

Abstract
Mutations in the endothelial cell (EC) tyrosine kinase receptor TIE2 cause inherited and sporadic forms of venous malformation. The recurrent somatic mutation L914F and common germline mutation R849W differ in terms of phosphorylation level, as well as sub-cellular localization and trafficking of the receptor. Previous studies have shed light on certain pathogenic properties of R849W, but the mechanisms of action of L914F are unknown. We used global gene expression profiling to study the effects of L914F on ECs. We found that L914F strongly dysregulates genes involved in vascular development, cell migration and extracellular matrix processing, while R849W has weak effects. We also demonstrate, for the first time, that TIE2-mutant ECs are deficient in the production of PDGFB, both in vitro and ex vivo in patient tissues. This defect is mediated by the chronic, ligand-independent activation of AKT by the mutant receptors. Inadequate secretion of the major mural cell attractant likely plays an important role in the development of abnormal vascular channels, contributing to the characteristic paucity of surrounding vascular smooth muscle cells.
INTRODUCTION
Mutations in the endothelial cell (EC) tyrosine kinase receptor TIE2 cause venous malformations (VM) (1–6). VM is a localized developmental defect of the vasculature characterized by enlarged EC-lined venous channels surrounded by sparse, irregularly distributed vascular smooth muscle cells (vSMCs). While germline substitutions cause the rare (<2% of VM) inherited form mucocutaneous venous malformation (VMCM; OMIM 600195), somatic mutations are present in the lesions of at least 50% of common sporadic VM (1–6). When overexpressed in vitro, all VM-causative mutations induce ligand-independent hyperphosphorylation of the receptor, to widely varying degrees (1,2,4–6). R849W, the most common germline mutation, has a far weaker effect than the recurrent somatic mutation, L914F (1,2,4–6).
Previous studies have shown that overexpression of R849W results in constitutive hyperactivation of the anti-apoptotic AKT kinase (7), as well as aberrant activation of the transcription factor STAT1 (8–10). L914F, however, differs from R849W, not simply in terms of more robust phosphorylation, but also sub-cellular localization: R849W, like wild-type (WT)-TIE2 (11,12), is evenly distributed on the cell surface of sparse human umbilical vein ECs (HUVECs), and translocates to the cell rear and retraction fibers upon ligand stimulation. L914F instead accumulates in an activated state in the Golgi apparatus and endoplasmic reticulum, and shows incomplete Angiopoietin-1 (ANGPT1) ligand-induced translocation (2). Moreover, R849W is identified frequently in familial VMCM, where it seems to require localized inactivation of the WT allele in order to cause a phenotype (2). L914F, in contrast, has never been identified in the germline despite its recurrence at the somatic level. This suggests it may be incompatible with life when ubiquitous. Thus, it is unclear whether the two forms share similar pathogenic mechanisms. We therefore set out to identify pathways dysregulated by L914F. When overexpressed in HUVECs, L914F profoundly altered the expression of genes implicated in vascular development, cell migration and extracellular matrix (ECM) processing. While R849W shared these targets, it had weaker (lower than 2-fold) effects, rendering it statistically indistinguishable from WT-TIE2 in global analyses. Furthermore, we showed, for the first time, that both VM-causative TIE2 mutants significantly hampered the ability of ECs to produce PDGFB, in confluent cultures as well as L914F-positive VM lesions, by exerting an AKT-mediated inhibition of the transcription factor FOXO1. Lack of the major vSMC attractant likely contributes to defective development and/or maintenance of the mural cell layer by mutant TIE2-expressing ECs, resulting in the sparse, patchy vSMC distribution characteristic of VMs.
