

Abstract
The photodissociation of disulfide bonds in model peptides containing Ala and Ala-d3 generates a series of photoproducts following the generation of a CysS• thiyl radical pair. These photoproducts include transformations of Cys to dehydroalanine (Dha) and Ala, as well as Ala to Dha. Intramolecular Michael addition of an intact Cys with a photolytically generated Dha results in the formation of cyclic thioethers. The conversion of Cys into Dha likely involves a 1,3-H-shift from the Cys αC-H bond to the thiyl radical, followed by elimination of HS•. The conversion of Dha into Ala most likely involves hydrated electrons, which are generated through the photolysis of Cys, the photoproduct of disulfide photolysis. Prior to stable product formation, CysS• radicals engage in reversible hydrogen transfer reactions with αC-H and βC-H bonds of the surrounding amino acids. Especially for the βC-H bonds of Ala such hydrogen transfer reactions are unexpected based on thermodynamic grounds; however, the replacement of deuterons in Ala-d3 by hydrogens in H2O provides strong experimental evidence for such reactions.
Keywords: alanine, cysteine, dehydroalanine, disulfide, hydrogen transfer, intramolecular, mass spectrometry, peptide, photolysis, thiol, thiyl radical, UV irradiation
1. Introduction
The biotechnology industry has seen a recent surge in the development of novel protein therapeutics (e.g., the immunoglobulins).1 The development of industrial protocols for the production and purification of proteins enables the mass production of a wide variety of immunoglobulins. However, specifically the purification processes and long-term storage can lead to chemical degradation,2 which could generate immunogenic products. Hence, a mechanistic understanding of the formation of potentially immunogenic products is mandatory. Proteins are sensitive to light-induced degradation,3–6 and proteins produced by the biotechnology industry are often exposed to UV-light during purification (UV-detectors), sterilization processes, inspection for particles, or during storage. We have recently established mechanistic details on product formation during the photodissociation of peptide disulfide bonds with light of wavelengths between 254 and 300 nm.7 Products are well rationalized by an initial formation of a pair of Cys thiyl radicals (CysS•), followed by hydrogen transfer and disproportionation processes.7,8 Specifically reversible hydrogen transfer reactions lead to intermediary carbon-centered radicals, which, at the Cα-position of alanine (Ala), cause L-Ala to D-Ala conversion.9 Such free radical pathways may ultimately lead to protein aggregation and fragmentation, two phenomena, which are frequently observed during antibody production and formulation.10–12 Moreover, these mechanisms may provide a chemical basis for adverse effects such as the potential for increased immunogenicity or loss of potency of antibody products. Several primary photoproducts observed during the photo-degradation of an antibody can be ascribed to initial CysS• radical formation.13
CysS• radicals are also engaged in a variety of biologically relevant redox,14–16 addition15,17,18 and atom abstraction reactions.19–23 Recently, the potential for hydrogen abstraction from nearby αC-H bonds in peptides7,8 and a protein24 has been recognized. We will show here that CysS• radicals will also react with the C-H bonds of amino acid side chains, for example that of Ala. Such processes are of great biological significance, because the facile generation of protein CysS• radicals may ultimately result in irreversible protein damage.25 Therefore, the chemistry of CysS• radical within proteins, especially with regard to reaction with other amino acids must be characterized. The current paper will demonstrate, using Cys and Ala containing model peptides, that i) hydrogen transfer reactions between CysS• and amino acids are not restricted to αC-H bonds, but also target amino acid side chains, ii) intermediary CysS• radicals appear to undergo a 1,3-H-shift followed by β-elimination of HS• to yield dehydroalanine, and iii) dehydroalanine is subject to photochemical reduction to Ala, mechanistically rationalizing the photochemical conversion of Cys to Ala. The formation of Ala during the photo-irradiation of cystine had been recognized several decades ago, but a satisfactory mechanism had not been postulated.26–29
2. Experimental section
2.1 Materials and Reactions
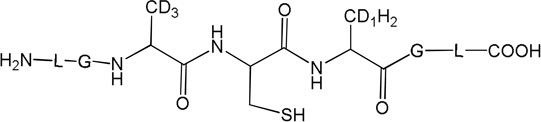
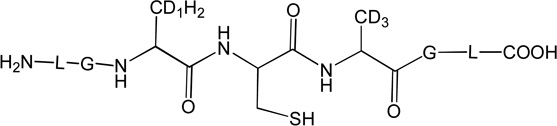
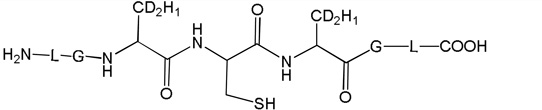
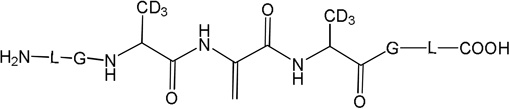
Three peptides were synthesized: (LGACAGL)2 (peptide 1a), LGACAGL (peptide 1b) and (LGAd3CAd3GL)2 (Ad3 represents a tri-deuterated Ala side chain –NH-CH(CD3)-CO-, peptide 1c) (Chart 1). The disulfide-linked peptides (LGACAGL)2 (peptide 1a), and (LGAd3CAd3GL)2 (peptide 1c) were synthesized by the Biochemical Resource Service Laboratory (BRSL) at the University of Kansas, purified to a purity level of >95% and characterized by mass spectrometry. Peptide 1b was obtained through dithiothreitol (DTT) reduction of peptide 1a, followed by HPLC purification (see below). The structures of all peptides are presented in Chart 1. Dithiothreitol (DTT), dichloromethane (CH2Cl2), tert-butanol (tert-BuOH), and nitrous oxide (N2O) were purchased from Sigma-Aldrich (St Louis, MO) at the highest available purity grade. Deuterium oxide (D2O, 99%) and tri-deuterated alanine (FMOC-Alad3 were purchased from Cambridge Isotope Laboratories (Andover, MA) at the highest purity grade. The peptides were dissolved in H2O (MilliporeQ) or in D2O at a concentration of 400 µM. Prior to UV-irradiation a 200 µL aliquot of each solution was placed in a quartz tube and saturated with Ar. The solutions were irradiated for up to 20 min with four UV lamps (Southern New England, Branford, CT, RMA-500) emitting light of λ = 253.7 nm light in a Rayonet™ photochemical reactor (Southern New England, Branford, CT).
Chart 1.

Representation of the Ala-containing model peptides.
2.2 Sample preparation for capillary LC-MS analysis
Immediately after photo-irradiation the samples were injected onto a Vydac column (25 cm x 0.5 mm C18, 3.5 µm), and eluted with a linear gradient delivered at the rate of 20 µL min−1 by a Capillary Liquid Chromatography system (Waters Corporation, Milford, MA, USA). Mobile phases consisted of water/acetonitrile/formic acid at a ratio of 99%, 1%, 0.08% (v:v:v) for solvent A and a ratio of 1%, 99%, 0.06% (v:v:v) for solvent B. The following linear gradient was set: 10–50% of solvent B within 30 min.
2.3 Nano-electrospray ionization time-of-flight mass spectrometry (ESI TOF MS) analysis
ESI-TOF-MS spectra were acquired on a Q-TOF-2 (Micromass Ltd., Manchester, U.K.) hybrid mass spectrometer operated in the MS1 mode and acquiring data with the time-of-flight analyzer. The instrument was operated for maximum resolution with all lenses optimized on the [M + 2H]2+ ion from the cyclic peptide Gramicidin S. The cone voltage was 35 eV and Ar was admitted to the collision cell at a pressure that attenuates the beam to about 20% and the cell was operated at 12 eV (maximum transmission). Spectra were acquired at 16,129 Hz pusher frequency covering the mass range 350–2,000 amu (amu = atomic mass unit) and accumulating data for 4 sec per cycle. Time to mass calibration was made with CsI cluster ions acquired under the same conditions.
2.4 MS/MS analysis
CID spectra were acquired by setting the MS1 quadrupole to transmit a precursor mass window of ±1.5 amu centered on the most abundant isotopomer. Ar was the collision gas admitted at a density that attenuates the beam to 20%; this corresponds to 16 psi on the supply regulator or 5.3 × 10−5 mbar on a penning gauge near the collision cell. The collision energy varied between 20–45 eV. Spectra were acquired for 2–3 min in 5 sec cycles as the peptides were eluted off a desalting column. The CID spectra acquired with the Q-TOF were re-analyzed by means of an LTQ-FT hybrid linear quadrupole ion trap Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (ThermoFinnigan, Bremen, Germany).30
2.5 Covalent H/D exchange and isotopic correction
The deuterium composition of peptide ions and their fragments was determined from the differences between the average mass of a covalently deuterated peptide and the average mass of the corresponding fully protonated peptide. The average masses were calculated from centroided isotopic distributions. The distribution of deuterium incorporation was obtained after isotopic correction by subtracting the isotope abundance distribution in the product formed during UV-irradiation in H2O from the isotope abundance distribution of the same product generated in D2O. This variation of the isotopic distribution between the experiments performed in deuterium oxide solution and water is given by the variation of the percent base peak intensity (%ΔBPI).
2.6 Purification of the thiol peptide LGACAGL
The thiol peptide LGACAGL (peptide 1b) was obtained after chemical reduction of the disulfide bond of peptide 1a. Peptide 1a at a concentration of 1 mM was exposed to 1.5 mM of DTT and incubated at 37 °C for 20 min. Peptide 1b was purified by chromatography on a Vydac column (25 cm x 4.5 mm, C18), monitored with a diode array detector (Shimadzu SPD-M10A), and eluted with a linear gradient delivered at the rate of 1 mL min−1 by an IPro-500 pump system (IRIS technologies, Olathe, KS). Mobile phases consisted of water/acetonitrile/TFA at a ratio of 99%, 1%, 0.08% (v:v:v) for solvent A and a ratio of 1%, 99%, 0.06% (v:v:v) for solvent B. The following linear gradient was set: 0–70% of solvent B within 20 min.
3. Results
3.1 Photo-irradiation of peptide 1a at 254 nm in H2O
3.1.1 Photo-irradiation at pH 3.5
Peptide 1a was dissolved in H2O at a concentration of 400 µM and pre-saturated with Ar prior to photo-irradiation. Solutions were photo-irradiated at 254 nm for 0, 2, 5, 10, and 20 min in quartz tubes at room temperature. The HPLC chromatograms of the irradiated solutions are presented in Figures 1A–1E. HPLC analysis reveals that UV-irradiation of peptide 1a (m/z 1205.6) generates eight major products, referred to as 2a, 2b, 2c (each with m/z 570.3), 3 (m/z 572.3), 4a, 4b (each with m/z 584.3), 5 (m/z 586.3), and 6 (m/z 604.3). Structures for all these products have been tentatively assigned by mass spectrometry (including chemical derivatization) and are listed in Table 1.
Fig. 1.

