

Abstract
Glutathione thiyl radicals (GS•) were generated in H2O and D2O by either exposure of GSH to AAPH#, photoirradiation of GSH in the presence of acetone, or photoirradiation of GSSG. Detailed interpretation of the fragmentation pathways of deuterated GSH and GSH-derivatives during mass spectrometry analysis allowed us to demonstrate that reversible intramolecular H-atom transfer reactions between GS• and C-H bonds at Cys[αC], Cys[βC], and Gly[αC] are possible.
Keywords: glutathione, thiyl radical, C-centered radical, hydrogen transfer, mass spectrometry
1. Introduction
The tripeptide glutathione (GSH) plays a critical role in cellular homeostasis.1 GSH is an essential component of (i) the glutathione-S-transferase-catalyzed conjugation affording signalling and regulation of biological pathways,2 xenobiotic detoxification3 and (ii) cellular redox homeostasis through reversible oxidation to glutathione disulfide (GSSG; reaction 1).4
| (1) |
GSH is also a suitable hydrogen/electron donor for one-electron reduction reactions, which lead to glutathione thiyl radicals, GS• (reaction 2).
| (2) |
Such one-electron reduction reactions of thiols are relevant for the protection of organisms against free radicals, generated, for example, through metabolic processes5 or the exposure to ionizing radiation.6 However, it was recognized that thiyl radicals may also involve in the reverse reaction, i.e. hydrogen abstraction from C-H bonds present in alcohols and ethers,7–10 carbohydrates,11 polyunsaturated fatty acids,12 and amino acids,13–15 and rate constants for such reactions have been provided by means of radiation chemical techniques. In peptides the resulting carbon-centered radicals can involve in hydrogen transfer reactions between amino acid side chains.16
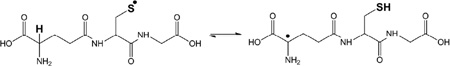 |
(3) |
Specifically GS• radicals feature a pH-dependent intramolecular hydrogen transfer between the thiyl radical moiety and the αC-H bond of the N-terminal γ-Glu residue (reaction 3).13,17,18 However, electron spin resonance (ESR)19,20 and pulse radiolysis studies21 have also demonstrated evidence for hydrogen transfer between the thiyl radical and additional C-H bonds. For example, Karoui et al.20 demonstrated that the oxidation of GSH resulted in the appearance of two types of carbon-centered radicals, monitored by ESR, and Neta and Fessenden19 interpreted their ESR data with the formation of a carbon-centered radical at the αC position of the Cys residue. The latter observation would suggest the possibility for a 1,3-hydrogen transfer in GS•, supported also by experimental observations on the oxidation of penicillamine.20 Additional ESR experiments obtained with small model thiols also suggest the possibility for a formal 1,2-hydrogen transfer in thiyl radicals, i.e. the observation of •CH2SH radicals after the photolysis of CH3SH,22 and the detection of CH3C•H-SH radicals after photolysis of CH3CH2SH.23
Theoretical calculations have examined activation energies for intramolecular hydrogen transfer reactions in GS•.24 Here, the hydrogen transfer between the thiyl radical and the αC-H bond of γ-Glu displays a rather low activation energy of ΔG≠ = 37–42 kJ mol−1, whereas the 1,3-hydrogen shift within the Cys residue shows ΔG≠ = 110 kJ mol−1 and hydrogen transfer from the C-H bonds of the C-terminal Gly residue shows ΔG≠ = 134 kJ mol−1 (in the (Z)-conformation). Nevertheless, experimental evidence for a 1,3-hydrogen transfer within thiyl radicals from Cys (CysS•) exists, both in the gas phase25 and in solution.13,26 In addition, covalent H/D exchange experiments have demonstrated the potential for CysS• radicals to abstract hydrogen atoms from neighbouring Gly and Ala residues in several model peptides,27–31 suggesting that such a hydrogen transfer reactions should, in principle, be experimentally possible also in GS•.
In this paper, we report experimental evidence that the thiyl radical in GS• enters reversible hydrogen transfer reactions not only with the γ-Glu residue, but to a significant extent with the C-H bonds of Cys (1,2- and 1,3-hydrogen transfer) as well as the C-terminal Gly residue. To demonstrate these hydrogen transfer reactions, we have generated GS• via three independent chemical methods, (i) the reaction of GSH with carbon-centered radicals generated through the thermal decomposition of (AAPH), (ii) the photolysis of GSSG, and (iii) the reaction of GSH with carbon-centered radicals (•CH3, CH3C•O) generated through the photolysis of acetone. Reversible hydrogen transfer reactions were monitored through covalent H/D exchange reactions of original C-H bonds, analysed by mass spectrometry, including MS2, MS3, and MS4 experiments#. An important experimental result is the ultimate conversion of the GSH Cys residue into dehydroalanine (Dha), likely via β-elimination following a 1,3-hydrogen transfer of the initial thiyl radical, a mechanism recently established for small Cys-containing model peptides.32 Dha represents an important electrophile with the potential to generate thioether cross-links with excess GSH or protein thiols.33
2. Materials and Methods
2.1 Materials
Reduced glutathione (GSH), oxidized glutathione (GSSG), ammonium bicarbonate (NH4HCO3), 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AAPH), N-ethyl-maleimide (NEM), iodoacetamide (IOA) and acetone were supplied by Sigma-Aldrich (St Louis, MO) at the highest purity grade. Stable isotope-labeled glutathione (GSH*, here the Gly residue is labeled with 15N, and 13C at carbons C1 and C2), deuterated acetone (acetone-d6, 99.9%) and deuterium oxide (D2O, 99.9%) were purchased at Cambridge Isotope Laboratories (Andover, MA). The structures of GSH, GSH*, and GSSG are presented in Chart 1.
Chart 1.

Structures of glutathione (GSH, GSH*) and GSSG.
2.2 Preparation of the deuterated buffer solutions
Ammonium bicarbonate buffers were prepared at a concentration of 50 mM at three different pH/pD, 7.1, 8.1, and 9.1. The deuterated buffers were obtained by dilution of NH4HCO3 salt (19.2 mg) in pure D2O (1 mL) and repetition (3 times) of the following procedure: i) vacuum drying of D2O, ii) reconstitution into 1 mL D2O. After the final vacuum drying step, ND4DCO3 was resolubilized into 5 mL D2O. In parallel, a stock solution of KOD (0.1 M) was prepared in D2O according to the same protocol. KOD stock solution was used to adjust the pD of ND4DCO3 buffer solutions.
2.3 Reaction between GSH and AAPH
GSH (750 µM) and AAPH (2 mM) were incubated for 1 hour at 37 °C under Ar in ammonium bicarbonate buffers (50 mM, pH/pD 7.1, 8.1 or 9.1). The stock solutions of GSH and AAPH were pre-saturated under Ar and placed on ice prior to be mixed. The reaction was stopped by alkylation of Cys with NEM. The latter was performed as follows: the samples were diluted in 50 mM ammonium bicarbonate, pH 8, in 50:50 (v/v) H2O/D2O (referred to as balanced ammonium bicarbonate; for a rationale to use balanced buffer, see section 2.9) containing 1 mM NEM. The reaction products were analyzed by capillary LC-MS/MS connected to a SYNAPT-G2 (Waters Corporation, Milford, MA, USA) mass spectrometer (for more details on LC-MS analysis, see section 2.7).
2.4 Reaction between GSH* and AAPH
GSH* (750 µM) and AAPH (2 mM) were incubated for 1 hour at 37 °C under Ar in a non-buffered solution. The amount of GSH* was limited. To run MSn experiments on the GSH*-derived products, we had to maintain a concentration of GSH* similar to that used with GSH. Experimentally, the latter led us to avoid dilution of the GSH* stock solution. The pH of the solution was therefore different from the pH of the other solution (GSH). Because in this pH range the decomposition rate of AAPH may vary, our experiments will not allow us to compare quantitatively the amount of covalent deuterium incorporation into GSH* (vs GSH). GSH* was only used, in parallel to GSH, to discriminate between the different fragment ions during the MSn experiments. By no means the GSH* solution could be used for quantitative measurements. The pH/pD of the final solutions was 3.5 (after addition of AAPH). The stock solutions of GSH* and AAPH were pre-saturated with Ar, and placed on ice prior to be mixed. The reaction was stopped by alkylation of Cys with either NEM or IOA. The chemical derivatizations were performed as follows: 100 µL of each sample were diluted either with 900 µL of milliQ H2O (if the sample was incubated in H2O) or with 900 µL D2O (if the sample was incubated in D2O) which both contained 1 mM of either NEM or IOA. The pH/pD of each solution was adjusted to 8.0 by addition of KOH (or KOD). The derivatized products GSH*-NEM and GSH*-IOA were analyzed by mass spectrometry using a Fourier Transform Ion Cyclotron Resonance (FT-ICR) instrument (for more details, see section 2.8).
2.5 UV-irradiation of GSSG
GSSG (500 µM) was photo-irradiated in H2O and D2O for 10 min at λ = 253.7 by means of a photo-irradiator (Rayonet™, Southern New England, Branford, CT, RMA-500) equipped with four UV lamps (RMR-2537Å) with a flux of 3.2 × 10−7 einstein/sec. Prior to UV-irradiation, a 300 µL aliquot of each H2O and D2O solution containing GSSG (500 µM) in ammonium bicarbonate buffer (50 mM, pH/pD 7.1, 8.1 or 9.1) was placed in a quartz tube and saturated with Ar. After photo-irradiation the samples were diluted into a balanced ammonium bicarbonate buffer containing 1 mM NEM (the balanced buffer is described in section 2.9). The products of UV-irradiation were analyzed by capillary LC-MS connected to a SYNAPT-G2 mass spectrometer (Waters Corporation, Milford, MA, USA).
2.6 Acetone-h6 and acetone-d6 photochemistry in the presence of GSH and GSH*
GSH and GSH* (500 µM) were prepared in 1 mL solutions consisting of acetone-h6:H2O (20%:80% v:v) or acetone-d6:H2O (20%:80% v:v), respectively. The solutions were pre-saturated with Ar prior to photo-irradiation for 10 min at λ = 253.7 by means of a photo-irradiator (Rayonet™, Southern New England, Branford, CT, RMA-500) equipped with four UV lamps with a flux of 3.2 × 10−7 einstein/sec. The photo-irradiated samples were diluted by adding 100 µL of each to 900 µL of milliQ H2O prior to FT-ICR mass spectrometry analysis.
2.7 Sample preparation for capillary LC-MS analysis connected to a SYNPAT-G2 mass spectrometer
10 µL of the sample were injected onto a Vydac column (25 cm × 0.5 mm C18, 3.5 µm), and eluted with a linear gradient delivered at a rate of 20 µL min−1 by a Capillary Liquid Chromatography System (Waters Corporation, Milford, MA, USA). Mobile phases consisted of water/acetonitrile/formic acid at a ratio of 99%, 1%, 0.08% (v:v:v) for solvent A and a ratio of 1%, 99%, 0.06% (v:v:v) for solvent B. The following linear gradient was set: 1–50% of solvent B within 15 min. Electrospray ionization (ESI) MS spectra of the photoproducts were acquired on a SYNAPT-G2 (Waters Corporation, Milford, MA), operated for maximum resolution with all lenses optimized on the [M + 2H]2+ ion from the [Glu]1-fibrinopeptide B. The cone voltage was 30 V and Ar was admitted to the collision cell. The spectra were acquired using a mass range of 50–2000 amu. The data were accumulated for 0.7 sec per cycle.
2.8 Sample preparation for FT-ICR analysis
The following procedure was applied to analyze products obtained from i) the acetylation and deuteron-acetylation of GSH and GSH* during photo-irradiation in the presence of acetone-h6 and acetone-d6, and ii) the covalent deuterium incorporation into GSH* during exposure to AAPH in D2O. Here the derivatization of GSH/GSH* with NEM or IOA was performed in 100% of D2O before dilution (1/100) into H2O:formic acid (99.9%:0.1%, v:v) to allow H/D-exchange of mobile protons/deuterons. Collision induced dissociation (CID) of fragment ions, referred to as MS3 and MS4, was performed by means of an LTQ-FT hybrid linear quadrupole ion trap Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (ThermoFinnigan, Bremen, Germany)34. MS3 and MS4 were acquired with an attenuation of the parent ion in the range of 25%-35%. The mass window to collect the parent ion was fixed to 0.2 Da. The samples were directly infused.
2.9 Covalent H/D exchange into GSH/GSH* and isotopic correction
After exposure of GSH to AAPH or formation of GSH during photoirradiation of GSSG, covalent deuterium incorporation into GSH (and GSH*) was determined as described below.
The formation of GSH during the photo-irradiation of GSSG and the mechanisms leading to covalent deuterium incorporation into GSH are described in Scheme 1. Briefly, UV-irradiation at λ = 253.7 nm of a disulfide bond (as in GSSG) leads to the formation of a pair of thiyl radicals (GS•). Ultimately, the disproportionation reaction of two GS• radicals leads to the formation of a thiol and a thioaldehyde. More details about the photochemistry of disulfide bonds are documented elsewhere.28,30,35 The reaction of GSH with AAPH (Scheme 1, reactions 4 and 5) leads to the formation of GS•.11 Intramolecular H-atom transfer between GS• and one of the several C-H bonds present in GS• (Scheme 1, reaction 7) generates C-centered radicals and a thiol, which converts into deuterothiol in D2O, and donates a deuteron back to the carbon-centered radical (Scheme 1, reactions 8 and 9).
Scheme 1.