RESULTS
VM-causative TIE2 mutations R849W and L914F activate AKT and STAT1
We began by characterizing the HUVEC lines used in this study, for the level of TIE2 overexpression (Fig. 1A; Supplementary Material, Fig. S1), and the degree of ligand-independent receptor phosphorylation (Fig. 1A). We also assessed whether chronic mutation-induced phosphorylation activates two major pathways that act downstream of ligand-induced phosphorylation of the WT receptor: PI3K/AKT and MAPK, important for cell survival, and for cell migration and proliferation, respectively (11–15). Overexpression of L914F in HUVECs induces a higher level of AKT phosphorylation than R849W (Fig. 1B), correlating with the strength of receptor phosphorylation (Fig. 1A). Neither TIE2 mutant significantly alters activation of ERK1/2 (p44/42) or the p38 MAPKs in confluent HUVEC cultures (Fig. 1C and D). R849W aberrantly activates the signal transducer and activator of transcription 1 (STAT1) (8–10); we observe that L914F induces a more pronounced hyperphosphorylation of STAT1 (Fig. 1E).
Figure 1.

Mutant TIE2 causes ligand-independent activation of AKT and STAT1. Western blots on lysates from confluent HUVECs: non-transfected (NT); or retrovirally transfected to overexpress WT-TIE2 (WT), R849W or L914F. Blots probed for pan phospho-tyrosine (phospho-Tyr) or phospho-AKT, -ERK1/2, -p38 and -STAT1; then stripped and reprobed for total TIE2, AKT, ERK1/2, p38 and STAT1, respectively. TIE2 blots were re-stripped and probed for β-actin.
We assessed the overall effects of the TIE2 mutations on the growth, proliferation and survival of HUVEC cultures. Mutant HUVECs show a significant increase in cell number with time (Fig. 2A), which does not however correlate with increased proliferation (Fig. 2B). The higher cell number in L914F and R849W instead correlates with resistance to cell death under both normal culture conditions (full growth media; Fig. 2C) and under serum starvation (Fig. 2D and E), in accordance with previous data on the effects of hyperactivation of anti-apoptotic AKT in R849W-overexpressing cells (7). Thus, altered cellular phenotypes associated with R849W hold true but are amplified in the case of L914F.
Figure 2.

Growth advantage of mutant-TIE2 expressing HUVECs is not due to increased cell proliferation. (A) Total cell numbers of NT, WT, R849W and L914F HUVECs measured at time points indicated. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 mutant versus NT; ††P ≤ 0.01, †††P ≤ 0.001 mutant versus WT. (B) BrdU ELISA showing proliferation rate of NT, WT, R849W and L914F HUVECs normalized to total cell number. Higher cell numbers in L914F HUVECs (A) do not correlate with higher proliferation rate (B); *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 mutant versus NT, †P ≤ 0.05, †††P ≤ 0.001 mutant versus WT. (A and B) n = 4 experiments run in triplicate per cell line. (C) Cell viability of NT, WT, R849W and L914F HUVECs grown in complete growth medium. R849W and L914F show lower rate of cell death. 1905–2365 cells included in each group calculated from 30 randomly selected fields from three cover slips. *P ≤ 0.05, ***P ≤ 0.001 versus NT. (D) Number of adherent NT, WT, R849W and L914F HUVECs after 18 h of serum starvation, relative to cells grown in complete growth medium (set to 100%, gray line). In NT and WT, but not in mutant-transfected HUVECs serum starvation leads to decreased cell number when compared with the respective controls. 1512–2567 cells included in each group calculated from 30 randomly selected fields from three cover slips. ***P ≤ 0.001 versus NT; †††P ≤ 0.001 versus WT. (E) Caspase 3/7 activation in NT, WT, R849W and L914F HUVECs after 12 h of serum starvation. Bars represent relation of caspase activity in serum-starved cells/cells grown in complete media normalized to cell number. WT set to 1.00 (gray line). ***P ≤ 0.001 versus NT; †††P ≤ 0.001 versus WT. n = 6 experiments run in triplicate per cell line. NS, not significant.
L914F, but not R849W, strongly dysregulates EC gene expression
We used microarray technology to identify genes that are differentially expressed when comparing confluent cultures of non-transfected (NT) HUVECs to HUVECs transfected with WT and mutant (R849W and L914F) TIE2 (Fig. 3). The inclusion of NT allowed us to track genes dysregulated simply due to overexpression of TIE2, rather than specific mutant forms of the receptor.