HPLC separation of the photoproducts generated after UV-irradiation at 253.7 nm of peptide 1a (400 µM) in Ar-saturated H2O solution at pH 3.5. A) non irradiated peptide, B) 2 min of irradiation, C) 5 min of irradiation, D) 10 min of irradiation, E) 20 min of irradiation.
Table 1.
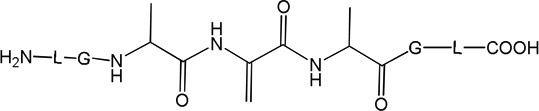
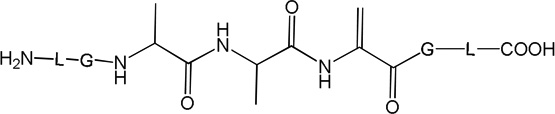
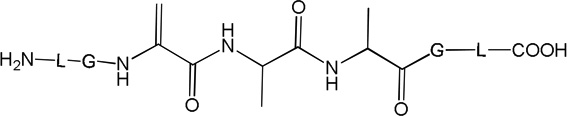
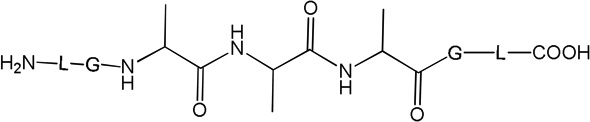
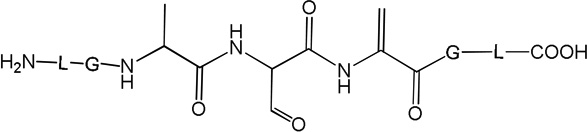
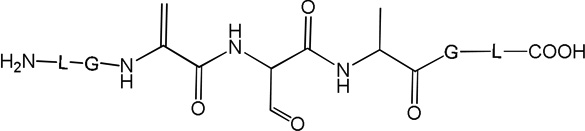
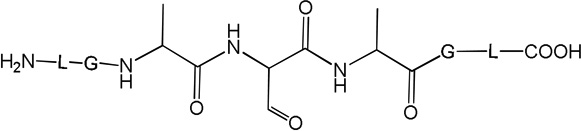
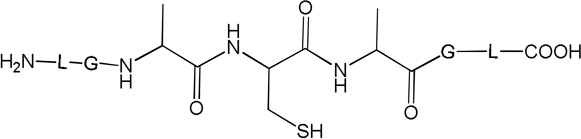
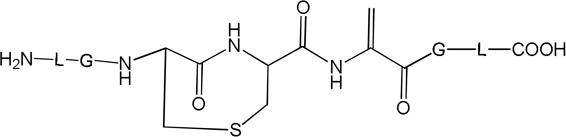
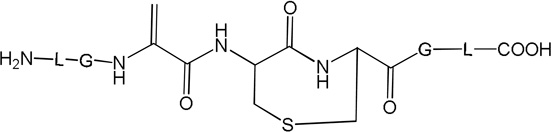
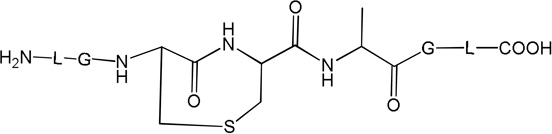
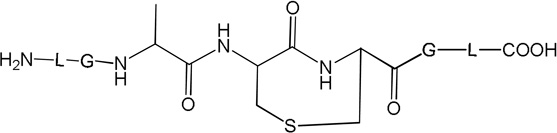
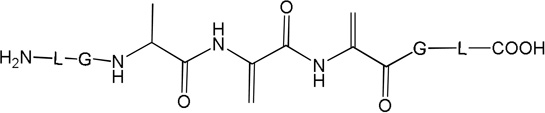
i) Product 2a (m/z 570.3)
Product 2a elutes with tel = 6.9 min (Fig. 1), co-elutes with 2b and 2c, and contains dehydroalanine (Dha) instead of Cys. The MS/MS fragmentation (Fig. S1) shows most of the b and y fragment ions expected for the structure of 2a. In particular, the b3 and b4 fragment ions demonstrate clearly the transformation of Cys into Dha. However, MS/MS fragmentation of the parent ion m/z 570.3 shows additional fragments (noted b’, b’’ and y’ in Fig. S1) which do not fit the structure of 2a. These fragments suggest the presence of isobaric products of 2a (such as 2b and 2c, see below). The time course for the formation of 2a (together with 2b and 2c) is described below. A comparison of the intensities of the b, b’ and b’’ fragments suggest that 2a represents the major product.
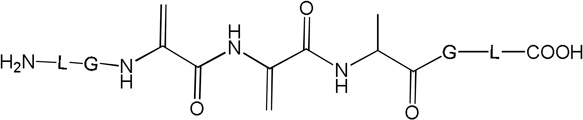
ii) Products 2b and 2c (m/z 570.3)
Products 2b and 2c are isobaric to 2a and co-elute with 2a, tel = 6.9 min (Fig. 1). This co-elution does not allow for the MS/MS fragmentation of 2b and 2c to be performed independently from that of 2a. However, specific fragments indicate the formation of 2b and 2c. For example, the b’4 and y’3 ions are characteristic for the structure of 2b (Table 1) which corresponds to the transformation of Cys into Ala and to the conversion of the C-terminal Ala into Dha. The b’’3 ion is the only direct evidence for the structure of 2c (Table 1), which corresponds to the transformation of Cys into Ala and the conversion of the N-terminal Ala into Dha. The conversion of peptide 1a into the isobaric products 2a,b,c reaches a maximum (8%, relative to the initial concentration of 1a) after 2 min of UV-irradiation (Fig. 2, ♦), followed by a slight time-dependent decrease of the yields.
Fig. 2.

Time course of the photolysis at 22 °C and pH 3.5 of peptide 1a and its transformation into 2 (m/z 570.3, ♦), 3 (m/z 572.3, ○), 4 (m/z 584.3, ■), 5 (m/z 586.3, ▲) and 6 (m/z 604.3, ●).
iii) Product 3 (m/z 572.3)
Product 3 elutes with tel = 4.9 min (Fig. 1). The b and y ions, in particular the b4 and y4 ions, displayed in the MS/MS spectrum of 3 (Fig. S2), show that in the structure of 3 (Table 1) the original Cys is converted into Ala. Product 3 evolves later than the other photoproducts, i.e. appears only after 5 min of UV-irradiation from which time point on its amount increases with the time of UV-exposure (Fig. 2, ○).
iv) Products 4a and 4b (m/z 584.3)
Products 4a and 4b are isobaric and co-elute with tel = 9.8 min (Fig. 1). The MS/MS fragmentation of products 4a and 4b is displayed in Figure S3. The b and y fragment ions indicate the transformation of the N-terminal Ala into Dha and the conversion of the thiol function into aldehyde for product 4a (Table 1). The presence of the b’3, b’4 and y’3 ions indicate the transformation of the C-terminal Ala into Dha and the conversion of the thiol function into an aldehyde for product 4b. The conversion of peptide 1a into the isobaric products 4a,b reaches a maximum (11%, relative to the initial concentration of 1a) after 2 min of UV-irradiation, followed by a slight decrease over 5–20 min of UV-irradiation (Fig. 2, ■).
v) Product 5 (m/z 586.3)
Product 5 elutes with tel = 6.4 min (Fig. 1). The b and y ions, in particular the b4 and y4 ions, displayed in the MS/MS spectrum of 5 (Fig. S4) show the conversion of the Cys thiol function into an aldehyde. The conversion of peptide 1a into 5 increases rapidly within 2 min of UV-irradiation (15%, relative to the initial concentration of 1a), followed by a slight increase over 5–20 minutes of UV-irradiation (Fig. 2, ▲).
vi) Product 6 (m/z 604.3)
Product 6 elutes with tel = 8.4 min (Fig. 1). The b and y ions displayed in the MS/MS spectrum (Fig. S5) demonstrate that 6 contains a reduced Cys instead of the disulfide bond (Table 1). The conversion of peptide 1a into 6 approximately reaches a plateau (52%, relative to the initial concentration of 1a) after 10 min of UV-irradiation. Product 6 is the major photoproduct formed over the time of UV-irradiation (Fig. 2, ●). An isobaric product of 6 elutes with tel = 11.8 min. The MS/MS fragmentation of this isobaric product is similar to the MS/MS fragmentation of 6. However, the amount of this isobaric product does not vary with the time of UV-exposure. In addition, a trace of this product is observed in the control (Fig. 1, A). These observations suggest that an impurity, structurally close to 6, is present in the sample but does not interfere with the photochemistry of peptide 1a. Product 6 is structurally equivalent to the synthetic peptide 1b.
3.1.2 Photo-irradiation at pH 7.5
The HPLC chromatograms obtained after photo-irradiation of peptide 1a at pH 7.5 at room temperature are presented in Fig. 3. The UV-irradiation of peptide 1a (m/z 1205.6) generates the same products 2a, 2b, 2c (each with m/z 570.3), 3 (m/z 572.3), 4a, 4b (m/z 584.3), 5 (m/z 586.3), and 6 (m/z 604.3) as observed at pH 3.5. In addition to these photoproducts, six other products are detected: 7a, 7b (each with m/z 600.3), 7c, 7d (each with m/z 602.3) and 8a, 8b (each with m/z 568.3). The structures of these products are also displayed in Table 1. The product yields for all products at pH 7.5 as a function of irradiation time are displayed in Figure 4.
Fig. 3.