Formation of glutathione thiyl radical using three different protocols, i) the thermal decomposition of AAPH, ii) the photochemistry of disulfide bond, iii) the photochemistry of acetone (X stands for H or D), and subsequent reactions of the thiyl radical.
After derivatization with NEM, the deuterium composition of GS-NEM and its fragment ions was determined from the differences between the average mass of a covalently deuterated peptide and the average mass of the corresponding fully protonated peptide. The average masses were calculated from centroided isotopic distributions. The distribution of deuterium incorporation is obtained after isotopic correction by subtracting the isotope abundance distribution in the product formed during either exposure to AAPH or UV-irradiation in H2O from the isotope abundance distribution of the same product generated in D2O. This variation of the isotopic distribution between the experiments performed in D2O and H2O will be given throughout this paper by the variation of the percent base peak intensity (%ΔBPI).
Importantly, the derivatization of GSH with NEM in D2O leads to the incorporation of one deuteron into the derivatized photoproduct (GS-NEM). In order to minimize quantitative errors, and in order to compare GSH/GSH* isolated from reactions in H2O and D2O, all NEM derivatizations were performed in balanced ammonium bicarbonate buffer (i.e., 50/50 H2O/D2O, v/v), where either H2O samples were supplemented with D2O, or D2O samples were supplemented with H2O (see section 2.3). This method of balanced buffer was successfully applied previously to monitor covalent H/D exchange of several peptides and proteins.27–30 Consequently, any differences in covalent deuterium content in GSH/GSH* are the result of the free radial reactions (and not the derivatization with NEM).
2.10 Computational details
Density Functional Theory (DFT) calculations presented were performed with the Gaussian-03 (G03) molecular orbital packages. The geometry optimization and frequency calculations were carried out at the B3LYP/6-311+G(d,p) level by the hybrid HF-DFT procedure implemented in the Gaussian software package.36 The geometries of the molecules were optimized in vacuum. The Gibbs free energies were calculated at 298°K (G0298) and are given in kcal/mol.
3. Results
Three chemical routes for the formation of glutathione thiyl radicals (GS•) were used (Scheme 1). The first route consisted of exposing GSH (or GSH*) to the C-centered radical resulting from the thermal decomposition of AAPH. Such reaction allows H-atom abstraction from the thiol to generate GS• (Scheme 1, reactions 4 and 5). The second and third routes are based on photochemical techniques. The photoirradiation of GSSG at λ=253.7 nm yields a pair of thiyl radicals (Scheme 1, reaction 6), analogous to processes characterized for various peptide disulfides.28,30,35 The photochemistry of acetone yields methyl and acetyl radicals (Scheme 1, reaction 10). GSH (and GSH*) reacts with •CH3/•CD3, and •COCH3/•COCD3 generated during the photochemistry of acetone-h6 or acetone-d6, respectively, which initially abstract the H-atom from the thiol function of GSH (or GSH*). Subsequently, covalent H/D exchange reactions (Scheme 1, reactions 7–9) were monitored, and, in addition, the final recombination products of glutathione-derived radicals (either C- or S-centered radicals) with the acetyl radical (Scheme 1, reactions 11–13). Using a combination of GSH/GSH* and CX3CO (X=H,D) in H2O provided a number of variables to identify specific products characteristic for radical formation on glutathione after initial formation of GS•.
3.1 GSH and the thermal decomposition of AAPH
GSH (m/z 308.1) was incubated under Ar for 1 hour in the presence of AAPH in H2O and D2O. Subsequently, GSH was derivatized with NEM in a balanced mixture of H2O/D2O at pH/pD 8.0 (See sections 2.3 and 2.9). The GS-NEM derivatives obtained after reaction of GSH with AAPH in H2O and with AAPH in D2O will be referred to as products 1-h and 1-d-X, respectively (summarized in Table 1). The label X in the series of products 1-d (X = Cys[αC], Cys[βC], or Gly[αC]) represents deuterium incorporation at Cys[αC], Cys[βC], or Gly[αC], respectively.
Table 1.
Overview of the products resulting from the derivatization of GSH with either NEM after reaction of GSH with AAPH . Products 1-d-Cys[αC], 1-d-Cys[βC], 1-d-Gly[αC] are products with a second deuteron located on Cys[αC], Cys[βC], Gly[αC], respectively. In the text, the labels Cys[αC], Cys[βC], and Gly[αC] are sometimes represented by the letter X.
| Reference | Structure | Reference | Structure |
|---|---|---|---|
| 1-h | 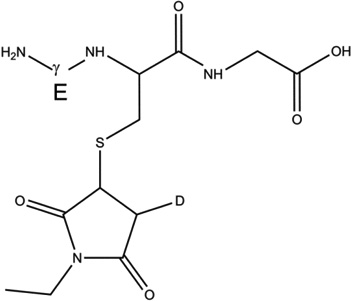 |
1-d-Cys[βC] | 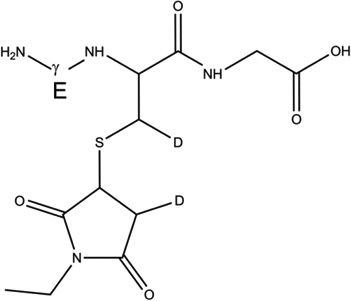 |
| 1-d-Cys[αC] | 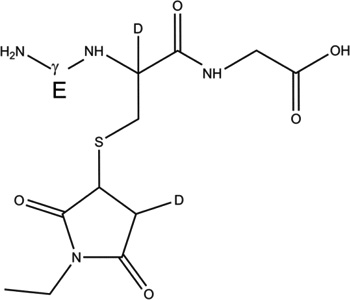 |
1-d-Gly[αC] | 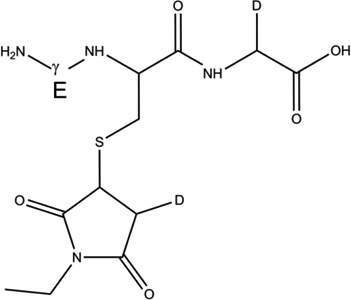 |
Analysis by MS1 and MS2
The comparison of the isotopic distributions of the parent ions of 1-h and 1-d-X (m/z 433.14) obtained after exposure of GSH to AAPH in H2O (Fig. 1, A) and in D2O (Fig. 1, B), respectively, shows a shift toward higher masses. This observation indicates that deuterium atoms are covalently incorporated into GSH during the exposure to AAPH in D2O. The deconvolution of the isotopic distributions of 1-h and 1-d-X obtained after reactions with AAPH in the range of pH/pD 7.1 – 9.1, shows that 37% – 41% of the molecules have incorporated one deuteron (Fig. 1, inserted table). The MS/MS analysis of 1-h and 1-d-X, and the comparison of the isotopic distributions of their b2 (m/z 358.1) and y2 (m/z 304.1) fragment ions, show that deuterium incorporation occurs to an equal extent in the subsequences γGlu-Cys and Cys-Gly, respectively (Fig. 2). The fragment ion with m/z 102.05, corresponding to the γGlu residue, shows covalent deuterium incorporation.
Figure 1.

