SNAREpins, until dunbinding = 9±2 nm at which the bilayers jump out as the SNAREpins are pried apart, and display a binding energy of ~ 35 kBT19. When a saturating concentration of CPX is added, full separation of the two SNARE bilayers now occurs at dunbinding = 16±2 nm (Fig. 1c). The distance at which the jump-out occurs is related to the degree of assembly of CPX-bound SNAREpins. To estimate this, we treat the unassembled part of SNARE proteins as random polymers, and determine the extent to which these unstructured sequences are stretched prior to the adhesive jump. A jump-out at 16 nm suggests that the number of C-terminal aminoacids which are not yet zippered into the SNAREpin structure is 24–44 on each of the v- and t-SNARE sides (see Supplementary Discussion). On the v-SNARE side, this would correspond to a region bounded by the hydrophobic layers −2 and +4 (ref. 20). On average, the SNAREpin is thus about 50% zippered (down to layer +1) when bound to CPX. This would be consistent with the pattern of sensitivity and resistance to tetanus and botulinum toxins that indicates a similar degree of zippering when SNAREs are functionally clamped in vitro11 and for the readily releasable pool of synaptic vesicles at the neuromuscular junction in vivo21. CPX thus acts as an inhibitor (a clamp) of SNAREpin assembly by blocking C-terminal zippering of membrane proximal domains of cognate v- and t-SNARE proteins.
The detailed behaviour of adhesion as a function of less than saturating concentrations of CPX allows us to estimate the affinity of CPX for assembling SNAREpins at the interface between the apposed membranes. Specifically, at intermediate CPX concentrations (Fig. 1b,d), a two-stage unbinding process is now observed: the bilayers first jump apart at 10 nm but remain adherent (first jump-out), and then completely separate at 16 nm (second jump-out). The average energy per SNAREpin associated with the first jump-out, e1, vanishes as the CPX concentration increases, while that associated with the second jump-out, e2, increases up to about 15 kBT. The concentration dependence indicates that the first jump-out corresponds to a sub-population of free SNAREpins, whereas the second jump-out corresponds to a sub-population of CPX-bound SNAREpins (of lower energy), and this allows us to estimate the affinity constant (~ 0.2 μM) for the binding of CPX to the SNAREpin (Supplementary Discussion and Fig. 4).
Figure 4. Affinity of CPX variants for the SNAREpin.

The dissociation constants K between the SNAREpin and the wild-type CPX (purple), the non-clamping CPX(K26A) mutant (green), or the super-clamping CPX(Q37A R41F Y44A Q48L) mutant (red) were estimated from the fraction of clamped SNAREpins (which is directly related to e2, the adhesion due to CPX-bound SNAREpins in Fig. 1d) at various CPX concentrations. For each mutant, at least 3 different concentrations were tested and the plot 1/e2 versus 1/[CPX] was fit by a straight line whose slope gives the dissociation constant K (Supplementary Discussion and Supplementary Table 1). In the case of the super-clamping mutant, only the two highest concentrations are displayed here for clarity (the fit was however deduced from 3 different protein concentrations). The non-clamping CPX mutant displays a slightly lower affinity for SNAREpins than wild-type CPX, whereas the super-clamping CPX mutant displays a much higher affinity for SNAREpins.
Importantly, the energy necessary to disassemble SNAREpins – which is also the energy released upon SNAREpin assembly and made available to drive close membrane apposition – is thus much smaller in the presence of CPX, which explains how CPX can clamp fusion.