Figure 3.
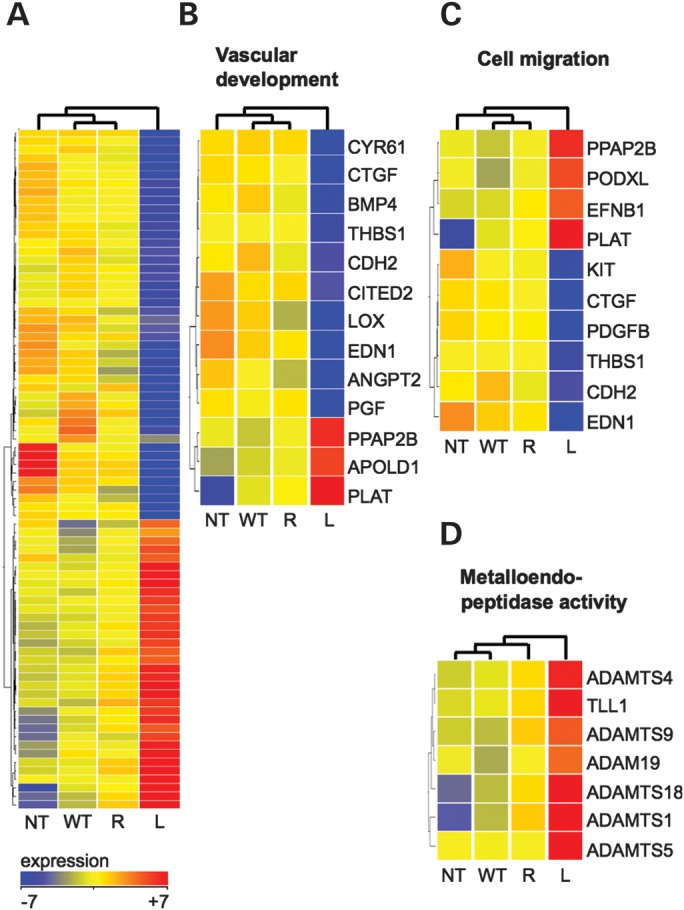
L914F dysregulates genes implicated in vascular development, cell migration and metalloproteinase activity. (A) Heat map of 80 genes differentially expressed between WT and L914F (L) HUVECs. The Mann–Whitney U-test, P ≤ 0.05, fold-change ≥ 2, no correction for multiple testing. NT and R849W (R) are included for comparison. (B–D) Subsets of the differentially expressed genes (WT versus L914F) that fall into significant GO-annotation categories (corrected P-value ≤ 0.01): (B) vascular development, (C) cell migration, (D) metalloendopeptidase activity. n = 3 per cell line.
A one-way analysis of variance (ANOVA) identified 743 genes differentially expressed between the four groups. In keeping with the ‘stronger’ molecular phenotypes associated with L914F when compared with R849W (Fig. 1), these are mainly attributable to differences between L914F and the other groups. R849W, in contrast, is similar to WT, with which it is grouped in non-supervised hierarchical clustering (Supplementary Material, Fig. S2A). A Tukey post hoc test revealed that 41% of the genes (n = 306) are different between L914F and WT cells, and 16% of the genes (n = 116) between NT and WT cells, with only 5% of the genes (n = 39) distinguishing between R849W and WT cells (Supplementary Material, Fig. S2B). A direct comparison of L914F to WT identified 80 genes with significant changes in expression (uncorrected P-value ≤ 0.05) of at least 2-fold (34 genes up, 46 genes down in L914F) (Fig. 3A; Supplementary Material, Table S1). The same cutoff yielded no genes that distinguish R849W from WT. Pathway analysis was therefore subsequently performed on genes differentially expressed between L914F and WT.