MS spectra of GSH-NEM derivatized (m/z 433.14) obtained after exposure of GSH (500 uM) to AAPH (2 mM) at 37 °C during 1 hour in H2O-buffer (A) and D2O-buffer (B). The table summarizes the percentage of GSH-NEM derivatized molecules having incorporated covalently one deuteron atom after reaction of GSH with AAPH at different pH/pD in H2O/D2O-buffers.
Figure 2.

Overlay of the CID spectra of GSH-NEM derivatized (m/z 433.14) obtained after exposure of GSH (500 µM) to AAPH (2 mM) at 37 °C during 1 hour in H2O-buffer (black) and D2O-buffer (gray). The table summarizes the percentage of fragment ions having incorporated covalently one deuteron atom after reaction of GSH with AAPH at different pH/pD in H2O/D2O-buffers.
3.2 GSH* and the thermal decomposition of AAPH
GSH* (m/z 311.1) was incubated under Ar for 1 hour in the presence of AAPH in H2O and D2O in non-buffered solutions. Subsequently, GSH* was derivatized with either NEM or IOA in either H2O or D2O at pH/pD 8.0 (See sections 2.4 and 2.9). The GS-NEM derivative products obtained after reaction of GSH with AAPH in H2O and D2O, respectively will be referred to as products 2-h and 2-d-X, respectively (Table 2). The GS-IOA derivative products obtained after reaction of GSH with AAPH in H2O and D2O will be referred to as products 3-h and 3-d-X, respectively (Table 3). The label X (= Cys[αC], Cys[βC], or Gly[αC]) in the series of products 2-d and 3-d represents deuterium incorporation at Cys[αC], Cys[βC], or Gly[αC], respectively.
Table 2.
Overview of the products resulting from the derivatization of GSH* with either NEM after reaction of GSH* with AAPH. Products 1-d-Cys[αC], 1-d-Cys[βC], 1-d-Gly[αC] are products with a second deuteron located on Cys[αC], Cys[βC], Gly[αC], respectively. In the text, the labels Cys[αC], Cys[βC], and Gly[αC] are sometimes represented by the letter X.
| Reference | Structure | Reference | Structure |
|---|---|---|---|
| 2-h | 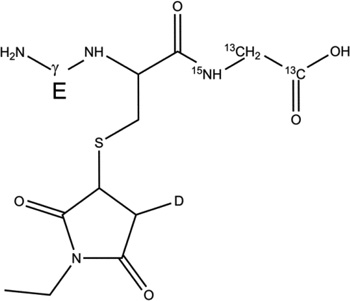 |
2-d-Cys[βC] | 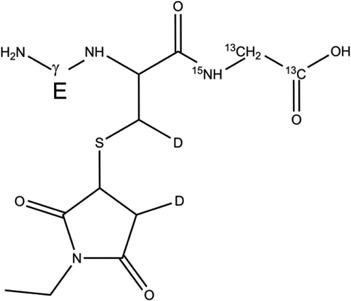 |
| 2-d-Cys[αC] | 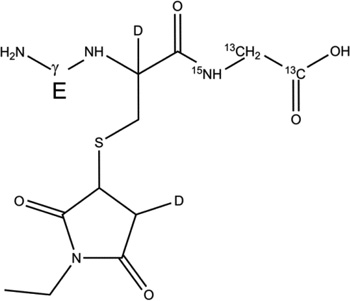 |
2-d-Gly[αC] | 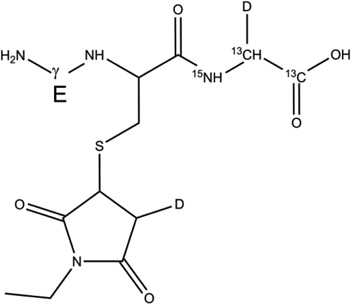 |
Table 3.
Overview of the products resulting from the derivatization of GSH with IOA after reaction of GSH with AAPH. Products 1-d-Cys[αC], 1-d-Cys[βC], 1-d-Gly[αC] are products with a second deuteron located on Cys[αC], Cys[βC], Gly[αC], respectively. In the text, the labels Cys[αC], Cys[βC], and Gly[αC] are sometimes represented by the letter X.
| Reference | Structure | Reference | Structure |
|---|---|---|---|
| 3-h | 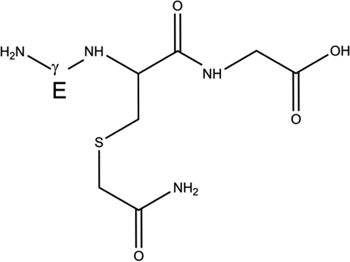 |
3-d-Cys[βC] | 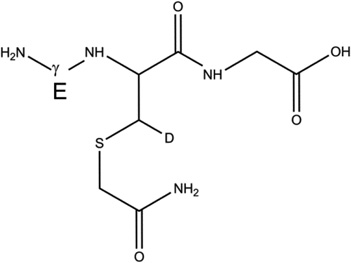 |
| 3-d-Cys[αC] |  |
3-d-Gly[αC] | 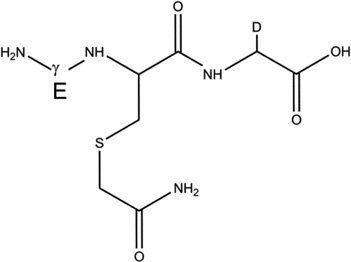 |
First, 15N and 13C labeling of the Gly* residue in GSH* allows several fragments to be distinguished during MS/MS analysis, such as the immonium ion from Cys (m/z 76.05) and the y1 fragment (= Gly*, m/z 79.05). Second, the 13C label in the carboxylic acid group of Gly* will allow us to detect the loss of 13CO (MW = 29 Da) during MS3 fragmentation.
MS3 analysis of product 2-d-X
After the reaction of GSH* with AAPH in H2O and D2O, and derivatization of GSH* with NEM, a comparison of the MS1 spectra of the products 2-h (m/z 436.1) and 2-d-X (m/z 438.1 and 439.1) indicate the incorporation of two (m/z 438.1) and three (m/z 439.1) deuterons into product 2-d-X. To localize the deuteron atoms in 2-d-X, an MS3 analysis of the y2 fragment ions was performed. The ions with m/z 309.1 and m/z 310.1, corresponding to the y2 fragment ions of 2-d-X after incorporation of two or three deuterons, respectively, were fragmented. The MS3 spectra are displayed in Figure 3.
Figure 3.

MS3 spectra of the y2 fragment ions of product 2-d-X after incorporation of A) two deuterons, B) three deuterons. X stands for deuterium incorporation at Cys[αC], Cys[βC], or Gly[αC].
i) Fragmentation of the y2 fragment ion of 2-d-X after incorporation of two deuterons (m/z 309.1)
The ion with m/z 309.1 was selected for MS3 fragmentation. It corresponds to the y2-fragment ion of 2-d after incorporation of two deuterons during the reaction with AAPH in D2O. The formation of the fragment ions with m/z 291.1, 262.1, and 203.1 (Fig. 3, A) is explained in the Supplementary Material, in section S1.1. The reaction mechanism is displayed in Scheme 2. This reaction mechanism is consistent with the incorporation of the second deuteron into product 2-d-Cys[αC] or 2-d-Cys[βC] (in Scheme 2, the fragmentation is representatively shown for the second deuteron covalently bound to the βC carbon of Cys, i.e. for 2-d-Cys[βC]).
Scheme 2.

Reaction pathways for the MS3 fragmentation of the y2 fragment ions of product 2-d-X after incorporation of two or three deuterons during the reaction of AAPH with GSH/GSH* in D2O. X stands for deuterium incorporation at Cys[αC], Cys[βC], or Gly[αC].
ii) Fragmentation of the y2 fragment ion of 2-d-X after incorporation of three deuterons (m/z 310.1)
The third deuteron can be incorporated either on the αC or βC carbons of the Cys residue or on the αC carbon of Gly*.
The ion with m/z 310.1 was selected for MS3 fragmentation. It corresponds to the y2-fragment ion of 2-d-X after incorporation of three deuterons during the reaction with AAPH in D2O. The formation of the fragment ions with m/z 292.1, 263.1, 203.1, and 204.1 (Fig. 3, B) is explained in the Supplementary Material, in section S1.2. The reaction mechanism is displayed in Scheme 2. The presence of the ion with m/z 204.1 (Fig. 3, insert B1) provides evidence that the third deuterium is also incorporated into Cys (rather than Gly*), either on the αC or the βC carbon. The concomitant observation of the ion with m/z 215.1 (Fig. 3, insert B2) suggests that both αC and βC of Cys contain one deuteron each. In such case, the fragmentation pathway is fully explained through reactions 18b-21b, invoking the same neutral losses as described in reactions 14– 17 (Scheme 2). In summary, the MS2 analysis of 2-d-X provides strong evidence for covalent H/D exchange at αC and βC of Cys.
MS3 analysis of product 3-d-X
After reaction of GSH* with AAPH in H2O and D2O and subsequent derivatization of GSH* with IOA, a comparison of the MS1 spectra of products 3-h (m/z 368.1) and 3-d-X (m/z 371.1) indicate the incorporation of three deuterons into product 3-d-X. To localize the deuteron atoms in 3-d-X, an MS3 analysis of the y2 fragment ion was performed. The ions with m/z 239.1 and 242.1 correspond to the y2 fragment ions of 3-h and 3-d-X, respectively, after incorporation of three deuterons. The MS3 spectra are displayed in Figure 4. The derivatization of Cys with IOA in D2O does not require any deuteron incorporation into the acetamide group. Therefore, the three deuterons incorporated into product 3-d-X are located on the αC carbon of Gly and/or the αC and/or βC carbons of Cys. In order to locate the deuterons we have compared the y2 fragments of 3-h and 3-d-X, after incorporation of three deuterons into 3-d-X.
Figure 4.