HPLC separation of the photoproducts generated after UV-irradiation at 253.7 nm of peptide 1a (400 µM) in Ar-saturated H2O solution at pH 7.5. A) no UV-irradiation B) 2 min of irradiation, C) 5 min of irradiation, D) 10 min of irradiation, E) 20 min of irradiation.
Fig. 4.

Time course of the photolysis at 22 °C and pH 7.5 of peptide 1a and its transformation into 2 (m/z 570.3, ♦), 3 (m/z 572.3, ○), 5 (m/z 586.3, ▲) and 6 (m/z 604.3, ●), 7 (m/z 600.3, Δ), 8 (m/z 568.3, □).
i) Products 7a and 7b (m/z 600.3)
The isobaric products 7a and 7b co-elute with products 4a and 4b (tel = 9.8 min, Fig. 3). The MS/MS fragmentation of 7a, 7b suggests Michael addition between the Cys residue and either an N-terminal Dha (7a) or a C-terminal Dha (7b). Evidence for such cyclization is the lack of the couples of specific fragment ions (b3, y5) and (b’4, y’3) for the structures 7a and 7b, respectively (Table 1, Fig. S6). Both products 7a and 7b display an additional Dha residue, which is not involved in the cyclization. Evidence for the latter is provided by the characteristic fragment ions (b5, y3) and (b’3, y’5) for structures 7a and 7b, respectively, and by chemical derivatization with β-mercaptoethanol (see below, section 3.1.4).
ii) Products 7c and 7d (m/z 602.3)
Products 7c and 7d co-elute with products 4a, 4b and 7a, 7b (tel = 9.8 min, Fig. 3). The MS/MS data for 7c suggest Michael addition of the Cys residue to an N-terminal Dha while the C-terminal Ala is maintained. Product 7d is formed through Michael-addition of the Cys residue to C-terminal Dha while the N-terminal Ala is maintained. The cyclic nature of products 7c and 7d is indicated by the lack of b3, y4 and b’4 and y’3 fragments, respectively (Table 1, Fig. S7).
iii) Products 8a and 8b (m/z 568.3)
The isobaric products 8a and 8b co-elute with tel = 8.9 min (Fig. 3). The MS/MS fragmentation (Fig. S8) suggests transformation of Cys into Dha combined with the conversion of either the C-terminal or N-terminal Ala into Dha, respectively (Table 1).
3.1.3 Product formation in the presence of electron scavengers
Especially the detection of Ala and Dha as photoproducts of Cys suggests the intermediary formation of highly reducing species such as the hydrated electron (eaq−). Therefore, two experiments were designed in which any eaq− would be scavenged efficiently by an electron scavenger, CH2Cl2 or N2O, which are described below.
i) Dichloromethane
Peptide 1a was dissolved at a concentration of 400 µM in 200 µL of H2O at pH 7.5 in the presence of 0.5% (v:v; equivalent to a final concentration of 3.9 × 10−2 M) of dichloromethane (CH2Cl2). Dichloromethane was saturated with Ar separately prior to addition to the Ar-saturated aqueous solution of 1a. The sample was irradiated at 254 nm for 10 min in a quartz tube.
| (1) |
Dichloromethane is an electron scavenger (reaction 1, k1 = 6.0 × 109 M−1 s−1).31 With 3.9 × 10−2 M CH2Cl2, the pseudo first order rate constant for reaction 1, k1 = 2.3 × 108 s−1, is 53-fold greater than the pseudo first order rate constant for the reaction between the electron and the disulfide bond (reaction 2)32 present in 1a. The addition of CH2Cl2 prevents the formation of products 2b, 2c and 3 (Fig. 5, A, B) while the yields of products 8a, 8b are not altered. Product 2a is formed but its yield is greatly reduced. These results are consistent with the formation of a solvated electron during the photo-irradiation of peptide 1a, which could reduce the Dha residues present in 2b, 2c, 8a, and 8b into Ala, i.e could be responsible for the formation of 2b, 2c, and 3. Whether the reduction of Dha would be via direct reaction with e−aq, or via the reaction of a disulfide with e−aq followed by electron transfer from the intermediary disulfide radical anion, cannot be concluded here. The presence of dichloromethane prevents reaction 2. Hence, the yields of thiyl radicals via reaction 2 are reduced, causing a general reduction of the yield of 2a (see below). The absence of products 2b, 2c and 3 demonstrate that the hydrated electron is likely at the origin of transformation of the Dha into Ala. The reactivity of Dha towards hydrated electrons may be compared to that of acrylamide, where one-electron reduction of acrylamide by hydrated electrons occurs with a rate constant of (1.5–3.3) x 1010 M−1s−1.33–35 Ultimately, the one-electron reduction of acrylamide is followed by protonation of the β-carbon. An analogous mechanism for Dha would yield an alanyl αC• radical, which could be transformed into Ala by reaction with a reduced Cys residue (such as present in product 6).
| (2) |
Fig. 5.

Detection by mass spectrometry of the products 2a,b,c (m/z 570.3), 3 (m/z 572.3) and 8a,b (m/z 568.3): A) products 8a,b is observed at pH 3.5 in the absence of CH2Cl2 and at pH 8.5 in the presence of CH2Cl2. B) products 2a,b,c and 3 are observed at pH 8.5 only in the absence of CH2Cl2.
ii) N2O and tert-butanol
Peptide 1a was dissolved at a concentration of 400 µM in 200 µL of H2O at pH 7.5 in the presence of 0.1 M of tert-butanol (tert-BuOH) and the solution was saturated with N2O. The sample was irradiated at 254 nm for 10 min in a quartz tube. N2O reacts with hydrated electrons with a rate constant36 k3 = 9.6 × 109 M−1 s−1, i.e. close to the rate constant for the reaction of hydrated electrons with the disulfide bond (1.1 × 1010 M−1 s−1).32 Thus, in N2O saturated solution hydrated electrons are nearly completely converted into HO• radicals (reaction 3). To avoid the reaction of HO• radicals with the disulfide bond present in peptide 1a (k = 2.1 × 109 M−1 s−1),37 tert-BuOH was added to the solution at a concentration of 0.1 M to scavenge the HO• radicals (reaction 4). Under these conditions, the pseudo first order rate constant for reaction 3, 6.0 × 108 s−1, is 60-fold greater than the pseudo first order rate constant for the reaction between e−aq radicals and the disulfide bond present in 1a. The reaction of HO• radicals with tert-BuOH results in the formation of a •CH2C(CH3)2OH radical.38 This radical is known to abstract an H-atom from CysSH with a rate constant of 5.0 × 107 M−1 s−1, but does not react efficiently with the disulfide bond.39
| (3) |
| (4) |
HPLC chromatograms (Fig. 6) recorded after photo-irradiation of 1a in the presence of N2O and tert-BuOH reveal the absence of product 3. In the absence of N2O/tert-BuOH, 21% of 1a is converted into 2a,b,c and 8a,b during 10 min of photo-irradiation. In the presence of N2O/tert-BuOH, 79% of 1a is converted into 2a, 7a,b and 8a,b. An additional product with m/z 676.4 is observed (Fig. 6). This product is the result of the recombination of a CysS• radical of product 6 with •CH2C(CH3)2OH, formed during the photoionization of the thiolate form of 6 (see below). In conclusion, the presence of electron scavengers prevent the reduction of Dha by hydrated electrons, providing mechanistic evidence that Dha is a key intermediate in the transformation of Cys to Ala.
Fig. 6.