L914F dysregulates genes involved in vascular development/angiogenesis, cell migration and ECM processing
Significant proportions of the genes differentially regulated by L914F are involved in vascular development and cell migration (Fig. 3; Supplementary Material, Table S2). Interestingly, the preponderance of affected transcripts in these Gene Ontology (GO) categories favor EC migration, proliferation or tube formation in vitro and/or in vivo, and are downregulated by L914F (Fig. 3B and C). Among these are the CCN-family growth factors cysteine-rich 61 (CYR61) and connective tissue growth factor (CTGF) (16); bone morphogenetic growth factor 4 (BMP4) and placental growth factor (PGF), the ligands for BMPRs and VEGFR1, respectively (17–19); and ANGPT2, the context-dependent antagonistic ligand of TIE2 (20). Expression changes are not exclusively pro-quiescence, however, as illustrated by the downregulation of anti-angiogenic thrombospondin-1 (THBS1) (21). Interestingly, dysregulated genes in the metalloendopeptidase category are all upregulated by L914F. They include members of the ADAMTS (A Disintegrin And Metalloproteinase with Thrombospondin Motifs) family (Fig. 3D), several of which (e.g. ADAMTS1, 4 and 5) cleave ECM components such as versican, predominant in the basement membrane of blood vessels (22,23).
Inhibition of the transcription factor FOXO1 by L914F and R849W
We performed TFactS analysis, which infers changes in transcription factor activity based on the up- or downregulation of significant proportions of their target genes (24). FOXO1, a member of the forkhead family, was predicted to be significantly inhibited by L914F when compared with WT (see Supplementary Material, Table S3 for a complete list). Known endothelial FOXO1 targets (25,26) (Fig. 4A) account for 12.5% of the 80 genes differentially expressed between L914F and WT cells. Real-time quantitative RT–PCR validation of a subset of transcripts positively (ANGPT2, BMP4, PDGFB) and negatively (ADAMTS1) regulated by FOXO1 showed that their expression is in fact also significantly, albeit more weakly, dysregulated by R849W (Supplementary Material, Fig. S3). Western blot confirmed that both TIE2 mutant forms phosphorylate (i.e. inhibit) FOXO1 (Fig. 4B). In agreement with the dysregulation of its transcriptional targets, the level of inhibition correlates with the degree of TIE2 phosphorylation mediated by the two mutations.
Figure 4.
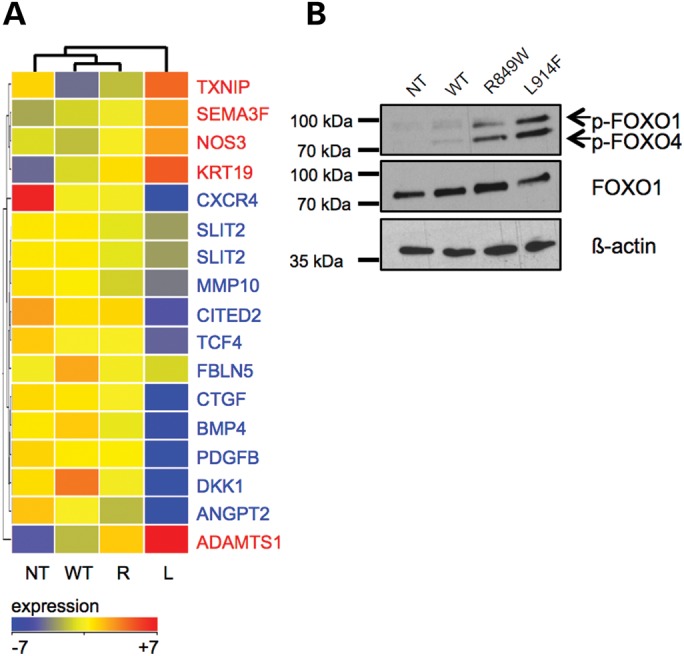
VM-causative TIE2 mutations cause FOXO1 repression. (A) Heat map of FOXO1 target genes significantly dysregulated in L914F (L) versus WT HUVECs. NT and R849W (R) included for comparison. The Mann–Whitney U-test, P ≤ 0.05, fold-change ≥ 1.5, no correction for multiple testing. Gene names in blue: positive targets, transcribed by FOXO1; in red: negative targets, repressed by FOXO1. (B) Western blot showing FOXO1 phosphorylation level in lysates from NT, WT, R849W and L914F HUVECs. Blots probed for phospho-FOXO1, then stripped and reprobed for total FOXO1 and β-actin.