MS3 spectra of the y2 fragment ions of A) product 3-h and B) product 3-d-X after incorporation of three deuterons. X stands for deuterium incorporation at Cys[αC], Cys[βC], or Gly[αC].
i) Fragmentation of the y2 fragment ion of 3-h (no deuteron incorporated)
The ion with m/z 239.1 was selected for MS3 fragmentation. It corresponds to the y2 fragment ion of 3-h. The fragmentation spectrum is displayed in Figure 4, A. The fragmentation mechanism is given in Scheme 3, with X1=X2=X3=X4=H. The ions with m/z 220.9, 194.0, and 148.0 are rationalized in section S2 of the Supplementary Material.
Scheme 3.

Reaction pathways for the MS3 fragmentation of the y2 fragment ions of product 3-h and 3-d-X after incorporation of three deuterons during the reaction of AAPH with GSH/GSH* in D2O. X stands for deuterium incorporation at Cys[αC], Cys[βC], or Gly[αC].
ii) Fragmentation of the y2 fragment ion of 3-d-X after incorporation of three deuterons (m/z 242.1)
The ion with m/z 242.1 was selected for MS3 fragmentation. It corresponds to the y2 fragment ion of 3-d-X. The fragmentation spectrum is displayed in Figure 4, B, and the fragmentation mechanism is given in Scheme 3. The fragmentation mechanism is similar to the one described above for the y2 fragment of 3-h. Three deuterons are located on the y2 fragment ion of product 3-d-X. These deuterons can occupy three of the positions X1, X2, X3, or X4, as outlined in Scheme 3. The smallest fragment detected is the ion with m/z 133.0. Such ion results from the loss of H2O from the ion with m/z 151.0 (Scheme 3, reactions 28–30). The fragmentation mechanism leading to the formation of the ion with m/z 151.0 involves reaction 27. Due to the low amount of material, none of the ions resulting from the further loss of 13CO and 15NH=13CHX are observed. Consequently, the MS3 fragmentation of trideuderated y2 cannot give any further information on whether deuterium is incorporated in Gly*. Nevertheless, the presence of three deuterons in y2 of 3-d-X confirms that at least one deuteron must be located on the Cys residue.
3.3 UV-irradiation of GSSG
UV-irradiation (λ = 253.7 nm) of GSSG leads to the formation of GSH via an initial pair of GS•. The isotopic distributions of GSH generated in H2O and D2O, respectively, and derivatized with NEM (products 2-h and 2-d-X) are compared in Figure 5. LC-MS analysis reveals that product 2-d-X has incorporated deuterons. The respective percentages of 2-d-X which have incorporated one and two deuterons increase over a pH/pD range of 7.1–9.1 (Fig. 5, inserted table).
Figure 5.

Overlay of the MS spectra of 2-h and 2-d-X (m/z 433.14) generated after 10 min of UV-irradiation (λ = 253.7 nm) of GSSG (500 µM) in H2O (black) and D2O (gray). X stands for deuterium incorporation at Cys[αC], Cys[βC], or Gly[αC].
3.4 Photochemistry of acetone in the presence of GSH and GSH*: analysis of the b2 ion
The 253.7 nm photolysis of acetone at room temperature leads to the formation of methyl (H3C•) and acetyl radicals (CH3C•O).37 These radicals are able to abstract an H-atom from GSH38 to yield GS• which subsequently enters reversible H-transfer reactions (Scheme 1, reactions 11–13). More details about the photochemistry of sulfur-containing peptides in the presence of acetone are given elsewhere.29 Ultimately, both H3C• and CH3C•O can combine either with C- and S-centered radicals of GSH and GSH*. Specifically, the recombination products with carbon-centered radicals of GSH and GSH* provide information about the original formation and location of intermediary carbon-centered radicals. Hence, the recombination products were analysed by mass spectrometry.
i) Acetylation of the thiol
Products 4-h-S (m/z 350.1, CH3C(O)-SG, Table 4) and 4-d-S (m/z 353.1, CD3C(O)-SG, Table 4) are the results of the acetylation of the thiol function during the photolysis of acetone-h6 and acetone-d6, respectively, in the presence of GSH (reactions 31 and 32, X stands for H or D, depending on the use of acetone-h6 or acetone-d6).
Table 4.
Overview of the products resulting from acetone photolysis in the presence of GSH and GSH*. The acetylation on either the αC carbon of Cys, the αC carbon of Gly or the βC carbon of Cys is in the text summarized as Y in the name of the product (e.g. 5-h-Y). When we will refer to one of these products, the Y label will be replaced with either the label Cys[αC], Gly[αC] (or Gly[αC13], in GSH*), or Cys[βC]. In this table, a full description of all the possible products 5-h-Y, 5-d-Y, and 7-h-Y is presented.
| Reference | Structure | Reference | Structure | Reference | Structure |
|---|---|---|---|---|---|
| 4-h-S | 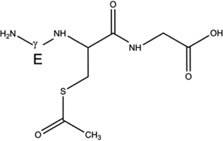 |
4-d-S | 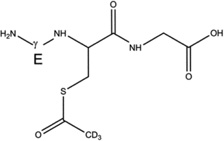 |
6-h-S | 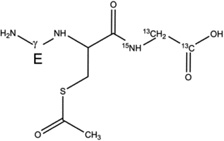 |
| 5-h-Cys[αC] | 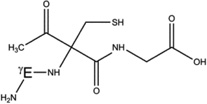 |
5-d-Cys[αC] | 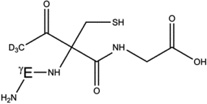 |
7-h-Cys[αC] | 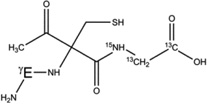 |
| 5-h-Cys[βC] | 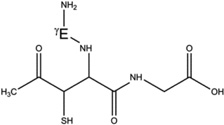 |
5-d-Cys[βC] | 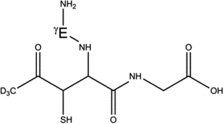 |
7-h-Cys[βC] | 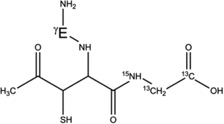 |
| 5-h-Gly[αC] | 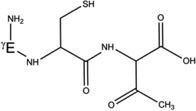 |
5-d-Gly[αC] | 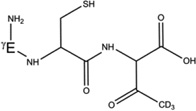 |
7-h-Gly[αC13] | 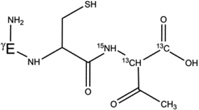 |
 |
(31) |
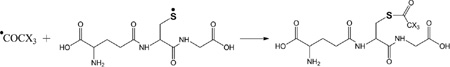 |
(32) |
The acetylation of Cys in GSH is demonstrated through MS3 analysis of the b2 fragment ions of products 4-h-S and 4-d-S, displayed in Figures 6A and 6B, where 4-h-S and 4-d-S display m/z 275.1 and m/z 278.1, respectively.
Figure 6.

MS3 spectra of the b2 fragment ions of A) product 4-h-S and B) product 4-d-S. MS4 of the fragment ion with m/z 199.0 is presented in panel C.
The b2 fragment ion is an amino-acylium ion that cyclizes through reaction 33 (Scheme 4, I1). This cyclic intermediate (I1) is used to explain the formation of all fragment ions observed during MS3 fragmentation of the b2 ions of products 4-h-S and 4-d-S (Figure 6, A and B). The structure of I1 was confirmed independently by MS4 analysis (see below). The fragmentation pathway of the b2 ions of 4-h-S and 4-d-S is explained in the Supplementary Material, section S3. The fragmentation mechanisms are displayed in Scheme 4 and 5.
Scheme 4.

Postulated fragmentation pathway for the MS3 fragmentation of the b2 fragment ions of products 4-h-S and 4-d-S. Color code for m/z: X=H (black), X=D (blue).
Scheme 5.

Postulated fragmentation pathways for the MS3 fragmentation of the b2 fragment ions of products 4-h-S and 4-d-S (continued). The dashed frame indicates the MS4 fragmentation mechanism of the ion with m/z 199.0. Color code for m/z: X=H (black), X=D (blue).
To confirm the structure of I1, we have further fragmented the ion with m/z 199 (Figure 6, C). The formation of ions with m/z 181.0, 153.0, and 135.0 during MS4 is explained in section S4 of the Supplementary Material. The formation of the cyclic intermediate I1 is also supported by calculations presented in the Supplementary Material (Scheme S1). The calculations show that the intermediate I1 is more stable than the b2 ion by 23.63 kcal/mol. The activation in the gas-phase of intermediate I1 allows further fragmentation of the b2 ion. Because the loss from I1 of the ketene group is more favourable than the loss of H2O (See calculations in Supplementary Material, Scheme S1), we believe that the fragmentation pathways described in Scheme 5 are more likely to occur than those described in Scheme 4.
ii) Acetylation of the γGlu residue
The presence of ions with m/z 172.0 (Fig. 6, A) and m/z 175.0 (Fig. 6, B) suggest also that the γGlu residue of GSH is acetylated during the photolysis of GSH in the presence of acetone-h6 (and acetone-d6). The low intensity of these ions did not allow further fragmentation to localize with more precision the acetylation site within γGlu.
3.5 Photochemistry of acetone in the presence of GSH and GSH*: analysis of the y2 ion
The acetylation of Cys and Gly/Gly* will be analyzed through fragmentation pathways of the y2 fragment ions of products 4-h-S, 4-d-S, 5-h-Y, 5-d-Y, 6-h-S and 7-h-Y (Table 4). Here, the labels h and d refer to the use of acetone-h6 and actone-d6, respectively. The label S refers to the acetylation of the thiol group. The label Y summarizes an acetylation on either the αC carbon of Cys, the αC carbon of Gly or the βC carbon of Cys. When we will refer to one of these products, the Y label will be replaced with either the label Cys[αC], Gly[αC] (or Gly[αC13], in GSH*), or Cys[βC].
In summary, products 4-h-S, 5-h-Y, 6-h-S, and 7-h-Y correspond to the acetylation products of GSH during the photolysis of acetone-h6. Products 4-d-S, and 5-d-Y correspond to the acetylation products of GSH during the photolysis of acetone-d6. The mechanism leading to the formation of 4-h-S and 4-d-S is described in reactions 31 and 32 (see above). The formation of products 5-h-Y and 5-d-Y is explained through reactions 56 to 58 (Scheme 6). The mechanism leading to the formation of 6-h-S proceeds through reactions 31 and 32 (see above, the reactions are representatively shown only for GSH). The formation of products 7-h-Y is explained by reactions 59 to 61 (Scheme 6).
Scheme 6.