HPLC separation of the photoproducts generated after UV-irradiation at 253.7 nm of peptide 1a (400 µM) in N2O-saturated H2O solution at pH 7.5 in the presence of 0.1 M tert-BuOH. The presence of tert-BuOH in the sample results in the shift of the times of elution of the photoproducts.
3.1.4 Additional evidence for Dha formation: Michael addition of β-mercaptoethanol
Peptide 1a (400 µM) was irradiated at 254 nm for 10 min in Ar-saturated solution at pH 7.5. Immediately after UV-irradiation a final concentration of 100 mM of β-mercaptoethanol was added to the solution, and the pH was increased to 8.5 by the addition of NaOH. The Michael-addition of thiolate to Dha has been reported for aqueous solution,40–42 so that we expect that the thiolate form of β-mercaptoethanol reacts with the Dha residues.43,44 The derivatization of photo-products from 1a resulted in the formation of seven new species consistent with our previous structural assignment of Dha formation: 2β (m/z 648.4), 4β (m/z 662.4), 5β (m/z 664.4), 6β (m/z 680.4), 7β (m/z 678.4), 8β (m/z 724.4), and 9β (m/z 756.3). Here, reaction products derivatized with β-mercaptoethanol are indicated by the additional label “β” after the original product number. A mass spectrometric scan displaying all products is presented in Figure 7. A detailed description of the seven new structures labeled 2β–9β is given in the Supplementary Material, together with their MS/MS spectra (Figures S9–S15). Products 2β, 4β, 7β, and 8β (and their respective derivatives labeled a,b,c) result from the addition of one molecule β-mercaptoethanol to the Dha residues present in products 2, 4, 7 and 8, respectively. Product 6β results from the reaction of β-mercaptoethanol with peptide 1a. Product 9β results from the formal addition of two molecules of β-mercaptoethanol to the Cys and Ala residues of 6, respectively. A precursor for 9β, i.e. peptide 1a with one Ala substituted by Dha, was not detected in the absence of β-mercaptoethanol derivatization. Hence, we cannot explain the formation of 9β mechanistically at this point in time. Product 3 is not affected by the presence of β-mercaptoethanol because of the absence of Dha residues. Thus, the total conversion of the products 2a,b,c, 4a,b, 7a,b and 8a,b into their β-mercaptoethanol derivatives provides further evidence for the photolytic transformation of Cys and Ala into Dha.
Fig. 7.

Detection by mass spectrometry of the photoproducts generated after UV-irradiation at 253.7 nm of peptide 1a (400 µM) in Ar-saturated H2O solution at pH 7.5 and followed by the addition of 100 mM of β-mercaptoethanol. The photoproducts observed are the following: 3 (m/z 572.3), 5 (m/z 586.3), 6 (m/z 604.3), 2β (m/z 648.4), 4β (m/z 662.4), 5β (m/z 664.4), 6β (m/z 680.4), 7β (m/z 678.4), 8β (m/z 724.4), and 9β (m/z 756.3):
3.1.5 Photochemical yields as function of temperature
Disulfide photolysis generates a thiyl radical pair, which initially exists within a solvent cage prior to dissociation apart. If product formation within the cage is different from product formation outside the cage, these pathways can be distinguished through variation of the lifetime of the cage. A variation of temperature in the range of 0–25°C is sufficient to modify the cage escape rate constant of the initial radicals, i.e. the lifetime of the cage.45 The Stokes-Einstein equation for the diffusion coefficient, D = kbT / 6πηr (η = dynamic viscosity of the solvent, r = radius of the spherical particles, kb = Boltzmann constant, T = absolute temperature), shows that the diffusion is directly proportional to the temperature of the solvent and inversely proportional to the viscosity. At 4°C and 22°C, the viscosity of H2O under atmospheric pressure is η(4°C)=1.569 g m−1 s−1 and η(22°C)=0.955 g m−1 s−1. Based on the Stokes-Einstein expression, a decrease of the temperature from 22°C to 4°C results in a decrease of the diffusion coefficient by a factor of 1.75, enhancing the lifetime of the cage. Thus, to monitor the effect of the temperature, peptide 1a (400 µM) was irradiated at 254 nm for 2, 5, 10, and 20 min in Ar-saturated solution at pH 7.5 at 4°C. A 20 minutes photo-irradiation leads to products 2a, 2b, 2c, 5 and 7a, 7b. The time course for these photoproducts is presented in Fig. 8. Hence, the photochemistry of peptide 1a at 4°C is significantly different from that at 22°C. First, product 6, 3 and 4a,b are not observed at 4°C. Second, the respective yields of 2a,b,c and 5 are 2-fold and 2.5-fold times lower, respectively, at 4°C compared to room temperature (Fig.4 vs Fig. 8, A). In contrast the yields of product 7a and 7b are not affected by the temperature. A 20 minute UV-exposure of peptide 1a yields 35% photoproducts (i.e., peptides different from 1a) at 4°C, but >65% at 22°C. The absence of product 6 at 4°C, and the lower efficient photo-conversion of 1a suggest a higher probability for the initial thiyl radical pair to regenerate peptide 1a in the solvent cage. Product 5 displays an almost linear increase of its yield between temperatures of 4°C and 26°C and 5 minutes of photo-irradiation (Fig. 8, B). The presence of 5 and the absence of 6, suggest that 6 is a substrate for degradation (see also section 3.2).
Fig. 8.

A) Time course of the photolysis at 4 °C and pH 7.5 of peptide 1c and its transformation into 2 (m/z 570.3, ♦), 5 (m/z 586.3, ▲) and 7a,b (m/z 600.3, Δ). B) Transformation of peptide 1c into 5 at various temperatures after 5 min of photo-irradiation at pH 7.5.
3.2 Photo-irradiation of peptide 1b at 254 nm
Peptide 1b was photo-irradiated at 254 nm in Ar-saturated H2O or D2O in the presence and absence of 39 mM CH2Cl2 (Fig. 9). Peptide 1b is identical to product 6, but we assigned a different number here to differentiate the synthetic peptide from the reaction product.
Fig. 9.

HPLC separation of the photoproducts generated after 10 min of UV-exposure at 253.7 nm of peptide 1b (100 µM) in Ar-saturated H2O solution at pH 7.5. A) peptide 1c non irradiated and obtained after DTT reduction of peptide 1a and HPLC purification, B) peptide 1c irradiated in the absence of CH2Cl2, C) peptide 1c irradiated in the absence of CH2Cl2.
3.2.1 Photo-irradiation at pH 7.5 in H2O in the absence of CH2Cl2
A 10 min photo-irradiation of peptide 1b yields predominantly product 3 (Fig. 9, B) and a new product with m/z 1173.2, which is consistent with a thioether formed between 2a and 1b and, therefore, referred to as 2a-1b (Fig. 9D). The fragmentation pattern observed during MS/MS analysis of 2a-1b (Fig. 9E) is consistent with this structure.
3.2.2 Photo-irradiation at pH 7.5 in H2O in the presence of CH2Cl2
In the presence of 39 mM CH2Cl2, the yield of 3 is significantly reduced and product 5 as well as a new product, 10 (characterized by m/z 562.3) are formed (Fig. 9, C). The MS/MS data of product 10 reveal an addition of +48 amu to the original Cys residue of peptide 1b (data not shown). There are several possibilities which could rationalize a gain of +48 Da: i) the oxidation of 1b into sulfonic acid, ii) a radical-radical recombination of •CH2Cl with the CysS• radical of 1b, and iii) the transformation of the original thiol of 1b through formal addition of SO. Potential structures for the latter would be thiosulfinates, such as R-S(O)SH or R-SS(O)H, or RSSOH, but our MS and MS/MS data are unable to provide such structural information. The isotopic composition of 10 does not fit the theoretical isotopic distribution expected from the presence of a Cl atom in a potential recombination product between thiyl radical and •CH2Cl, and such product can therefore be excluded. The reduction with DTT to the original thiol 1b excludes the formation of sulfonic acid. Hence, a perthiol with an added oxygen remains as a possibility.
3.2.3 Photo-irradiation at pH 3.5 in H2O in the absence of CH2Cl2
In contrast to experiments at pH 7.5, photo-irradiation of peptide 1b at acidic pH did not result in photodecomposition, as expected from the protonation state of the thiol group and in accordance with earlier results.46
3.2.4 Photo-irradiation at pD 7.5 in D2O in the absence of CH2Cl2
While photo-irradiation of peptide 1b in H2O in the absence of CH2Cl2 generates 3 with m/z 572.3, photo-irradiation in D2O yields 3 with m/z centered at 573.3, indicating the covalent incorporation of deuterium into 3 (Fig. S20). The number of covalently incorporated deuterons into 3 will be discussed in Section 4.2.
3.3 Photo-irradiation of peptide 1c at 254 nm
3.3.1 Photo-irradiation in H2O at pH 3.5
Peptide 1c (400 µM) was photo-irradiated at 254 nm for 2, 5, and 10 minutes in quartz tubes. The HPLC chromatograms of the irradiated solutions are presented in Figure 10. LC-MS analysis revealed that the photo-irradiation of peptide 1c (m/z 1217.5) generates ten major products: 11 (m/z 592.7), 12 (m/z 610.7), 13a, 13b, 13c (m/z 608.7), 14 (m/z 576.7), 15a, 15b (m/z 606.7), 16 (m/z 574.7), and 17 (m/z 575.7). The tentative structures of these products were assigned through MS/MS analysis and are summarized in Table 2.
Fig. 10.