L914F and R849W cause an AKT-dependent deficiency of the FOXO1 target PDGFB
Among the FOXO1 target genes affected by the VM-causative TIE2 mutations is the vSMC attractant PDGFB. The transcriptional decrease in PDGFB (Supplementary Material, Fig. S3) translates into significantly lower levels of protein secreted by mutant (L914F and R849W) when compared with WT TIE2-bearing (NT, WT) cells (Fig. 5). The low level of FOXO1 inhibition (phosphorylation) that occurs upon overexpression of WT TIE2 (Fig. 4B) is echoed by lowered PDGFB production by WT when compared with NT cells (Fig. 5). ANGPT1 stimulation, which has been shown to cause inhibition of FOXO1 (25), results in a significant decrease in PDGFB secretion by NT and WT cells. The magnitude of this effect did not match that of the mutant forms, as treated NT and WT cells nevertheless secreted significantly more PDGFB than untreated mutant (L914F or R849W) cells.
Figure 5.

Mutant TIE2 receptors downregulate PDGFB secretion by HUVECs. PDGFB secretion, in the absence (dark grey bars) or presence (light grey bars) of ANGPT1 (500 ng/ml, 6 h stimulation), measured by ELISA on media from confluent NT, WT, R849W and L914F HUVECs. Lower limit of detection of the assay indicated by a dashed line. ***P ≤ 0.001 versus -ANGPT1; +++P ≤ 0.001 versus NT; ###P ≤ 0.001 versus WT, all -ANGPT1. n = 2 per group.
FOXO1 can be inhibited due to phosphorylation by activated AKT (25) in response to ANGPT1 stimulation of the WT receptor, or chronically due to the VM-causative TIE2 mutant forms (Fig. 1B, Supplementary Material, Fig. S4). We confirmed a role for AKT in the impairment of PDGFB production, by showing a dose-dependent effect of the AKT inhibitor MK2206 (Supplementary Material, Fig. S5). MK2206 (2 µmol/l) caused a significant decrease in AKT phosphorylation by the TIE2 mutants (Fig. 6A and B, left panels), which was accompanied by a decrease in phosphorylated (inhibited) FOXO1 (Fig. 6A and B, right panels) and a concomitant increase in PDGFB secretion (Fig. 6C). The increase in secretion in MK2206-treated cells relative to non-treated controls was comparable between the different TIE2 genotypes (4.5-fold for WT, 6.9-fold for R849W and 5.8-fold for L914F) (Fig. 6C, right panel). Interestingly, the level of PDGFB secretion correlated inversely with the level of AKT and FOXO1 phosphorylation, across (regardless of) TIE2 genotype (Fig. 6D).
Figure 6.

TIE2 activation downregulates PDGFB-secretion via AKT-mediated inhibition of FOXO1. NT, WT, R849W and L914F HUVECs treated with 2 µmol/l MK2206 (MK) or 0.002% DMSO. (A) Representative western blots showing AKT and FOXO1 phosphorylation in non-treated versus MK2206- or DMSO-treated HUVECs. Blots probed for phospho-AKT or phospho-FOXO1, and then reprobed for β-actin. (B) Quantification of the same. NT, not quantifiable due to the absence of visible bands. Phosphorylation-band intensities of WT, R849W and L914F normalized to β-actin bands. (C) PDGFB ELISA on conditioned media from confluent HUVECs. (Left) Absolute amounts secreted. (Right) Relative values, normalized to non-treated samples per HUVEC-type, set at 1. n = 3 per group. DMSO versus non-treated of same HUVEC type: not significant; ***P ≤ 0.001 versus non-treated of same HUVEC-type. (D) Correlation of AKT (dark grey) and FOXO1 (light grey) phosphorylation levels, with PDGFB secretion. Intensity of phosphorylation normalized to β-actin (i.e. all values shown in panel B), versus PDGFB secretion by these same samples (i.e. all values shown in panel C left, with the exception of ‘NT’).