CPX creates an activated and clamped intermediate state
Overall, CPX therefore appears to have a dual role in fusion, being both an activator and an inhibitor of SNAREpin assembly. It is an activator of SNAREpin early during its N-terminal assembly while it prevents C-terminal completion of SNAREpin zippering and therefore clamps fusion by retaining SNAREpins in a lower energy state. The thermodynamic mechanism at the origin of the dual activatory-inhibitory function of CPX is illustrated with the energy landscape representation in Fig. 5. In the absence of CPX, once cognate v- and t-SNAREs have overcome a non-specific polymer-like repulsion due to their confinement between lipid bilayers, they start binding (d = 9±1 nm) and their assembly then spontaneously progresses down to an interbilayer distance d = 4±1 nm to form a highly energetic (~ 35 kBT) SNAREpin. In this case, the energy landscape of SNAREpin folding thus consists of a single repulsion barrier (for N-terminal assembly) followed by a deep adhesion well. In the presence of CPX, cognate SNAREs begin to assemble at a larger distance (d = 15±1 nm) to form a SNAREpin of lower energy (~ 15 kBT) whose membrane-proximal zippering is inhibited. On the energy landscape of SNAREpin folding, this translates into the appearance of an intermediate binding state flanked with (i) a first repulsion barrier at a larger distance (N-terminal assembly is facilitated) and (ii) a second repulsion barrier at short distance (C-terminal assembly is prevented).
Figure 5. CPX reshapes the energy landscape of SNAREpin folding.

CPX digs an adhesion well in the pathway of cognate SNARE assembly. In the absence of CPX (black), v- and t-SNAREs need to overcome a non-specific repulsion barrier (ending at an interbilayer distance d ~ 9 nm) to begin their assembly and form highly energetic (~ 35 kBT) SNAREpins. In the presence of CPX (purple), an intermediate energetic state of lower energy (~ 15 kBT) appears on the folding pathway of SNAREpin. The non-specific repulsion barrier still exists but is displaced further away, thus allowing v- and t-SNAREs to bind at a larger distance (~ 15 nm). This barrier at large distance is generated by the central helix of CPX, which facilitates N-terminal assembly of SNAREpins most likely by increasing the exposure of t-SNAREs (see text and Fig. 6 for details). It is followed by a second repulsion barrier at short distance generated by the accessory helix of CPX, which prevents further SNAREpin assembly by competing with the v-SNARE for C-terminal binding to the t-SNARE. The dashed lines indicate the regions that are not observed in the SFA. Since the intermediate state is stable over an hour (typical duration of the contact time between two SNARE bilayers in the SFA), the energy difference between the binding energy and the barriers height (i.e. the activation energy) must be higher than 30 kBT (see Supplementary Discussion).
The seemingly contradictory activatory-inhibitory effects of CPX on membrane fusion thus come from a common mechanism: the creation of an intermediate energetic state in the folding pathway of SNAREpins, which makes the SNAREs find each other at a larger distance, but traps the SNAREpin in this state until an external action makes it overcome the repulsion barrier towards further zippering.
In these studies we employ lipid-anchored SNAREs from which the trans-membrane (TM) anchors have been removed, for two reasons. First, neither we nor other SFA users have succeeded in reproducibly forming bilayers containing functional TM proteins. Second, and more fundamentally, including the TM anchors would preclude establishing reproducible measurements of pre-fusion SNARE assembly, because the approaching bilayers would probably fuse on their first close approach; as a result we could never do the pull-away portion which actually reveals the binding energy. Using lipid anchors instead of TMs prevents fusion22 and therefore allows us to measure adhesion and to take the bilayers through repetitive cycles of approach-separation to establish the reproducibility of measurements. It is certainly true that the absence of TM domains might have some quantitative effect on the energetic landscape, but this is expected to be small because the TM domains zipper last, after the cytoplasmic domains.