Acetylation of GSH and GSH*.
The MS3 spectra of the y2 fragments ions are displayed in Figure 7.
Figure 7.

MS3 spectra of the y2 fragment ions of A) products 4-h-S and 5-h-Y, B) products 4-d-S and 5-d-Y, and C) products 6-h-S and 7-h-Y. The label Y refers to the acetylation of GSH or GSH* at the αC carbon of Cys, at the αC carbon of Gly/Gly* or at the βC carbon of Cys.
i) Acetylation of the thiol
The acetylation of the sulfhydryl group during the photolysis of acetone in the presence of GSH and GSH* is confirmed by the analysis of the y2 fragment ions of the products 4-h-S (m/z 221.0), 4-d-S (m/z 224.0) and 6-h-S (m/z 224.0) (Figure 7, A, B and C). To follow the fragmentation routes of the ions, the y2 ions and their fragments derived from 4-h-S, 4-d-S and 6-h-S are colored in black, blue and red, respectively, and presented in Scheme 7.
Scheme 7.

Postulated fragmentation pathways for the MS3 fragmentation of the y2 fragment ions of the following products: 4-h-S (m/z are black colored, and X=H), 4-d-S (m/z are blue colored, and X=D), 6-h-S (m/z are red colored, and X=H). The red-colored atoms indicate the position of 15N and 13C when GSH* is used to explain the fragmentation mechanism.
During MS analysis, products 4-h-S and 6-h-S generate y2 ions with m/z 221.0 and 224.0, respectively (Scheme 7, Figure 7, A, C). The reactions explaining the proton migration occurring during the fragmentation of the y2 ions are given in the Supplementary Material, in section S5.
The photo-irradiation of GSH in the presence of acetone-d6 yields product 4-d-S. During MS analysis, product 4-d-S generates the y2 ion with m/z 224.0 (Scheme 7, blue, Fig. 7, B). The fragmentation of this y2 ion according to reactions 62b-64b (Scheme 7) generates I2 with m/z 162.0. Reaction 64b (Scheme 7), cleaving off the deuterated acetamide (NH2COCD3), supports the mechanism depicted in Scheme 7 and is mechanistically described in the Supplementary Material (section S5).
ii) Acetylation of Gly[αC]
The products 5-h-Gly[αC], 5-d-Gly[αC], 7-h-Gly[αC] arise from the recombination of CH3C•O with a carbon-centered radical on Gly/Gly*, and can be analyzed through further fragmentation of the y2 ion. In fact, it will be shown that only the acylation of Gly/Gly* can fully explain the nature of the fragment ions derived from y2. Our proposed reaction schemes for the fragmentation of the y2 ions of the products 5-h-Cys[αC], 5-h-Cys[βC], 5-d-Cys[αC], 5-d-Cys[βC], 7-h-Cys[αC], and 7-h-Cys[βC] (Table 4) are presented in Scheme 8 and in the Supplementary Material (Schemes S1 and S2).
Scheme 8.