Paucity of PDGFB is observed ex vivo in L914F-positive VMs
We performed a semi-quantitative minisequencing (SNaPshot, Applied Biosystems) assay [detailed in the study of Limaye et al. (2)] on cDNA from nine surgically resected L914F (c.2740C>T)-positive VM tissues. This allowed us to assess for the relative expression levels of the two TIE2 alleles, as reflective of the composition of these heterogeneous samples in terms of mutant versus normal ECs (Supplementary Material, Figs S6 and S7 and Table S4). For eight tissues, in which the mutant allele constituted at least 10% of the TIE2 expressed (Supplementary Material, Figs S6 and S7 and Table S4), we carried out real-time quantitative RT–PCR for PDGFB expression, relative to VE-Cadherin expression in order to normalize for total EC content. VE-Cadherin was chosen as its expression does not seem to be affected by TIE2 mutant type, in contrast to other commonly used EC markers (Supplementary Material, Fig. S8). L914F VMs were compared with surgically resected glomuvenous malformations (GVMs), clinically and genetically distinct VMs with abundant, maldifferentiated vSMCs that harbor a complete loss of function of the gene glomulin (27). We found significantly lower levels of PDGFB mRNA in VMs when compared with GVMs (Supplementary Material, Fig. S7). We performed immunohistochemistry on paraffin-embedded tissue sections from seven VM patients with the L914F mutation. Malformed veins, characterized by distended channels and irregular vSMCs, showed lower levels of PDGFB than adjacent normal veins and arteries (Supplementary Material, Fig. S9). Immunofluorescence staining demonstrated an overlap between secreted PDGFB and endothelium as well as surrounding vSMCs (Fig. 7A and B). Quantification of anti-PDGFB staining, normalized to lumen length, confirmed that VMs are indeed surrounded by significantly less PDGFB, than are normal veins, GVMs and arteries (Fig. 7C).
Figure 7.

PDGFB is downregulated in human VM lesions. Representative immunofluorescence staining on an L914F-positive VM and a GVM tissue, showing normal arteries (AR) and veins (V), and malformed vessels (VM, GVM) characterized by distended channels. (A) Hematoxylin and eosin (VM: left panel, GVM: right panel). Scale bars: 500 µm. (B) Smooth muscle α-actin (SMA), PDGFB and DAPI staining of indicated areas (boxed). Scale bars: 50 µm. (C) Box- and whiskers plot showing intensity of PDGFB staining normalized to length of the lumen border. Boxes: 25th and 75th percentile; whiskers: 5th and 95th percentile; horizontal bar: median. The Kruskal–Wallis test (four groups: arteries, veins, VMs, GVMs): P ≤ 0.001. Mann–Whitney between two groups: ***P ≤ 0.006; *P ≤ 0.05.