HPLC separation of the photoproducts generated after UV-irradiation at 253.7 nm of peptide 1c (400 µM) in Ar-saturated H2O solution at pH 7.5. A) no UV-irradiation B) 2 min of irradiation, C) 5 min of irradiation, D) 10 min of irradiation.
Table 2.
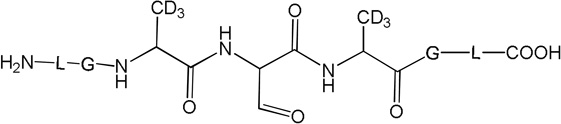
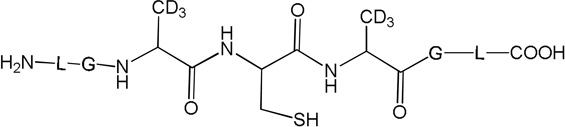
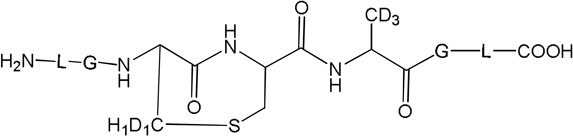
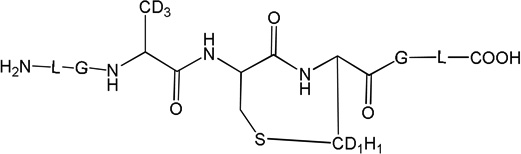
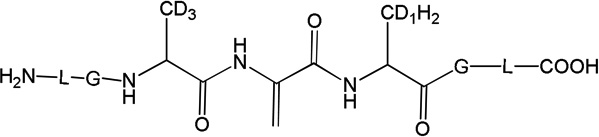
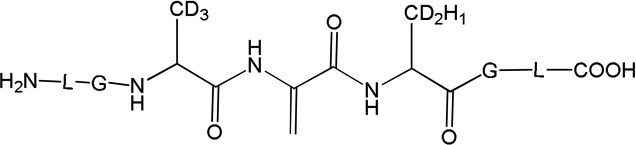
i) Product 11 (m/z 592.7)
Product 11 elutes with tel = 6.8 min (Fig. 10). The b and y ions, in particular the b4 and y4 ions, presented in the MS/MS spectrum (Fig. S16) show that the structure of 11 (Table 2) arises from the formal conversion of a peptide Cys residue into an aldehyde.
ii) Product 12 (m/z 610.7)
Product 12 elutes with tel = 8.8 min (Fig. 10). The b and y ions presented in the MS/MS spectrum (Fig. S17) demonstrate that 12 arises from the conversion of the disulfide bond of peptide 1c to free thiol (Table 2).
iii) Products 13a, 13b, and 13c (m/z 608.7)
The isobaric products 13a,b,c co-elute with tel = 8.3 min (Fig. 10). The three structures 13a, 13b and 13c are distinguished from product 12 through covalent H/D exchange at the Alad3 side chains. The MS/MS spectrum displayed in Figure 11, through the fragment ions b3, b4, y3 and y4, evidences the replacement of two deuterons of the C-terminal Alad3 by two protons (product 13a). The ions a’3, b’4, y’3 and y’4 indicate the presence of the isobaric product 13b where two deuterons of the N-terminal Alad3 are replaced by two protons. The replacement of one deuteron from each Alad3 residue by one proton (product 13c) is evidenced by the presence of the ions a’’3, a’’4, y’’3 and y’’4. A comparable H/D exchange at the βC-positions of Ala is observed for the monoisotopic masses and the MS/MS fragmentation spectra of products 14 (m/z 576.7), 16 (m/z 574.7) and 17 (m/z 575.7) (See section 3.3.2).
Fig. 11.

CID mass spectrum obtained by means of a FT-MS mass spectrometer of the isobaric products 13a,b,c (m/z 608.7) generated by UV-irradiation of an Ar-saturated aqueous solution containing peptide 1c.
iv) Product 14 (m/z 576.7)
Product 14 elutes with tel = 7.8 min (Fig. 10). Product 14 contains a Dha residue in place of Cys. The MS/MS (Fig. S16) shows most of the b and y fragments ions expected for the structure of 14. In particular the b3 and b4 fragment ions indicate the transformation of Cys into Dha.
v) Products 15a and 15b (m/z 606.7)
The isobaric products 15a and 15b co-elute with tel = 9.7 min (Fig. 10). The MS/MS of 15a,b suggests a cyclization of the Cys residue with Dha in place of either the N-terminal Ala (15a) or the C-terminal Ala (15b). The cyclic nature of these products is suggested by the lack of the fragment couples b3, and y4, and b’4, and y’3 for 15a and 15b, respectively (Table 2, Fig. S19). Internal fragments (indicated by the dashed frames) provide further support for our structural assignments.
3.3.2 The isotopic envelope of product 14 (m/z 576.7)
Theoretically, product 14 (m/z 576.7) should display a ratio between the peak areas for the first and the second isotope peaks of 27.8%. However, the experimental ratio is 51%, suggesting the presence of co-eluting products with monoisotopic masses close to m/z 576.7. In fact, the experimentally observed isotopic envelope of all products co-eluting at tel = 7.8 min can be deconvoluted into a mixture of at least three products with m/z 574.7, 575.7, 576.7 for their monoisotopic masses (Fig. 12), i.e. two additional products with Δ = −1 amu (product 17) and Δ = −2 amu (product 16), respectively, compared to product 14. The use of an LTQ-FT hybrid linear quadrupole ion trap Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer allows to measure the signal of all these ions in one experiment producing a complex frequency vs. time spectrum containing all the signals. The deconvolution of this signal by FT methods results in the deconvoluted frequency vs intensity spectrum which is then converted to the mass vs intensity spectrum. Furthermore, the FT-ICR has the capability to switch rapidly from the MS to the MS/MS mode. Thus, even if the ions are different by only 1 Da and are from co-eluting components, the signal of each ion can be extracted to obtain their MS/MS fragmentation spectra. The MS/MS spectra of products 14 (m/z 576.7), 16 (m/z 574.7) and 17 (m/z 575.7) were obtained using the FT-ICR instrument and were compared (Fig. 13). The detected fragment ions b3, and b4 are identical for all products 14, 16 and 17. However, the fragment ions b5 and y4 of the products 14, 16 and 17 are different. The mass of the fragment ion b5 increases by an increment of 1 amu from product 16 (m/z 574.7, b5 = 386.3) (Fig. 13a and 13b, the ion with m/z 387.3 is also observed), to 17 (m/z 575.7, b5 = 387.3) (Fig. 13d) and to 14 (m/z 576.7, b5 = 388.3) (Fig. 13f). An analogous observation is made for the y4 fragments. Indeed, the m/z of the fragment y4 of 17 is different by 2 amu in comparison to the y4 fragment of product 14. These observations indicate the possibility of replacement of deuterons of the C-terminal Ala in product 14 by protons from the solvent. The origin of these protons is discussed below.
Fig. 12.

Isotopic envelope of the combination of the products 14, 17 and 18 generated after 10 min of UV-exposure of peptide 1c in H2O solution. The dots represent the theoretical isotopic envelope for the mixture of the three isotopic distributions of the monoisotopic masses 574.7, 575.7 and 576.7.
Fig. 13.

Comparison of the CID mass spectrum obtained by means of a FT-MS mass spectrometer of the products 14 (m/z 576.7), 16 (m/z 574.7), and 17 (m/z 575.7) generated by UV-irradiation of an Ar-saturated aqueous solution containing peptide 1c.
3.4 Photo-irradiation of peptide 1c at 254 nm in D2O
3.4.1 Deuterium incorporation into product 12
Peptide 1c was dissolved at 400 µM concentration in Ar-saturated D2O, pD 3.5. The samples were photo-irradiated at 254 nm for 2, 5, and 10 minutes in quartz tubes. Covalent deuterium incorporation into product 12 was monitored by LC-MS (Fig. 14). The percentage of product 12 which incorporated one additional deuteron was plotted versus the time of photo-irradiation (Fig. 14, insert). The method to deconvolute the isotopic distributions presented in Fig. 14 is summarized in the Supplementary Material. A similar pattern of deuterium incorporation was observed in product 6 when peptide 1a was irradiated in D2O (data not shown).
Fig. 14.

Time courses of the variation of the isotopic envelopes of the products 12 during the UV-irradiation of peptide 1c in D2O solution. Insert: plot of the percentage of molecules of 12 having incorporated one deuteron vs the time of irradiation.
3.4.2 Deuterium incorporation into products 14, 16 and 17
In section 3.3.2, we described the isotopic distribution in product 14 after photo-irradiation in H2O. We rationalized the observed difference between the envelopes predicted for 14 only and the experiment by the presence of two additional products, products 16 and 17, corresponding to the covalent replacement of two and one deuterons by two and one protons, respectively compared to 14. The losses of 2 and 1 amu are related to the replacement of 2 and 1 deuterons by 2 and 1 protons, respectively, at the C-terminal Ala position in product 14. Consistent with this rationale, the ions with m/z 574.7 and 575.7 are not observed during photo-irradiation in D2O (where formally deuterons would be replaced by deuterons), confirming that products 16 and 17 are not formed in D2O. However, the isotopic distribution of 14 shows components of higher m/z during photo-irradiation in D2O (Fig. 15), consistent with covalent H/D exchange at the αC-positions of Ala and Gly.
Fig. 15.