Postulated fragmentation pathways for the MS3 fragmentation of the y2 fragment ions of the following products: 5-h-Gly[αC] (m/z are black colored), 5-d-Gly[αC] (m/z are blue colored), 7-h-Gly[αC] (m/z are red colored). The red-colored atoms indicate the position of 15N and 13C when GSH* is used to explain the fragmentation mechanism. The blue-colored atoms indicate either the use of a protonated acetyl radical (X=H) or a deuterated acetyl radical (X=D).
During MS2 fragmentation the products 5-h-Gly[αC], 5-d-Gly[αC], 7-h-Gly[αC] generate the y2 fragments ions with m/z 221.0, 224.0, and 224.0, respectively. These ions were selected for MS3 analysis (Figure 7, A, B, C, and Scheme 8). The m/z of the fragment ions resulting from the collision-induced dissociation of the y2 ions of 5-h-Gly[αC], 5-d-Gly[αC], and 7-h-Gly[αC] are displayed in black, blue and red, respectively (Scheme 8). The initial fragmentation (Scheme 8, reactions 68–73) of the y2 ions of the three products 5-h-Gly[αC], 5-d-Gly[αC], 7-h-Gly[αC] is similar. After proton transfer from the N-terminal amine to the ketone function, the N-terminal amine cyclizes to a six-membered ring (Scheme 8, reaction 68). The subsequent proton transfer reactions allowing for the description of the fragmentation profile of the y2 ions (Figure 7) are detailed in the Supplementary Material, in section S6.
iii) Acetylation of Cys[αC] or Cys[βC]
The products 5-h-Cys[αC] and 5-h-Cys[βC] are isobaric to the product 5-h-Gly[αC], the products 5-d-Cys[αC] and 5-d-Cys[βC] are isobaric to the product 5-d-Gly[αC], and the products 7-h-Cys[αC] and 7-h-Cys[βC] are isobaric to the product 7-h-Gly[αC]. Thus, during the fragmentation of the y2 ions of products 5-h-Gly[αC], 5-d-Gly[αC] and 7-h-Gly[αC] some of the fragment ions (Fig. 7, A, B, C) could be related to the fragmentation of the y2 ions of products 5-h-Cys[αC], 5-h-Cys[βC], 5-d-Cys[αC], 5-d-Cys[βC], 7-h-Cys[αC] and 7-h-Cys[βC].
In the following, we demonstrate that the acetylation of the Cys[αC] or Cys[βC] positions are less likely.
a) The acetylation of Cys[αC]
The fragmentation mechanism of the hypothetical y2 ions of products 5-h-Cys[αC], 5-d-Cys[αC], and 7-h-Cys[αC] can start with a cyclization between the thiol and the ketone as describe in reaction 81 (Scheme S2). Such cyclization allows for the loss of a first molecule of H2O (Scheme S2, reactions 82–83) and the formation of the ions with m/z 203.0, 206.0 and 206.0 (Fig. 7, A, B, C). Subsequently, the loss of 12CO (MW = 28 Da) or 13CO (MW = 29 Da) through reaction 84 (Scheme S2), and the loss of the imine group (Scheme S2, reaction 85) could yield ions with m/z 175.0, 178.0, and 177.0, and 146.0, 149.0, and 146.0, respectively. However, such combinations of ions are not observed. Other potential cyclizations were tested but none of them could explain the entire fragmentation pattern presented in Figure 7 (A, B, C).
b) The acetylation of Cys[βC]
The acetylation of Cys[βC] appears sterically more probable than the acetylation of Cys[αC]. In order to match as many fragments ions as possible observed during the fragmentation of the hypothetical y2 ions of products 5-h-Cys[βC], 5-d-Cys[βC], and 7-h-Cys[βC], the fragmentation mechanism must start with a cyclization between the nitrogen of the amide bond and the ketone as describe in reaction 87 (Scheme S3). The mobile proton on the nitrogen can be used to protonate the alcohol function to allow the loss of the first molecule of H2O (Scheme S3, reactions 88–89) and the formation of a new set of ions with m/z 203.0, 206.0 and 206.0 (Figure 7, A, B, C). The carbocation resulting from the loss of the first molecule of H2O, can be used to cyclize the primary amine (Scheme S2, reaction 90). Such cyclization would permit the formation of a mobile proton that could be used to protonate the carboxylic acid function to allow the loss of the second molecule of H2O (Scheme S3, reactions 91–92). The second loss of H2O permits the formation of a set of ions with m/z 185.0, 188.0, and 188.0, as observed in Figure 7 (A, B, C). The subsequent loss of 12CO (MW = 28 Da) or 13CO (MW = 29 Da) (Scheme S3, reaction 93) can explain the formation of the set of ions with m/z 157.0, 160.0, and 159.0, respectively. However, we cannot rationalize further fragmentations from these set of ions to yield new fragments with m/z 128.0, 131.0, and 128.0 and m/z 100.0, 103.0, and 100.0, respectively. These latter ions can only be rationalized by acetylation on Gly[αC] (See above, Scheme 8).
iv) Acetylation of Cys[αC], Cys[βC] or Gly[αC]: Ab initio calculation of the Gibbs energies of the y2 ions of products 5-h-Cys[αC], 5-h-Cys[βC], 5-h-Gly[αC]
The recombination of the acetyl radical (CH3C•O) with a carbon-centered radical on Cys[αC], Cys[βC] or Gly[αC] leads to the formation of new chiral carbons. During mass spectrometry analysis, the formation of the y2 fragment ions of 5-h-Cys[αC], 5-h-Cys[βC] and 5-h-Gly[αC] generates cyclic ions which lead to further fragmentation. Our analysis of MS3 and MS4 spectra show that the only way to fully rationalize the formation of all fragment ions of the y2 ions is to start the fragmentation from the six-membered ring generated from y2 of 5-h-Gly[αC] (Table 5, Scheme 8). Our calculations show that for all the combinations of absolute configurations of the cyclic y2 ions presented in Table 5, the most stable configuration under vacuum is the hexacyclic ion obtained from product 5-h-Gly[αC] (if the absolute configuration is (S,R,R)). Thus, these calculations provide additional support for the formation of such ions in the gas phase and support our proposed fragmentation mechanisms suggesting the acetylation of Gly[αC].
Table 5.
Structure of the y2 ions of products 5-h-Cys[αC], 5-h-Cys[βC], 5-h-Gly[αC]. (S) and (R) stand for the absolute configuration chirality of the carbon center.
| Product | y2 ion after cyclization | Absolute Configuration |
G0298 (kcal/mol) |
|---|---|---|---|
| 5-h-Cys[αC] | 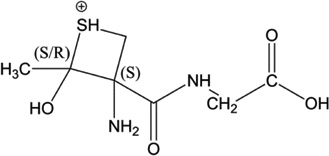 |
(S,S) (S,R) |
No convergence |
| 5-h-Cys[βC] | 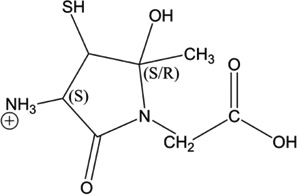 |
(S,S) (S,R) |
29.49 15.69 |
|
5-h-Gly[αC] Gly[αC] has a S configuration after addition of the acetyl radical |
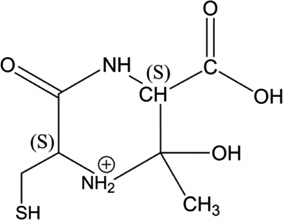 |
(S,S,S) (S,S,R) |
1.88 1.25 |
|
5-h-Gly[αC] Gly[αC] has a R configuration after addition of the acetyl radical |
 |
(S,R,S) (S,R,R) |
3.14 0.00 |
3.6 Formation of dehydroalanine during the reaction of GSH with AAPH
The exposure of GSH to AAPH generates a glutathione-thioether (GSG, m/z 581.2), corresponding to the formal loss of one sulfur atom from GSSG. The isotopic distribution is given in the Supplementary Material (Figure S1). The formation of the thioether is rationalized by the transformation of Cys into dehydroalanine, followed by the addition of the thiol of a second molecule of GSH (Scheme S4). The transformation of Cys into dehydroalanine (Dha) results from a 1,3-H-transfer between the sulfur-centered radical and the αC-H bond of Cys.32 The formation of such product further supports the formation of a carbon-centered radical through intramolecular hydrogen transfer reactions of GS•.
4. Discussion
Earlier experimental13,17,18 and computational24 studies supported an intramolecular hydrogen transfer reaction between the thiyl radical GS• and the αC-H bond of the γ-glutamyl moiety of glutathione. The bond dissociation enthalpies (BDE) of the different αC-H bonds in GSH, based on the method of isodesmic reactions, decrease in the following order: αC-H (Cys): 346 kJ mol−1, αC-H (Gly): 344 kJ mol− 1, and αC-H (Gln): 320 kJ mol−1.24,39 The low BDE of αC-H (Gln) is consistent with the experimental observation of the formation of an αC-centered radical at the Gln moiety.18 On the other hand, the calculated free energy of activation for the H-atom transfer reaction between GS• and the αC-H bond at Gly is hindered by a barrier of 134 kJ mol−1.24
Here, we provide evidence for additional intramolecular hydrogen transfer reactions in GS•, namely reversible hydrogen transfer between the thiyl radical in GS• and (i) the C-terminal Gly (or Gly*) residue, and (ii) the αC-H and βC-H bonds in Cys itself. The latter reactions correspond to 1,3- and 1,2-H atom transfers of the Cys thiyl radical, CysS•, in GS•. We hypothesize that the formation of the C-centered radicals are the results of reversible hydrogen atom transfer reactions between GS• and C-H bonds present in GSH. Recently, Nauser et al.26 have provided pulse radiolysis data supporting a 1,2- and 1,3-H atom transfer reactions in thiyl radical of model compounds. These reactions are most likely solvent-assisted. A proton-coupled electron transfer (PCET) is, therefore, a possibility, but in this paper we cannot experimentally distinguish PCET from hydrogen atom transfer (HAT).
A brief statement to exclude any intermolecular H-atom abstraction reactions between C-H bonds, in GSH, and the C-centered radicals or GS•, is required.
For intermolecular H-atom transfer reactions, the rate constants k94 and k−94 are estimated to be k94 = 3 × 103 – 3 × 104 M−1s−1,7,9,11 and k−94 = 107 – 108 M−1s−1,40 respectively.
| (94) |
Thus, under our conditions, the pseudo-first order rate constant for an intermolecular H-atom abstraction reaction between GS• and a C-H bond of GSH (k94) would be on the order of 2.3 - 23 s−1. In comparison, the rate constant for an intramolecular 1,2-H atom transfer within CysS• is several orders of magnitude faster (ca. 105 s−1).26 Therefore, an intermolecular hydrogen abstraction reaction between GS• and any C-H bond should be kinetically negligible. The C-centered radicals generated from the thermolysis of AAPH should react with the thiol of GSH (k−94) with a pseudo-first order rate constant of ca. 5 × 103 – 8 × 104 s−1. Moreover, the rate constants for H-atom abstraction reactions between methyl radicals and an S-H bond (in a thiol) and a C-H bond in glycine (or glycine anhydride) are 4.0–7.4 × 107 M−1 s−1 and 1.2 × 102 - 4 × 104 M−1 s−1, respectively, i.e. different by about three orders of magnitude.38,41,42 Assuming that AAPH-derived carbon-centered radicals, AAPH[C•], react with C-H bonds and the S-H bond of GSH with rate constants on the order of 4 × 104 and 4 × 107 M−1s−1, we can calculate the expected yield of deuterium incorporation only through an initial reaction of AAPH[C•] with the GSH C-H bonds. At 37°C, AAPH[C•] radicals are generated with a rate of AAPH[C•] = 1.36 × 10−6 [AAPH] (Ms−1).43 Hence, a one hour exposure of 2 mM AAPH yields ca. 10 µM AAPH[C•], of which ca. 0.1% are expected to react with the C-H bonds in GSH. However, over one hour ca. 40% of GSH at an initial concentration of 750 µM incorporated deuterium (Figure 1), i.e. a significantly higher yield compared to that expected if deuterium incorporation were initiated via reaction of AAPH[C•] with the C-H bonds of GSH. In fact, the yield is significantly higher than the initial yields of