The t-SNARE is the primary target for CPX action
The targets of regulatory factors can include individual SNARE proteins (to regulate their accessibility or their intracellular targeting), partially assembled fusion machines SNAREpins (to regulate their rate or extent of assembly), or fully assembled post-fusion cis-SNARE complexes (to regulate their disassembly). The efficiency of synaptic vesicle exocytosis can be modulated by manipulating the pools of SNAREs in any of these three states. What, then, is the primary target for CPX action? SFA experiments performed on symmetrical t-SNARE versus t-SNARE or v-SNARE versus v-SNARE systems (Fig. 6) show that CPX targets the t-SNARE but not the v-SNARE. CPX increases both the polymer decay length and the distance at contact between t-SNARE bilayers while it does not change the interaction profile between v-SNARE bilayers. Isothermal Titration Calorimetry (ITC) experiments performed on soluble SNARE domains further show that CPX binds to t-SNARE (but not to v-SNARE) with an affinity of about 2 μM (Supplementary Fig. 1). This is also in good agreement with single-molecule fluorescence and co-floatation experiments performed on membrane-embedded SNARE proteins, which showed that complexin-1 binds to and stabilizes the (1:1) Syntaxin1A-SNAP25 acceptor complex23,24. CPX would thus prepare and facilitate the association of cognate SNAREs through its interaction with the t-SNARE.
Figure 6. CPX directly interacts with membrane-anchored t-SNAREs.

Interaction energy versus distance profile between two t-SNARE (a) or between two v-SNARE (b) bilayers in the absence (black) or the presence (purple) of 1 μM CPX (squares: approach; circles: separation). No adhesion is observed in both cases, either with or without CPX (during the separation phase, the interaction force continuously decreases until reaching the zero baseline). CPX affects the interaction profile of t-SNARE but not v-SNARE bilayers. In the presence of CPX, the long-range repulsive forces between t-SNARE bilayers begin 10 nm further away (the t-SNAREs see each other at a larger distance) and the distance at contact is about 1 nm larger (the protein layer between the two compressed bilayers is now thicker), suggesting that CPX binds to t-SNARE and increases its exposure toward solution (makes it more erected on the bilayer surface). Note that the repulsion profile between two v-SNARE bilayers is stronger than that between two t-SNARE bilayers, which is consistent with the fact that v-SNARE is largely unstructured.
CPX domains responsible for SNAREpin activation and clamping
Complexin proteins consists of four structurally distinct domains, which are, in the case of the human complexin-3 studied here: an N- terminal domain (residues 1-25), an accessory helix (residues 26-57), a central helix (residues 58-80), and a C-terminal domain (residues 81-158). Structural studies show that CPX binds in an anti-parallel manner to the fully assembled post-fusion state of the SNARE complex through its central helix22–24. In the X-ray structure, the central helix binds in the groove between VAMP and Syntaxin helices, while the accessory helix does not contact the SNARE complex.
Structure-function analysis in a cell-cell fusion assay in vitro28 and by electrophysiology in vivo16,18 suggest that each domain of CPX contributes uniquely. The N-terminal domain was shown to have an activating effect, whereas the accessory helix was inhibitory. Furthermore, it was proposed that the central helix, in addition to stabilizing the SNARE complex27, could strategically position the N-terminal domain and the accessory helix for their interaction with SNAREs and/or lipid membranes. The role of the C-terminal domain is not clear as both activatory and inhibitory functions have been reported16,13.
To elucidate how functional domains of CPX work on SNAREpin assembly to activate or inhibit fusion, we have introduced various CPX mutants into the SFA experiment. A CPX mutant containing only the accessory and the central helices, CPX(26-93), previously identified in the cell-cell fusion assay as the minimum clamping domain28 (Supplementary Fig. 2), behaves exactly like wild-type CPX when added to SNARE bilayers (Fig. 3 and Table 1). This shows that the most extreme N- and C- terminal sequences of CPX are not required for the activating and clamping effects as observed in our SFA system. When the accessory helix is deleted [CPX(58-158)] or mutated [CPX(K26A)] so as to lose clamping function28 (Supplementary Fig. 2), activation (assembly at d > 9 nm) is retained but the intermediate state becomes unstable and the more zippered state now prevails at dmin < 9 nm (Fig. 3 and Supplementary Fig. 3). These mutants are thus able to create the intermediate state, but the energetic barrier towards further zippering (barrier at short distance) is now too low (less than 25 kBT; Supplementary Discussion and Supplementary Fig. 4), and can easily be overcome on the time-scale of SFA measurements (~ 1 min).