Isotopic envelopes of product 14 after 10 min of UV-exposure of peptide 1c in H2O and D2O solutions. Insert: Percentage of molecules of product 14, which have incorporated one deuteron over the time of irradiation in D2O. The deconvolution protocol of the isotopic envelopes is described in the Supplementary Material.
4. Discussion
The photolysis of an interchain disulfide bond in the Ala-containing peptides 1a and 1c leads to a series of transient and stable products identified by MS/MS analysis. Notably, we detect the transformation of Cys into Dha and Ala, and of Ala into Dha, as well as covalent H/D exchange at both the αC-H and βC-H positions of Ala. The nature of the stable products is consistent with the initial generation of CysS• radicals, and CysS• radicals have been commonly detected during the UV photolysis of cystine and cysteine-containing peptides.8,47 The latter is not unexpected based on the broad absorption maximum of disulfides around 250 nm, which contains two transitions where the higher energy transition initially leads to a singlet radical pair of CysS•.48 Rapid intersystem crossing (ISC) will efficiently generate a triplet radical pair of CysS•.49
Initially, photolysis will generate a thiyl radical pair within a solvent cage, from which the radicals can diffuse into the bulk solution (Scheme 1, reaction 6). The CysS• radical pair can disproportionate to form a thiol and a thioaldehyde (Scheme 1, reaction 7), or recombine to disulfide.8,47 In H2O, the thioaldehyde is transformed into an aldehyde (Scheme 1, reaction 13). During the lifetime of the thiyl radicals, there is an opportunity for reversible hydrogen transfer reactions (reactions 9 and 10). The temperature effect on product yields appears to indicate an effect of cage lifetime on product distribution. A less efficient conversion of the native disulfide into the disproportionation products at 4°C suggests that the primary geminate radical pair recombines with a higher probability in the cage (assuming a similar activation energy for disproportionation and recombination). This appears to be also consistent with the results from photo-irradiation of 1b. The absence of a disulfide bond in 1b excludes the possibility of any solvent cage effect on the reaction of two CysS• radicals. Thus, the formation of 5 during the photo-irradiation of 1b in the presence of CH2Cl2 indicates the possibility for disproportionation of CysS• outside the cage.50,51
Scheme 1.

Reaction scheme of photolytic processes of peptides 1a and 1c.
A comment on the stereochemistry is warranted. The initial thiyl radical pair formed according to reaction 6 (Scheme 1) will contain exclusively L-amino acids. Hence, reaction 10 (hydrogen transfer from the Ala Cβ-H/D bond) would be geometrically more favorable compared to reaction 9 (hydrogen transfer from the Ala Cα-H bond). However, through equilibrium 16a in Scheme 2 (see below) the initial L-configuration of the CysS• radical may convert into the D-configuration so that reaction 9 would become geometrically more favorable. Therefore, we did not indicate any particular stereochemistry in Scheme 1.
Scheme 2.

Transformation of Cys into Ala.
4.1 The conversion of Cys into Ala
The photochemical formation of Ala from cystine has been observed as early as 4–5 decades ago26–29 but no mechanistic analysis was provided. Considering the potential importance for light-induced protein degradation, and the chemical modification of protein therapeutics such a mechanistic analysis is warranted. Figures 2 and 4 show that 1a is converted to 3 at both pH 3.5 and 7.5, though the yields are higher at pH 7.5. At both pH the formation of 3 shows a delay compared with products 2a, 2b, 2c and 6 (peptide 1b). Especially at pH 7.5, the time-dependent formation and decay of 6 suggest a potential intermediacy of 6 for the formation of 3. This is corroborated by our result obtained at 4°C, where product 6 and, 3, consequently, were not formed. However, it seems that 2a, 2b, 2c were formed at 4°C suggesting the elimination of H2S. Therefore, we prepared 6 (=1b) independently and subjected it to photolysis both at pH 3.5 and pH 7.5. As expected, no photo-conversion of 6 occurred at pH 3.5 due to the protonation state of the thiol. However, 6 converted into 3 at pH 7.5. Chemically, the conversion of 6 into 3 requires a multistep pathway, which we propose in reactions 15–20 in Scheme 2. Several steps in Scheme 2 require discussion, but prior to that additional experimental support for the reactions in Scheme 2 will be provided. First, when 6 was photo-irradiated at pH 7.5 in the presence of 39 mM CH2Cl2, no conversion of Cys into Ala (i.e. product 3) was observed. This is consistent with the efficient removal of eaq− by reaction 1 (section 3.1.3). Hence, the addition of electron scavengers to solutions of 1a inhibited the conversion of Cys to Ala in accordance with the mechanism proposed in reactions 18–20. Next, we photo-irradiated 6 at pD 7.5 in the absence of electron scavengers in order to monitor deuterium incorporation into product 3. We observed significant incorporation of deuterium into product 3 (27% of the molecules of 3, incorporated two deuterons), where MS/MS analysis indicated incorporation into Leu1, Gly2, Ala3, *Ala4, Ala5, and Gly6 (*Ala4 represents the former Cys4 residue). This result indicates the intermediary formation of •Cα radicals also at the Gly residues (and possibly at the Leu residues; however, also the possibility for side chain radicals at Leu exists). A discussion of the origin and the mechanisms leading to the incorporation of deuterons in product 3 after photo-irradiation of 6 in D2O is given below (Section 4.2).
Importantly, 3 was the predominant product during photo-irradiation of 6 in the absence of electron scavengers while products 5 and 10 were formed only in the presence of CH2Cl2. The formation of 5 indicates disproportionation of the initial thiyl radical outside the solvent cage (i.e. no thiyl radical pair in the cage is formed during photolysis of 6). In addition, 5 may also form through reactions 5 and 13:
| (5) |
| (13) |
A comparable effect of electron scavengers was observed during the photolysis of 1a. Here, the presence of 39 mM CH2Cl2 prevented the formation of products 2b, 2c and 3. The MS/MS analysis of the product with m/z = 570.3 (2a,b,c) formed after photo-irradiation of 1a in the presence of CH2Cl2, indicates that only the formation of 2b and 2c is prevented by the presence of an electron scavenger while 2a is formed, albeit at a low yield. MS/MS data show that the major fragments detected (y4 and b4) originate from product 2a while the fragments characteristic for products 2b and 2c are not observed (Fig. S21). When 1a was photo-irradiated at pH 7.5 in the presence of N2O and 0.1 M tert-BuOH, product 3 was not formed. Instead, a recombination product of the initial thiyl radical with •CH2C(CH3)2OH was detected, where •CH2C(CH3)2OH is the product of reactions 3 and 4 (section 3.1.3). Further support for reactions 16–20 is derived from the detection of product 2a during the photo-irradiation of 1a. In Scheme 2, reaction 16a represents a 1,3-H-shift converting a thiyl radical into a capto-datively stabilized αC•-radical. Normally, a 1,3-H-shift is associated with a relatively high activation energy.52–54 Nevertheless, a rate constant of 2.5 × 104 s−1 for such 1,3-H-shift has been reported for the anionic form of the thiyl radical of Cys (H2N-CH(CO2−)-CH2-S•)53,55,56, which suggests that reaction 16a may be reasonably fast despite the theoretically predicted high activation energy. To what extent such 1,3-H-shift may be facilitated by a protic solvent (such as observed for a 1,2-H-shift in alkoxyl radicals)57,58 remains to be shown. The elimination of HS• (reaction 17) is supported by the formation of H2S which has been measured during photo-irradiation of several disulfide-containing peptides.7 We propose that H2S serves as a source for the photochemical generation of H•-atoms,59 which can reduce Dha via addition to Cβ, which formally represents a combination of reactions 18 and 19 (Scheme 2). Such a reduction mechanism would rationalize the formation of product 3 also at pH 3.5.
4.2 The isotope distribution in product 3
The isotopic distribution of product 3 generated in D2O is the result of the convolution of the four isotopic envelopes of product 3 after incorporation of 0 (21%), 1 (45%), 2 (27%), and 3 (6%) deuterons (the percentage indicates the weight of each isotopic envelope in the overall isotopic distribution of 3 generated in D2O). The presence of two deuterons in 3 is rationalized by the reduction of Dha in D2O according to reactions 18–20 (Scheme 2). The incorporation of three deuterons is the result of two different mechanisms. A first deuteron is incorporated into the peptide backbone according to the reversible reaction 16b (Scheme 2). At pD 7.5, we cannot exclude that two thiyl radicals will recombine to a disulfide bond, for example via formation of an intermediary thiyl radical-thiolate complex. Therefore a fraction of the cysteine thiyl radicals may also result from the photolysis of a product disulfide bond, where the first deuteron can be incorporated after the formation of a radical pair, as described in reactions 6, 9, 11 and 12 (Scheme 1). The two additional deuterons can again be explained by the reduction of Dha into Ala (reactions 18–20, Scheme 2). The 21% of product 3 containing a single deuteron can be explained by the presence of a trace of H2O in the D2O solvent, and a potentially large kinetic isotope effect of reaction 20 (Scheme 2).41 Assuming a KIE of 7 for reaction 20,41 we calculate that a (v:v) D2O:H2O ratio of 97:3 would lead to 21% of product 3 containing one deuteron (this was separately confirmed for covalent H/D exchange reactions in thiyl radicals from glutathione using D2O spiked with known amounts of H2O; data not shown).
4.3 The conversion of Ala into Dha
The photo-irradiation of 1a (and 1c) resulted in the conversion of Ala into Dha. Importantly, such Ala to Dha conversion was not detected during photo-irradiation of the thiol-containing peptide 1b at pH 7.5. This result suggests that a radical pair (potentially in the cage) rather than a CysS• radical alone could be responsible for the conversion of Ala to Dha. We propose reactions 6, 9, 11 and 12, displayed in Scheme 1 as a basis for Dha formation and will provide an analysis of this mechanism based on experimental support in the following. In reaction 9, a thiyl radical of the initial thiyl radical pair abstracts a hydrogen from the αC-H bond of Ala, generating a αC• radical. Evidence for such a process was obtained earlier through the measurement of L-Ala to D-Ala epimerization during photo-irradiation of peptide 1a.9 In the present paper we have obtained additional evidence for the formation of the αC• radical through photo-irradiation of peptide 1c in D2O, which led to H/D exchange at the only C-H bond available in the original Alad3 residue, the αC-H bond. Reaction 9 may be followed by electron transfer (reaction 11) and proton transfer (reaction 12), generating Dha. The oxidation potentials for linear peptide •Cα radicals, such as formed in equilibrium 9, can be approximated through values obtained with •Cα radicals from diketopiperazines. For glycine and alanine anhydride, E°ox = 0.18 and 0.09 V (vs. NHE), respectively, for the reaction R1-NH+=CR2(C=O)-R3 + e− → R1-NH-•C-(C=O)-R3.60 In addition, the glycine anhydride •Cα radical, R1-NH-•C-(C=O)-R3, reduces [IrCl6]2− with k = 3.1 × 109 M−1s−1, i.e. in a diffusion-controlled process,61 where E° = 0.87 V for the process [IrCl6]2− + e− → [IrCl6]3−.62 Considering that for RS• + e− + H+ → RSH, E° = 1.33 V,63 and for RS• + e− → RS−, E° = 0.73 V,63 we expect a very fast oxidation of R1-NH-•C-(C=O)-R3 by CysS• thiyl radicals, i.e. the process displayed in reaction 11 (Scheme 1).
4.4 The formation of cyclic thioethers 7a, 7b, 7c and 7d
Dha-containing products such as formed according to reactions 6, 9, 11 and 12 in Scheme 1 will likely constitute the precursor for products 7a, 7b, 7c, and 7d. Cyclization of Cys represents an intramolecular Michael-addition of thiol to Dha. Michael-addition reactions of thiols to Dha have been shown to be efficient in water.40 A representative cyclization is displayed in reaction 21. Such Michael-addition is also consistent with our experimental results where Dha residues in products 2a, 2b, 2c, 4a, 4b, 7a, 7b, 8a, and 8b were derivatized with β-mercaptoethanol subsequent to photo-irradiation.
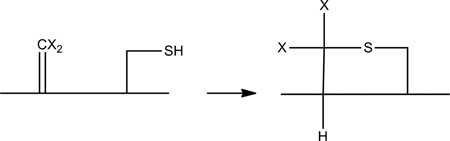 |
(21) |
4.5 The reaction of CysS• thiyl radicals with the βC-H bond of Ala
An important experimental observation is the covalent D/H exchange at the βC-D bonds originally present in peptide 1c, upon photo-irradiation of 1c in H2O. While the exchange of one deuterium by hydrogen in one Alad3 residue can theoretically be rationalized through a radical pair reaction (Scheme 1, reactions 11 and 12), the exchange of more than one deuterium by hydrogen requires additional mechanisms. It is possible that thiyl radicals cannot only abstract hydrogen atoms from the αC-H bond but also from the βC-H/D bond (Scheme 1, reaction 10). Such radical reaction could rationalize the formation of products 13a, 13b, 13c, for example through reactions 22–24 (Scheme 3), and 15a, 15b. Especially, the detection of covalent D/H exchange in products 15a and 15b suggests that a reversible hydrogen transfer reaction can precede the formation of Dha. While hydrogen transfer from the αC-H/D bond should be thermodynamically more favorable, the three βC-H bonds of the Ala side chain present a statistic advantage. Notably, in reaction 10 with peptide 1c (X=D) thiyl radicals abstract a deuterium atom from a βC-D bond. Based on our earlier results on primary kinetic isotope effects,41 we expect the abstraction of a deuterium atom to proceed about 7-times slower compared to that of a hydrogen atom. Nevertheless, our observations are consistent with earlier reports on the reaction of thiyl radicals with the βC-H bonds of amino acid residues, for example in N-acetyl-valine amide.42 In addition, recent pulse radiolysis data on thiyl radicals from glutathione suggest an intramolecular hydrogen transfer reaction with C-H bonds different from αC-H bonds.64
Scheme 3.