AAPH[C•], pointing to a chain reaction, which may be propagated through the reaction of a thiol radical with thiol, transferring the thiyl radical from an already deuterated GSH to a non-deuterated GSH. The reaction of C-centered radicals with the C-H bonds can therefore be neglected. We also discussed, and demonstrated experimentally, that neither H3C• nor CH3C•O radicals, resulting from the photolysis of acetone, could efficiently abstract hydrogen atoms from αC-H bonds in a model peptide, which did not contain Cys:29 experimentally, we could neither observe product formation nor deuterium incorporation during the photolysis of hexaglycine in the presence of acetone.29 Thus, the C-centered radicals resulting from i) the thermolysis of AAPH (AAPH[C•]), or ii) the photolysis of acetone (H3C•, CH3C•O) are more likely to react with the thiol moiety of GSH than with any C-H bond in GSH. We can, therefore, consider that any intermolecular hydrogen atom abstraction reaction either between GS• and a C-H bond or AAPH[C•] and a C-H bond would be largely negligible. Moreover, when GSSG was photolyzed, we observed deuterium incorporation into the final product, GSH-NEM. Especially for low turnovers (0.1<[GSH]/[GSSG]<0.3) an intermolecular H-atom transfer from the product GSH is kinetically unfavorable compared to that from the reactant GSSG, which is initially present at much higher concentration. Nauser et al.26 also showed specifically for the 1,2-H atom transfer in thiyl radicals from cysteamine, that the reaction is concentration-independent, i.e intramolecular. All these experimental observations and kinetic data support the notion that intramolecular hydrogen atom transfer reactions are more favorable than intermolecular reactions under our experimental conditions.
The formation of a C-centered radical at Gly[αC] was demonstrated through the observation of deuterium incorporation at the Gly[αC] position, but also through the recombination of CH3C•O with Gly[αC•]. Indeed, when GS• radicals were generated through photolysis of acetone-containing solutions of GSH, recombination products of CH3C•O with radicals from GSH were detected. Specifically the recombination products 5-h-Gly[αC], 5-d-Gly[αC] and 7-h-Gly[αC] provide evidence for the recombination of CH3C•O with a carbon-centered radical of the Gly/Gly* residue, generated through intramolecular hydrogen abstraction from Gly/Gly* by the thiyl radical of GS•.
The occurrence of 1,2- and 1,3-H atom transfers in Cys is specifically evident from the presence of up to three deuterons in the y2 fragments of products 2-d-X and 3-d-X as a result of GS• radical formation in D2O. Here, MS3 fragmentation of the y2 fragment of product 2-d-X clearly provides evidence for the incorporation of up to a total of two deuterons into the original C-H bonds of αC and βC of the Cys residue. Based on the structure of the Cys residue, these results demonstrate that at least one deuteron must be incorporated into the βC-H bond. The MS3 fragmentation of the y2 fragment of product 3-d-X provides additional evidence for the incorporation of deuterium into the Cys residue.
Specifically the 1,2-H atom transfer, depicted in reaction 56 in Scheme 6, leads to an α-mercaptoalkyl radical, R-C•H-SH. Based on theoretical calculations for 1,2-H atom transfer equilibria of small organic radicals, such as CH3S• 44 and HOCH2CH2S•,45 we can expect that equilibrium 56 is located far on the side of the thiyl radical. For Cys with a polypeptide sequence, theoretical calculations provide homolytic bond dissociation energies at 298 K of 380.9 and 367.0 kJ/mol for the βC-H and the S-H bonds, respectively, corresponding to a difference of ca. 13.9 kJ/mol.46 These calculations suggest that in equilibrium 56, ca. 0.4% of the radicals may be present as α-mercaptoalkyl radicals. On the other hand, recent pulse radiolysis experiments on thiyl radicals from Cys and related model compounds suggest, that in aqueous solution a significant fraction of these thiyl radicals undergo a 1,2-H atom transfer.26 Generally, activation energies for 1,2-H- and 1,3-H-shifts are high.47 On the other hand, detailed mechanistic studies with alkoxyl radicals (RCH2O• → RC•HOH) demonstrate that 1,2-H atom transfer reactions may be assisted by protic solvents, suggesting that solvent effects may also play a role in 1,2-H atom transfer reactions of thiyl radicals.48 Calculations for Cys αC-H and S-H bonds at 298 K provide homolytic bond dissociation energies of 345.5 and 367 kJ/mol, clearly demonstrating that a 1,3-H atom transfer should be thermodynamically feasible.
Based on a homolytic bond dissociation energy of 344 kJ/mol for Gly αC-H,39 also the hydrogen transfer from Gly to the thiyl radical of GS• appears thermodynamically feasible. However, conformational restraints must be taken into account. A very high barrier of 134 kJ/mol was calculated for the (Z)-conformation of GS•.24 However, reversible 1,3-H atom transfer within GS• may convert the original L-Cys into D-Cys (such conversion was recently observed for αC radicals of Ala in small model peptides29), suggesting that additional conformations of GS• may have to be considered for hydrogen transfer. Importantly, reversible hydrogen transfer reactions between Cys thiyl radicals and adjacent Gly residues, in positions n+1 and n+2, have been observed for a series of model peptides.28
The use of MSn experiments for the identification of deuterated sites
The important features used to identify the positions of covalently incorporated deuterons during the MSn experiments are not the structure of the different intermediates, but the nature of the neutral losses. This is, the comparison of the different changes of masses observed for each of the MSn experiments, which allowed to determine when deuterons were removed from the ion, and, therefore, included in the neutral loss. The structures of the ions presented in the different Schemes were designed to explain as clearly as possible how a specific neutral loss converts an ion into a successor ion. We understand that some of these structures might be speculative in the absence of calculations. Even though the structures displayed in Schemes 4 and 5 are not based on calculations, they are the result of logical deduction from the comparison of multiple experimental results (MSn spectra). In an effort to rationalize these structures, we have performed representative calculations to justify the formation of the cyclic structure I1 (Schemes 4 and 5). The results of the calculations are presented in the Supplementary Material (Scheme S1). The results show that the intermediate I1 is about 23 kcal/mol more stable than the b2 ion and can be used for further fragmentation as described in Schemes 4 and 5. Moreover, the calculations indicate that the fragmentation pathways in Scheme 5 are likely to be more favorable than the ones given in Scheme 4, since the ion resulting from I1 after the loss of the ketene is more stable than the ion obtained after the loss of H2O (Scheme S1).
The observed hydrogen transfer reactions are of biological significance. First, the conversion of GS• into carbon-centered radicals may provide the possibility for an irreversible formation of peroxyl radicals through reaction with molecular oxygen.49 Second, especially after the 1,3-H atom transfer of GS•, the resulting αC• radical at the Cys residue can undergo β-elimination of HS• to yield dehydroalanine. This reaction has been characterized in some detail for thiyl radicals of small Cys-containing model peptides.32 In accordance with such reaction, we detected the formation of thioether-linked glutathione dimers after the exposure of GSH to AAPH (see Section 3.6). These thioether-linked dimers are likely the result of dehydroalanine formation followed by a Michael-type addition of a second GSH molecule. In vivo, dehydroalanine-containing products of GSH cause irreversible glutathiolation of proteins such as observed during cataract formation in the human lens.50
5. Conclusion
A detailed mass spectrometry study of the fragment ions of deuterated and derivatized GSH allows the detection of intramolecular H-atom transfer reactions between GS• and the αC-H bond at Gly. Additionally, covalent H/D exchange, within Cys provides evidence for a 1,2- and 1,3-H atom transfers. The latter is of particular interest, since subsequent β-elimination from the Cα-centered radical at Cys can generate DHA. The transformation of Cys into DHA in GSH is a potential source of thioether crosslinks in proteins.
Supplementary Material
Acknowledgments
Funding Support
We gratefully acknowledge financial support from the NIH (P01AG12993).
Footnotes
Abbreviations: NEM: N-ethylmaleimide; IOA: Iodoacetamide; AAPH: 2,2′-azobis(2-methylpropionamidine) dihydrochloride; GSH*: Reduced-glutathione, where Gly residue is labeled with 15N and 13C at carbons C1 and C2.
MSn fragmentation (n=1,2,3,4): An ion trap mass spectrometer equipped with multiple quadrupoles can perform multiple stage mass spectrometry (MSn). Therefore, this symbol refers to multi-stage MS/MS experiments designed to record product ion spectra where n is the number of product ion stages. The latter is so-called, progeny ion, and corresponds to a charged product of a series of consecutive reactions that includes product ions, at the 1st generation, 2nd generation and etc… According to the sequential fragmentation: M1+ → M2+ → M3+ → M4+ → M5+, M4+ is the precursor ion of M5+, a 1st generation product ion of M3+, but also a 2nd generation product ion of M2+ and a 3rd generation product ion of M1+. Thus, the fragmentation of the M4+ ion is obtained at the MS4 stage.
Associated content
Supporting Information. Sections S1-S6, describing MS3 and MS4 fragmentation pathways, DFT calculations of the fragmentation pathway of intermediate I1, Schemes S1-S4, and Figure S1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fujii J, Ito J, Zhang XH, Kurahashi T. Unveiling the roles of the glutathione redox system in vivo by analyzing genetically modified mice. J Clin Biochem Nutr. 2011;49:70–78. doi: 10.3164/jcbn.10-138SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free radical biology & medicine. 2011;51:299–313. doi: 10.1016/j.freeradbiomed.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oakley A. Glutathione transferases: a structural perspective. Drug Metab Rev. 2011;43:138–151. doi: 10.3109/03602532.2011.558093. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert HF. Thiol/Disulfide Exchange Equilibria and Disulfide Bond Stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 5.Ross D, Albano E, Nilsson U, Moldeus P. Thiyl Radicals - Formation during Peroxidase-Catalyzed Metabolism of Acetaminophen in the Presence of Thiols. Biochem Biophys Res Commun. 1984;125:109–115. doi: 10.1016/s0006-291x(84)80341-1. [DOI] [PubMed] [Google Scholar]
- 6.Von Sonntag C. The chemical basis of radiation biology. London: Taylor & Francis; 1987. [Google Scholar]