Results obtained with the CPX(58-158) mutant show that the central helix is sufficient for the activation function of CPX and for inducing the intermediate state. They also show that the accessory helix is required to have a repulsion barrier at short distance which is high enough to trap the SNAREpin in this intermediate state and thus stabilize clamping (even when the v- and t-SNAREs are forced to be in very close contact with the SFA; see Supplementary Discussion). As suggested previously18,29, the accessory helix probably does so by binding to C-terminal residues of the t-SNARE, therefore occupying the v-SNARE binding site (Supplementary Figs. 4 and 5).
In the CPX(K26A) mutant experiment, the altered accessory helix could thus be displaced from the t-SNARE at short interbilayer distance. This suggests that binding of the native accessory helix of CPX to the t-SNARE is not very strong and mainly controlled by the Lys26 residue. SFA titration data indeed show that the overall affinity of the SNAREpin for the wild-type CPX or the CPX(K26A) mutant are similar: KCPX ~ KCPX(K26A) ~ 0.2 μM (Supplementary Discussion and Fig. 4), and ITC experiments show that binding of the accessory helix of CPX to the C-terminus of t-SNARE is substantially weakened by the mutation K26A (Supplementary Fig. 5). In addition, since both the CPX(58-158) and the CPX(K26A) mutants display the same distance at contact (dcontact ~ 5–6 nm) as wild-type CPX (Table 1), the central helix of CPX probably stays bound to the SNAREpin at short interbilayer distance, after displacement of the accessory helix30 (the distance at contact is directly related to the thickness of protein complexes between two compressed bilayers).
The final CPX mutant tested, CPX(Q37A R41F Y44A Q48L), was designed to improve the sequence similarity between the accessory helix of CPX and the C-terminus of VAMP2 (ref. 29), and thus to increase the affinity of the accessory helix for membrane-proximal residues of the t-SNARE. In the cell-cell fusion assay, this mutant displayed a much higher capacity to inhibit fusion and, as such, was identified as a “super-clamping” CPX mutant29 (Supplementary Fig. 2). When added to the SFA experiment, this super-clamping mutant creates the same intermediate energetic state as does wild-type CPX (Fig. 3 and Supplementary Fig. 3), but does so at about 10 times lower concentration (Ksuper-clamp ~ 10 nM; Supplementary Discussion and Fig. 4). This explains energetically how increasing the affinity of the accessory helix for the C-terminus of t-SNARE improves the clamping efficiency of CPX.
It is important to note that the CPX specific nature of the observed features is clear and alternate explanations such as non-specific aggregation of CPX are not tenable because: i) approach and separation curves can be quantitatively repeated many times without hysteresis (hysteresis is a hall mark of non-specific binding in the SFA); ii) CPX mutations affect the SFA curves in a manner fitting with functional analysis; iii) the specific features we study only occur when CPX is added.
DISCUSSION
Variations in the SFA interaction profiles of SNARE bilayers obtained in the presence of CPX mutants allow us to assign distinct features to the accessory and central helical domains of CPX. The central helix is responsible for activating SNAREpin assembly by stabilizing an intermediate energetic state (~ 50% zippered, 15 kBT) at large separations (up to ~ 15 nm) that are afforded by the ~ 50% of the SNARE motif sequence (membrane-proximal C-terminal half) that is unzippered. We assume that this state is stabilized by the known contacts between the central helix of CPX and the interface between VAMP and Syntaxin helices (in layers −3 to +3 of the SNARE complex). In the absence of the accessory helix, this intermediate state is only metastable at separations < ~ 9 nm at which the more fully zippered, highly energetic (~ 35 kBT) SNAREpin is permitted to assemble. The accessory helix precludes this transition and stabilizes the intermediate energetic state by binding to the unassembled C-terminal portion of the t-SNARE thereby preventing the v-SNARE from completing its assembly18,29. The X-ray study presented in the accompanying paper from Kuemmel et al.31 suggests that, in this process, the central and accessory helices come from two neighboring CPX molecules in a zig-zag array that forms at the interface of the apposed bilayers. Disruption of the bonds between accessory helices and the SNAREpins to which they bind in the array leads to a conformation switch in CPX and triggers membrane fusion (see accompanying paper by Krishnakumar et al.32).