Hydrogen transfer from the Ala side chain to the thiyl radical.
4.6 Generation of CysS• via photolysis of 1a or 1b
An important experimental difference between the photolysis of 1a and 1b is that during photolysis of 1b, CysS• radicals are generated in the presence of thiol while during the photolysis of 1a CysS• radicals are initially generated in the absence of thiol (here, the thiol product 6 = 1b builds up over time). This difference rationalizes the formation of 2a (and 2b and 2c) during the photolysis of 1a, while 2a was not detected during the photolysis of 1b. However, during the photolysis of 1b we detected a thioether product 2a-1b, which most likely formed through Michael addition of excess 1b to 2a, suggesting the formation of 2a also through CysS• radicals generated during the photolysis of 1b. This Michael addition product is absent when 1b was photo-irradiated in the presence of CH2Cl2, which can be rationalized through radical anion dimer, [CysSSCys]•−, formation between CysS• and CysS−, followed by electron transfer to CH2Cl2. Consistent with this rationale, the photo-irradiation of 1a gives maximal yields of 2a (and 2b and 2c) for short photo-irradiation times (i.e., 2 minutes; see Figure 2), i.e. conditions where only small yields of product 6 (= 1b) are present. With increasing yields of 6, an increasing fraction of initial CysS• radicals can generate [CysSSCys]•−, which can transfer its electron to 2a initiating reduction of 2a to 3. When the photolysis of 1a is carried out in the presence of CH2Cl2, we observe no formation of 3 but also a reduction of the yields of 2a. These observations are consistent with the fact the CH2Cl2 inhibits the reduction of 2a (either through hydrated electrons or [CysSSCys]•−, but also removes CysS• from the reaction mixture via oxidation of intermediary [CysSSCys]•−.
4.7 Other photoproducts
Some other photoproducts, more hydrophobic than the native disulfides, were observed during the photo-irradiation of peptides 1a and 1c. These products account for no more than 10–12% of the total products based on the integration of chromatographic peak areas of the mass spectrometric total ion current. Because the structures of these products remain presently unclear, we decided not to include any detailed description of such products in the present paper.
5. Conclusion
The present paper provides mechanistic information on the reactivity of Cys thiyl radicals with amino acid residues within model peptides. Notably, the mechanism of Cys to Ala transformation involves Cys thiyl radical formation, likely followed by a 1,3-H-shift, β-elimination, and reduction of an intermediary Dha residue. The reduction of Dha is the result of a photochemical generation of hydrated electrons from deprotonated Cys residues. The photochemical transformation of Ala into Dha appears to require radical pair formation, rather than the generation of an isolated Cys thiyl radical. Such radical pair can play a significant role in the photochemical degradation of protein disulfides, for example in protein pharmaceuticals. The intramolecular hydrogen transfer reactions of cysteine thiyl radicals are relevant also for biological conditions of oxidative stress, where they may preceed protein fragmentation and aggregation reactions.
Supplementary Material
Acknowledgement
We gratefully acknowledge financial support from Amgen, Inc. and from the NIH (P01AG12993).
Footnotes
7. Supporting Information
Supplemental CID mass spectra of photoproducts are displayed in Figures S1–S21. This information is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Vitetta ES, Ghetie VF. Science. 2006;313:308. doi: 10.1126/science.1130482. [DOI] [PubMed] [Google Scholar]
- 2.Tous GI, Wei Z, Feng J, Bilbulian S, Bowen S, Smith J, Strouse R, McGeehan P, Casas-Finet J, Schenerman MA. Anal. Chem. 2005;77:2675. doi: 10.1021/ac0500582. [DOI] [PubMed] [Google Scholar]
- 3.Volkin DB, Mach H, Middaugh CR. Mol. Biotechnol. 1997;8:105. doi: 10.1007/BF02752255. [DOI] [PubMed] [Google Scholar]
- 4.Ellison D, Stalteri MA, Mather SJ. Biotechniques. 2000;28:318. doi: 10.2144/00282rr02. [DOI] [PubMed] [Google Scholar]
- 5.Lam XM, Yang JY, Cleland JL. J. Pharm. Sci. 1997;86:1250. doi: 10.1021/js970143s. [DOI] [PubMed] [Google Scholar]
- 6.Kerwin BA, Remmele RL. J. Pharm. Sci. 2007;96:1468. doi: 10.1002/jps.20815. [DOI] [PubMed] [Google Scholar]
- 7.Mozziconacci O, Kerwin B, Schöneich C. J. Phys. Chem. B. 2010;114:3668. doi: 10.1021/jp910789x. [DOI] [PubMed] [Google Scholar]
- 8.Mozziconacci O, Sharov V, Williams TD, Kerwin BA, Schöneich C. J. Phys. Chem. B. 2008;112:9250. doi: 10.1021/jp801753d. [DOI] [PubMed] [Google Scholar]
- 9.Mozziconacci O, Kerwin BA, Schöneich C. J. Phys. Chem. B. 2010;114:6751. doi: 10.1021/jp101508b. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, Singh S, Zeng DL, King K, Nema S. J. Pharm. Sci. 2007;96:1. doi: 10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 11.Cordoba AJ, Shyong BJ, Breen D, Harris RJ. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005;818:115. doi: 10.1016/j.jchromb.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 12.Gaza-Bulseco G, Liu H. Pharm. Res. 2008;25:1881. doi: 10.1007/s11095-008-9606-3. [DOI] [PubMed] [Google Scholar]
- 13.Mozziconacci O, Kerwin BA, Schöneich C. Chem. Res. Toxicol. 2010;23:1310. doi: 10.1021/tx100193b. [DOI] [PubMed] [Google Scholar]
- 14.Guerra M. Sulfur-centered reactive intermediates in chemistry and biology. Vol. 197. New York: Series A Life Sciences; Plenum Press; 1991. [Google Scholar]
- 15.Ferreri C, Costantino C, Landi L, Mulazzani Q, Chatgilialoglu C. Chem. Commun. (Camb.) 1999;(No. 5):407. [Google Scholar]