- 7.Akhlaq MS, Schuchmann HP, von Sonntag C. The reverse of the 'repair' reaction of thiols: H-abstraction at carbon by thiyl radicals. Int J Radiat Biol. 1987;51:91–102. doi: 10.1080/09553008714550531. [DOI] [PubMed] [Google Scholar]
- 8.Schöneich C, Bonifačić M, Asmus K. Reversible H-atom abstraction from alcohols by thiyl radicals: determination of absolute rate constants by pulse radiolysis. Free Radic. Res. Commun. 1989;6:393–405. doi: 10.3109/10715768909087923. [DOI] [PubMed] [Google Scholar]
- 9.Schöneich C, Asmus K, Bonifačić M. Determination of absolute rate constants for the reversible hydrogen-atom transfer between thiyl radicals and alcohols or ethers. J. Chem. Soc. Faraday Trans. 1995;91:1923–1930. [Google Scholar]
- 10.Roberts BP. Understanding the rates of hydrogen-atom abstraction reactions: empirical, semi-empirical and ab initio approaches. J. Chem. Soc. Perkin Trans. 1996;2:2719–2725. [Google Scholar]
- 11.Pogocki D, Schöneich C. Thiyl radicals abstract hydrogen atoms from carbohydrates: reactivity and selectivity. Free Radic. Biol. Med. 2001;31:98–107. doi: 10.1016/s0891-5849(01)00559-7. [DOI] [PubMed] [Google Scholar]
- 12.Schöneich C, Asmus KD, Dillinger U, Von Bruchhausen F. Thiyl Radical Attack on Poly-Unsaturated Fatty-Acids - a Possible Route to Lipid-Peroxidation. Biochem Biophys Res Commun. 1989;161:113–120. doi: 10.1016/0006-291x(89)91568-4. [DOI] [PubMed] [Google Scholar]
- 13.Zhao R, Lind J, Merényi G, Eriksen TE. Kinetics of One-Electron Oxidation of Thiols and Hydrogen Abstraction by Thiyl Radicals from α–Amino C-H Bonds. J. Am. Chem. Soc. 1994;116:12010–12015. [Google Scholar]
- 14.Nauser T, Schöneich C. Thiyl radicals abstract hydrogen atoms from the αC-H bonds in model peptides: absolute rate constants and effect of amino acid structure. J. Am. Chem. Soc. 2003;125:2042–2043. doi: 10.1021/ja0293599. [DOI] [PubMed] [Google Scholar]
- 15.Nauser T, Casi G, Koppenol W, Schöneich C. Reversible Intramolecular Hydrogen Transfer between Cysteine Thiyl Radicals and Glycine and Alanine in Model Peptides: Absolute Rate Constants Derived from Pulse Radiolysis and Laser Flash Photolysis. J. Phys. Chem. B. 2008;112:15034–15044. doi: 10.1021/jp805133u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raffy Q, Buisson DA, Cintrat JC, Rousseau B, Pin S, Renault JP. Carbon-centered radicals can transfer hydrogen atoms between amino acid side chains. Angew Chem Int Ed Engl. 2012;51:2960–2963. doi: 10.1002/anie.201108856. [DOI] [PubMed] [Google Scholar]
- 17.Grierson L, Hildenbrand K, Bothe E. Intramolecular transformation reaction of the glutathione thiyl radical into a non-sulphur-centred radical: a pulse-radiolysis and EPR study. International journal of radiation biology and related studies in physics, chemistry, and medicine. 1992;62:265–277. doi: 10.1080/09553009214552111. [DOI] [PubMed] [Google Scholar]
- 18.Zhao R, Lind J, Merényi G, Eriksen TE. Significance of the intramolecular transformation of glutathione thiyl radicals to α-aminoalkyl radicals. Thermochemical and biological implications. J. Chem. Soc. Perkin Trans. 1997;2:569–574. [Google Scholar]
- 19.Fessenden RW, Neta P. Electron Spin Resonance Study of Radicals Produced in Irradiated Aqueous Solutions of Thiols. J . Phys. Chem. 1971;75:2277–2283. doi: 10.1021/j100676a003. [DOI] [PubMed] [Google Scholar]
- 20.Karoui H, Hogg N, Frejaville C, Tordo P, Kalyanaraman B. Characterization of sulfur-centered radical intermediates formed during the oxidation of thiols and sulfite by peroxynitrite - ESR-SPIN trapping and oxygen uptake studies. The Journal of biological chemistry. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 21.Hofstetter D, Nauser T, Koppenol WH. Hydrogen Exchange Equilibria in Glutathione Radicals: Rate Constants. Chem. Res. Toxicol. 2010;23:1596–1600. doi: 10.1021/tx100185k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volman DH, Wolstenholme J, Hadley SG. Photochemical formation of free radical from hydrogen sulfide, mercaptans, and cysteine. J. Phys. Chem. 1967;71:1798–1802. doi: 10.1021/j100865a037. [DOI] [PubMed] [Google Scholar]
- 23.Skelton J, Adam FC. The Photolysis and Radiolysis of Simple Mercaptans in Dilute Glassy Matrices. Can. J. Chem. 1971;49:3536–3543. [Google Scholar]
- 24.Rauk A, Armstrong DA, Berges J. Glutathione radical: intramolecular H abstraction by thiyl radical. Can. J. Chem. 2001;79:405–417. [Google Scholar]
- 25.Osburn S, Berden G, Oomens J, O’Hair RAJ, Ryzhov V. Structure and Reactivity of the N-Acetyl-Cysteine Radical Cation and Anion: Does Radical Migration Occur? J. Am. Soc. Mass Spectrom. 2011;22:1794–1803. doi: 10.1007/s13361-011-0198-5. [DOI] [PubMed] [Google Scholar]
- 26.Nauser T, Koppenol W, Schöneich C. Reversible Hydrogen Transfer Reactions in Thiyl Radicals From Cysteine and Related Molecules: Absolute Kinetics and Equilibrium Constants Determined by Pulse Radiolysis. The Journal of Physical Chemistry. B. 2012 doi: 10.1021/jp210954v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mozziconacci O, Williams T, Kerwin B, Schöneich C. Reversible intramolecular hydrogen transfer between protein cysteine thiyl radicals and αC-H bonds in insulin: control of selectivity by secondary structure. J. Phys. Chem. B. 2008;112:15921–15932. doi: 10.1021/jp8066519. [DOI] [PubMed] [Google Scholar]
- 28.Mozziconacci O, Sharov V, Williams TD, Kerwin BA, Schöneich C. Peptide cysteine thiyl radicals abstract hydrogen atoms from surrounding amino acids: the photolysis of a cystine containing model peptide. J. Phys. Chem. B. 2008;112:9250–9257. doi: 10.1021/jp801753d. [DOI] [PubMed] [Google Scholar]
- 29.Mozziconacci O, Kerwin BA, Schöneich C. Reversible hydrogen transfer between cysteine thiyl radical and glycine and alanine in model peptides: covalent H/D exchange, radical-radical reactions, and L- to D-Ala conversion. J. Phys. Chem. B. 2010;114:6751–6762. doi: 10.1021/jp101508b. [DOI] [PubMed] [Google Scholar]
- 30.Mozziconacci O, Kerwin B, Schöneich C. Photolysis of an intrachain peptide disulfide bond: primary and secondary processes, formation of H2S, and hydrogen transfer reactions. J. Phys. Chem. B. 2010;114:3668–3688. doi: 10.1021/jp910789x. [DOI] [PubMed] [Google Scholar]
- 31.Mozziconacci O, Haywood J, Gorman EM, Munson E, Schöneich C. Photolysis of Recombinant Human Insulin in the Solid State: Formation of a Dithiohemiacetal Product at the C-Terminal Disulfide Bond. Pharm. Res. 2012;29:121–133. doi: 10.1007/s11095-011-0519-1. [DOI] [PubMed] [Google Scholar]
- 32.Mozziconacci O, Kerwin BA, Schöneich C. Reversible Hydrogen Transfer Reactions of Cysteine Thiyl Radicals in Peptides: the Conversion of Cysteine into Dehydroalanine and Alanine, and of Alanine into Dehydroalanine. J. Phys. Chem. B. 2011;115:12287–12305. doi: 10.1021/jp2070453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Younis IR, Elliott M, Peer CJ, Cooper AJL, Pinto JT, Konat GW, Kraszpulski M, Petros WP, Callery PS. Dehydroalanine Analog of Glutathione: An Electrophilic Busulfan Metabolite That Binds to Human Glutathione S-Transferase A1-1. J. Pharmacol. Exp. Ther. 2008;327:770–776. doi: 10.1124/jpet.108.142208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikehata K, Duzhak TG, Galeva NA, Ji T, Koen YM, Hanzlik RP. Protein targets of reactive metabolites of thiobenzamide in rat liver in vivo. Chem. Res. Toxicol. 2008;21:1432–1442. doi: 10.1021/tx800093k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fung YM, Kjeldsen F, Silivra OA, Chan TW, Zubarev RA. Facile disulfide bond cleavage in gaseous peptide and protein cations by ultraviolet photodissociation at 157 nm. Angewandte Chemie (International ed. 2005;44:6399–6403. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]
- 36.Frisch MJTGW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Wallingford CT: Gaussian, Inc; 2004. [Google Scholar]
- 37.Gandini A, Hackett PA. Electronic relaxation processes in acetone and 1,1,1-trifluoroacetone vapor and the gas phase recombination of the actyl radical at 22°C. J. Am. Chem. Soc. 1977;99:6195–6205. [Google Scholar]
- 38.Huston P, Espenson JH, Bakac A. Kinetics of formation and reactions of thiyl radicals in aqueous solution. Inorg. Chem. 1992;31:720–722. [Google Scholar]
- 39.Rauk A, Yu D, Taylor J, Shustov GV, Block DA, Armstrong DA. Effects of structure on alpha C-H bond enthalpies of amino acid residues: relevance to H transfers in enzyme mechanisms and in protein oxidation. Biochemistry. 1999;38:9089–9096. doi: 10.1021/bi990249x. [DOI] [PubMed] [Google Scholar]
- 40.von Sonntag C. Sulfur-Centered Reactive Intermediates in Chemistry and Biology. In: Chatgilialoglu C, Asmus K-D, editors. NATO-ASI SerA. Vol. 197. New York and London: Plenum; 1990. p. 359. [Google Scholar]
- 41.Moger G, Dobis O. The reaction of free methyl radicals with glycine in aqueous solution. Magy. Kem. Foly. 1970;76:328–332. [Google Scholar]
- 42.Reid DL, Armstrong DA, Rauk A, von Sonntag C. H-atom abstraction by thiyl radicals from peptides and cyclic dipeptides. A theoretical study of reaction rates. Phys. Chem. Chem. Phys. 2003;5:3994–3999. [Google Scholar]
- 43.Niki E. Methods in Enzymology. In: Lester Packer ANG, editor. Free radical initiators as source of water- or lipid-soluble peroxyl radicals. Vol. 186. Academic Press; 1990. pp. 100–108. [DOI] [PubMed] [Google Scholar]
- 44.Fossey J, Sorba J. HO and HS substituent effect on alkyl radicals: an ab-initio moleuclar orbital study. J. Mol. Struct. 1989;186:305–319. [Google Scholar]
- 45.Naumov S, Von Sonntag C. UV/Vis absorption spectra of alkyl-, vinyl-, aryl- and thiylperoxyl radicals and some related radicals in aqueous solution. A quantum-chemical study. J. Phys. Org. Chem. 2005;18:586–594. [Google Scholar]
- 46.Rauk A, Yu D, Armstrong DA. Oxidative Damage to and by Cysteine in Proteins: An ab Initio Study of the Radical Structures, C-H, S-H, and C-C Bond Dissociation Energies, and Transition Structures for H Abstraction by Thiyl Radicals. J. Am. Chem. Soc. 1998;120:8848–8855. [Google Scholar]
- 47.Viskolcz B, Lendvay G, Körtvélyesi T, Seres L. Intramolecular H Atom Transfer Reactions in Alkyl Radicals and the Ring Strain Energy in the Transition Structure. J. Am. Chem. Soc. 1996;118:3006–3009. [Google Scholar]
- 48.Konya KG, Paul T, Lin S, Lusztyk J, Ingold KU. Laser flash photolysis studies on the first superoxide thermal source. First direct measurements of the rates of solvent-assisted 1,2-hydrogen atom shifts and a proposed new mechanism for this unusual rearrangement. J. Am. Chem. Soc. 2000;122:7518–7527. [Google Scholar]
- 49.Davies MJ. The oxidative environment and protein damage. Biochim. Biophys. Acta. 2005;1703:93–109. doi: 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 50.Linetsky M, LeGrand RD. Glutathionylation of lens proteins through the formation of thioether bond. Cell. Biochem. 2005;272:133–144. doi: 10.1007/s11010-005-6908-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.