To regulate exocytosis, clamp-activator proteins grapple with SNAREs to sequentially facilitate and then inhibit their assembly across two fusing membranes1. Our SFA data show that CPX accomplishes its dual activatory-inhibitory task by way of a single elegant mechanism, reshaping the energy landscape of cognate SNARE assembly to create a single intermediate energetic state. SNAREpin zippering is first facilitated at large separations and then prevented at small separations when SNAREpins are about half assembled and the bilayers are within molecular contact range. We speculate that in vivo Ca2+-bound SYT mechanically provides the energy required to lower the repulsion barrier towards further zippering by physically displacing the accessory helix of CPX, thus triggering membrane-proximal assembly of SNAREpins and synaptic vesicle fusion.
METHODS
Protein purification
Soluble t- and v-SNARE proteins
The soluble t-SNARE complex – made of the cytoplasmic domain of rat Stx1a (residues 1-265) containing a single C-terminal Cysteine residue (C145S) and of mouse His6-SNAP25b (residues 1-206) with all 4 Cysteines in the loop region mutated to Serines – was produced by co-expression of pJM57 and pJM72 plasmids in the BL21 gold (DE3) Escherichia coli bacterial strain, and purified as described before19. The cytoplasmic domain of mouse His6-VAMP2 (residues 1-94) containing a C-terminal Cysteine residue was expressed and purified from pJM51 as previously described19. Protein concentrations (typically 1.5–5 mg.mL−1) were determined by a Bradford protein assay with Bovine Gamma Globulin (BGG) as the standard or by the Thermo Scientific Pierce Bicinchoninic Acid (BCA) protein assay with Bovine Serum Albumin (BSA) as the standard.
CPX variants
The soluble forms of human complexin 3 (CPX) and its mutants were cloned into a pET SUMO vector containing a His6 tag. The accessory helix deletion CPX(58-158) mutant and the minimum clamping domain CPX(26-93) mutant were cloned using the wild-type CPX plasmid as the template. The single point CPX(K26A) mutant and the super-clamping CPX(Q37A R41F Y44A Q48L) mutant were site-specific mutated using the QuickChange site directed mutagenesis kit from Stratagene and the wild-type CPX plasmid as the template. All CPX variants were expressed in the BL21-CodonPlus (DE3)-RIPL Escherichia coli bacterial strain. The purification procedure is available in the online Supplementary Information. The protein concentrations were typically 2.5–4 mg.mL−1 as determined by a Bradford protein assay with BGG as the standard.
SNARE bilayers reconstitution
Lipid bilayers (inner layer: DMPE; outer layer: DOPC, DOPS, DOPE-Maleimide) were prepared by Langmuir-Blodgett deposition on mica surfaces. A first monolayer of DMPE lipids was transferred at a constant pressure of 38 mN.m−1 onto two separate mica surfaces (previously glued to cylindrically curved glass lenses). DMPE surfaces were then dried for 15 minutes. A second monolayer of a lipid mixture containing 89 mol% DOPC, 10 mol% DOPS and 1 mol% DOPE-Maleimide lipids was transferred at a constant pressure of 35 mN.m−1 onto the hydrophobic DMPE surfaces19. The two glass lenses supporting the lipid bilayers were next transferred into two ~ 5 mL beakers which were moved into a dish containing 1 liter of coupling buffer (25 mM HEPES, pH 7.6, 100 mM KCl, 0.25 mM TCEP). Buffer exchange proceeded for 15 minutes and then the beakers were moved out of the dish. About 100 μL of each SNARE was then added to its corresponding beaker for a final concentration of soluble t- and v-SNARE proteins of ~ 1 μM and ~ 2 μM respectively; the reaction was incubated overnight at 4°C. Unreacted DOPE-Maleimide lipids were quenched by introducing Mercaptoethanol in the beakers. Then, extensive rinsing was done to remove unbound proteins before the beakers were transferred into the SFA chamber.