- 16.Tamba M, O’ Neil P. J. Chem. Soc., Perkin Trans. 1991;2:1681. [Google Scholar]
- 17.Sprinz H, Adhikari S, Brede O. Adv. Colloid Interface Sci. 2001;89–90:313. doi: 10.1016/s0001-8686(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 18.Sprinz H, Schwinn J, Naumov S, Brede O. Biochim. Biophys. Acta. 2000;1483:91. doi: 10.1016/s1388-1981(99)00175-4. [DOI] [PubMed] [Google Scholar]
- 19.Akhlaq MS, Schuchmann HP, von Sonntag C. Int. J. Radiat. Biol. 1987;51:91. doi: 10.1080/09553008714550531. [DOI] [PubMed] [Google Scholar]
- 20.Pogocki D, Schöneich C. Free Radic. Biol. Med. 2001;31:98. doi: 10.1016/s0891-5849(01)00559-7. [DOI] [PubMed] [Google Scholar]
- 21.Schöneich C, Bonifačić M, Asmus K. Free Radic. Res. Commun. 1989;6:393. doi: 10.3109/10715768909087923. [DOI] [PubMed] [Google Scholar]
- 22.Schöneich C, Asmus K. Radiat. Environ. Biophys. 1990;29:263. doi: 10.1007/BF01210407. [DOI] [PubMed] [Google Scholar]
- 23.Schöneich C, Dillinger U, von Bruchhausen F, Asmus K. Arch. Biochem. Biophys. 1992;292:456. doi: 10.1016/0003-9861(92)90016-p. [DOI] [PubMed] [Google Scholar]
- 24.Mozziconacci O, Williams T, Kerwin B, Schöneich C. J. Phys. Chem. B. 2008;112:15921. doi: 10.1021/jp8066519. [DOI] [PubMed] [Google Scholar]
- 25.Schöneich C. Chem. Res. Toxicol. 2008;21:1175. doi: 10.1021/tx800005u. [DOI] [PubMed] [Google Scholar]
- 26.Asquith RS, Shah AV. Biochim. Biophys. Acta. 1971;244:547. doi: 10.1016/0304-4165(71)90071-7. [DOI] [PubMed] [Google Scholar]
- 27.Dose K, Rajewski B. Photochem Photobiol. 1962;2:181. [Google Scholar]
- 28.Forbes W, Savige W. Photochem. Photobiol. 1962;1:1. [Google Scholar]
- 29.Risi S, Dose K, Rathinasamy TK, Augenstein L. Photochem. Photobiol. 1967;6:423. doi: 10.1111/j.1751-1097.1967.tb08889.x. [DOI] [PubMed] [Google Scholar]
- 30.Ikehata K, Duzhak TG, Galeva NA, Ji T, Koen YM, Hanzlik RP. Chem. Res. Toxicol. 2008;21:1432. doi: 10.1021/tx800093k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balkas TI. Int. J. Radiat. Phys. Chem. 1972;4:199. [Google Scholar]
- 32.Hoffman MZ, Hayon E. J. Am. Chem. Soc. 1972;94:7950. [Google Scholar]
- 33.Kapoor SK. Gopinathan Int. J. Chem. Kinet. 1995;27:535. [Google Scholar]
- 34.Cercek B. Nature. 1969;223:491. [Google Scholar]
- 35.Madhavan V, Lichtin NN, Hayon E. J. Am. Chem. Soc. 1975;97:2989. [Google Scholar]
- 36.Elliot AJ. Radiat. Phys. Chem. 1989;34:753. [Google Scholar]
- 37.Masuda T, Nakano S, Kondo M. J. Radiat. Res. (Japan) 1973;14:339. doi: 10.1269/jrr.14.339. [DOI] [PubMed] [Google Scholar]
- 38.Wolfenden BS, Willson RL. J. Chem. Soc., PerkinTrans. 2. No. 7. 1982. p. 805. [Google Scholar]
- 39.Eriksen TE, Fransson G. Radiat. Phys. Chem. 1988;32:163. [Google Scholar]
- 40.Naidu BN, Sorenson ME, Connolly TP, Ueda Y. J. Org. Chem. 2003;68:10098. doi: 10.1021/jo034762z. [DOI] [PubMed] [Google Scholar]
- 41.Nauser T, Casi G, Koppenol W, Schöneich C. J. Phys. Chem. B. 2008;112:15034. doi: 10.1021/jp805133u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nauser T, Pelling J, Schöneich C. Chem. Res. Toxicol. 2004;17:1323. doi: 10.1021/tx049856y. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Y, van dDWA. Org. Lett. 2001;3:1189. doi: 10.1021/ol015648a. [DOI] [PubMed] [Google Scholar]
- 44.Snow JT, Finley JW, Friedman M. Int. J. Pept. Protein Res. 1976;8:57. doi: 10.1111/j.1399-3011.1976.tb02481.x. [DOI] [PubMed] [Google Scholar]
- 45.Stickrath AB, Carroll EC, Dai X, Harris DA, Rury A, Smith B, Tang K-C, Wert J, Sension RJ. J. Phys. Chem. A. 2009;113:8513. doi: 10.1021/jp9017986. [DOI] [PubMed] [Google Scholar]
- 46.Alfassi ZB. General Aspects of the Chemistry of Free Radicals. Chichester: John Wiley & Sons; 1999. [Google Scholar]
- 47.Fung YM, Kjeldsen F, Silivra OA, Chan TW, Zubarev RA. Angew. Chem. Int. Ed. Engl. 2005;44:6399. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]
- 48.Thompson SD, Carroll DG, Watson F, O’ Donnell M, McGlynn SP. J. Chem. Phys. 1966;45:1367. [Google Scholar]
- 49.Autrey T, Devadoss C, Sauerwein B, Franz JA, Schuster GB. J. Phys. Chem. 1995;99:869. [Google Scholar]
- 50.Mezyk S. J. Phys. Chem. 1995;99:13970. [Google Scholar]
- 51.Mezyk SP, Madden KP. J. Phys. Chem. A. 1999;103:235. [Google Scholar]
- 52.Naumov S, Von Sonntag C. J. Phys. Org. Chem. 2005;18:586. [Google Scholar]
- 53.Zhang X, Zhang N, Schuchmann H-P, von Sonntag C. J. Phys. Chem. 1994;98:6541. [Google Scholar]
- 54.Lam AKY, Ryzhov V, O’Hair RAJ. J. Am. Soc. Mass Spectrom. 2010;21:1296. doi: 10.1016/j.jasms.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 55.Viscolcz B, Lendvay G, Körtvélyesi T, Seres L. J. Am. Chem. Soc. 1996;118:3006. [Google Scholar]
- 56.Zhao R, Lind J, Merenyi G, Eriksen TE. J. Am. Chem. Soc. 1994;116:12010. [Google Scholar]
- 57.Avila DV, Ingold KU, Di NAA, Zerbetto F, Zgierski MZ, Lusztyk J. J. Am. Chem. Soc. 1995;117:2711. [Google Scholar]
- 58.Snelgrove DW, Lusztyk J, Banks JT, Mulder P, Ingold KU. J. Am. Chem. Soc. 2001;123:469. [Google Scholar]
- 59.Volman DH, Wolstenholme J, Hadley SG. J. Phys. Chem. 1967;71:1798. doi: 10.1021/j100865a037. [DOI] [PubMed] [Google Scholar]
- 60.Jonsson M, Wayner DDM, Armstrong DA, Yu DK, Rauk A. J. Chem. Soc., Perkin Trans. 1998;2:1967. [Google Scholar]
- 61.Mieden OJ, Von SC. J. Chem. Soc., Perkin Trans. 2. 1989:2071. [Google Scholar]
- 62.Weast RC, editor. Handook of Chemistry and Physics. 66th ed. Boca Raton: CRC Press, Inc; 1985. [Google Scholar]
- 63.Surdhar PS, Armstrong DA. J. Phys. Chem. 1987;91:6532. [Google Scholar]
- 64.Hofstetter D, Nauser T, Koppenol WH. Chem. Res. Toxicol. 2010;23:1596. doi: 10.1021/tx100185k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.