Surface force measurements
Force measurements were carried out with a Surface Forces Apparatus (SFA)33. The SFA technique measures the force, F, between thin films confined to the surfaces of two crossed-cylindrical lenses as a function of their separation distance, d, measured with a resolution of 1 Å34. The force F is normalized by the mean radius of curvature, R, of the two cylinders (R ≈ 2 cm), which leads to F(d)/R with a resolution of 0.1 mN.m−1. The ratio F(D)/R is related to the interaction free energy per unit area, E(D), between two equivalent planar surfaces by the Derjaguin approximation35:
The interaction energy E(D) can be further normalized into interaction energy per SNAREpin after dividing by the surface density of SNAREs, which can be deduced from the repulsive parts of the interaction profiles19. All SNARE bilayers force measurements were carried out at 21°C in degassed coupling buffer (25 mM HEPES, pH 7.6, 100 mM KCl, 0.25 mM TCEP).
Isothermal Titration Calorimetry (ITC) experiments
ITC experiments were performed on a Microcal ITC200 instrument. Typically, about 200 μL of ~ 10 μM SNARE solution was loaded into the sample cell and about 40 μL of ~ 100 μM CPX solution was loaded into the syringe. An initial 0.2 μL injection of CPX was followed by several 1–2 μL injections. 180-second equilibration time was used after each injection to ensure complete binding. The heat change per injection was integrated and normalized by the moles of CPX in the injection. All ITC experiments were carried out at 37°C and at least twice. Microcal Origin ITC200 software package was used to analyze the titration calorimetric data and obtain the stoichiometric number (N), the molar binding enthalpy (ΔH), and the association constant (Ka). A simple one site chemical reaction was assumed. The affinity constant (Kd), the binding free energy (ΔG), and the binding entropy (ΔS) were calculated using the thermodynamic equations:
More details about these protocols are included in the online Supplementary Methods.
Supplementary Material
Acknowledgments
This work was supported by the Human Frontier Science Program, the ANR-08-PCVI-0014 grant to Frédéric Pincet, NIH grants to James E. Rothman and a Partner University Funds exchange grant between the Yale and Ecole Normale Supérieure laboratories. David Tareste is funded by the ANR-09-JCJC-0062-01 grant. We thank Dr. Thomas Melia for many helpful discussions, as well as Dr. Jeff Coleman, Will Eng and Alejandro Garcia-Diaz for technical help.
Footnotes
AUTHOR CONTRIBUTIONS
FL and CGG made constructs and did protein purification. FL performed SFA and ITC measurements. CGG performed cell-cell fusion assay. FL, FP and DT analyzed the data. FL, FP, EP, DT and JER interpreted the results and prepared the manuscript.
References
- 1.Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–7. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sørensen JB. Conflicting views on the membrane fusion machinery and the fusion pore. Annual review of cell and developmental biology. 2009;25:513–37. doi: 10.1146/annurev.cellbio.24.110707.175239. [DOI] [PubMed] [Google Scholar]
- 3.Hobson RJ, et al. Complexin Maintains Vesicles in the Primed State in C. elegans. Current Biology. 2011;21:106–113. doi: 10.1016/j.cub.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sollner T, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 5.Weber T, et al. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 6.Brose N. For better or for worse: complexins regulate SNARE function and vesicle fusion. Traffic. 2008;9:1403–13. doi: 10.1111/j.1600-0854.2008.00758.x. [DOI] [PubMed] [Google Scholar]
- 7.Reim K, et al. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 8.Tang J, et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 9.Maximov A, et al. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–21. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaub JR, et al. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nature structural & molecular biology. 2006;13:748–50. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- 11.Giraudo CG, et al. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–680. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- 12.Yoon TY, et al. Complexin and Ca 2+ stimulate SNARE-mediated membrane fusion. Nat Struct Mol Biol. 2008;15:707–713. doi: 10.1038/nsmb.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malsam J, et al. The carboxy-terminal domain of complexin I stimulates liposome fusion. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2001–6. doi: 10.1073/pnas.0812813106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue M, et al. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7875–80. doi: 10.1073/pnas.0803012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–1237. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- 16.Xue M, et al. Tilting the Balance between Facilitatory and Inhibitory Functions of Mammalian and Drosophila Complexins Orchestrates Synaptic Vesicle Exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho RW, Song Y, Littleton JT. Comparative analysis of Drosophila and mammalian complexins as fusion clamps and facilitators of neurotransmitter release. Molecular and Cellular Neurosciences. 2010;45:389–397. doi: 10.1016/j.mcn.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue M, et al. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol. 2007;14:949–958. doi: 10.1038/nsmb1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, et al. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat Struct Mol Biol. 2007;14:890–896. doi: 10.1038/nsmb1310. [DOI] [PubMed] [Google Scholar]
- 20.Sutton RB, et al. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 21.Hua SY, Charlton MP. Activity-dependent changes in partial VAMP complexes during neurotransmitter release. Nat Neurosci. 1999;2:1078–1083. doi: 10.1038/16005. [DOI] [PubMed] [Google Scholar]
- 22.McNew JA, et al. Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J Cell Biol. 2000;150:105–117. doi: 10.1083/jcb.150.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weninger K, et al. Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP-25 complex. Structure. 2008;16:308–20. doi: 10.1016/j.str.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan R, Dai H, Rizo J. Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry. 2008;47:1474–81. doi: 10.1021/bi702345m. [DOI] [PubMed] [Google Scholar]
- 25.Pabst S, et al. Rapid and selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. The Journal of Biological Chemistry. 2002;277:7838–48. doi: 10.1074/jbc.M109507200. [DOI] [PubMed] [Google Scholar]
- 26.Bracher A, et al. X-ray structure of a neuronal complexin-SNARE complex from squid. J Biol Chem. 2002;277:26517–26523. doi: 10.1074/jbc.M203460200. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, et al. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/s0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- 28.Giraudo CG, et al. Distinct domains of complexins bind SNARE complexes and clamp fusion in vitro. The Journal of Biological Chemistry. 2008;283:21211–9. doi: 10.1074/jbc.M803478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giraudo CG, et al. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science. 2009;323:512–6. doi: 10.1126/science.1166500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue M, et al. Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nature structural & molecular biology. 2010;17:568–75. doi: 10.1038/nsmb.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kümmel D, et al. Complexin cross-links pre-fusion SNAREs into a zig-zag array: a structure-based model for complexin clamping. Nat Struct Mol Biol. doi: 10.1038/nsmb.2101. Accepted (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krishnakumar SS, et al. A Conformational Switch in Complexin is Required for Synaptotagmin to Trigger Synaptic Fusion. Nat Struct Mol Biol. doi: 10.1038/nsmb.2103. Accepted (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Israelachvili JN, Adams GE. Measurement of Forces between 2 Mica Surfaces in Aqueous-Electrolyte Solutions in Range 0–100 Nm. J Chem Soc Farad Trans. 1978;74:975–1001. [Google Scholar]
- 34.Israelachvili J. Thin-Film Studies Using Multiple-Beam Interferometry. J Colloid Interface Sci. 1973;44:259–272. [Google Scholar]
- 35.Derjaguin BV, Muller VM, Toporov YP. Effect of Contact Deformations on Adhesion of Particles. J Colloid Interface Sci. 1975;53:314–326. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.