DISCUSSION
We set out to dissect the pathogenic effects of the most prevalent VM-causative TIE2 mutation: somatic L914F, identified in a large proportion (>70%) of common sporadic VM. Germline R849W, which causes the majority of rare, inherited VMCM, has previously been shown to induce ligand-independent hyperphosphorylation of AKT and STAT1 (7–10). We now show that L914F induces stronger activation of both. We also show it does not significantly alter the phosphorylation of the p38 or p42/44 MAPKs. As has been demonstrated for R849W, we found that L914F confers greater resistance to cell death, likely due to hyperphosphorylation of AKT. Expression profiling of confluent cells revealed that genes that affect cell migration, vascular development and ECM synthesis and/or turnover are strongly dysregulated by L914F, which seems to have a pro-quiescence effect reminiscent of ANGPT1 stimulation of TIE2. These data correlate with the in vivo phenotype of R894W and L914F expressing VMs, which are not proliferative and do not expand rapidly; nor do they ‘seed’ new lesions. Interestingly, several commonly used EC markers are downregulated by L914F. The levels of dysregulation for a subset of these reach significance by real-time qPCR; however, none does so at the whole-transcriptome level (Supplementary Material, Fig. S8). Targeted validation of individual genes and pathways of interest revealed that they are also perturbed by R849W, albeit more weakly, suggesting certain shared pathogenic mechanisms. Importantly, we demonstrate that both mutations inhibit FOXO1, and repress the production of its targets, including the vSMC attractant PDGFB. This may account for the irregularity of the mural cell layer characteristic of VMs, although the contribution of other factors certainly cannot be ruled out.
TFactS analysis, which interrogates more than 60 transcription factors and their target genes, predicted multiple transcription factors to be differentially regulated by L914F. Only the FOXOs (1 and 3, which largely share target genes; http://www.tfacts.org) remained significant after correction for multiple testing, however. In addition, the use of an antibody that detects both phospho-FOXO1 and 4 showed that the latter is also phosphorylated downstream of the TIE2 mutations (Fig. 4B). We focused on FOXO1, as neither FOXO3 nor 4 null mice exhibit vascular defects, present in FOXO1 knockouts (28). Furthermore, published evidence suggests that FOXO4, unlike FOXO1, may not play as significant a role in the vasculature (26). Somewhat surprisingly, STAT1 was not among the factors predicted to be activated in mutant HUVECs based on target gene dysregulation. This may in part be because its known targets are largely drawn from immune cells in response to inflammatory stimuli, and may differ from those in ECs, downstream of the mutated receptor. Alternatively, the pathogenic effects of STAT1 may only become evident under specific conditions of cell culture and/or stimulation (10). Its contribution to the L914F-mediated phenotype has yet to be determined.
A target gene of FOXO1 secreted by ECs, platelets and other cell types, PDGFB signals through its receptor PDGFRβ on mural cell precursors to induce their proliferation and migration during vessel maturation (29,30). Lack of (EC-secreted) Pdgfb or Pdgfrβ in mice causes impaired mural cell coverage, with dilated vascular channels and endothelial hyperplasia as secondary effects (29–32). This would support a role for decreased PDGFB production in the vSMC-layer defect of VMs. Given the vein-specific phenotype of PDGFB-reducing TIE2 mutations in humans, it is intriguing that certain studies suggest that PDGFB may affect human venous SMCs more strongly than arterial SMCs, perhaps due to differential receptor distribution (33,34).
PDGFB production was found to be dependent not only on TIE2 mutant forms, but also on cell confluence and the presence of serum: ‘normal’ (NT, WT) HUVECs produce minimal amounts of PDGFB when sparse, or when incubated overnight in serum-free medium (data not shown). Upon forming a cobblestone monolayer at confluence, higher levels of PDGFB are secreted into media by NT and WT cells, whereas mutant cells remain incapable of doing so. It has been demonstrated that AKT activity is elevated in sparse HUVECs and diminishes with confluence (11). In keeping with our data, this would dampen FOXO1 activity and PDGFB expression in sparse cells, with the effect being lifted upon reduction of AKT activity at confluence. This may imply that normal ECs, once they are in cell–cell contact upon tube-formation, secrete PDGFB in order to recruit vSMC. The TIE2 mutation-induced persistence of high AKT phosphorylation even in confluent cells would interfere with this process, keeping PDGFB levels inappropriately low. Stimulation of WT TIE2 by ANGPT1 also causes AKT phosphorylation and a concomitant decrease in PDGFB production (Fig. 5; Supplementary Material, Fig. S4) (25). Thus, once recruited, ANGPT1-producing vSMCs may dampen EC production of PDGFB, to levels that are compatible with the maintenance, but not the continued proliferation and/or attraction of mural cells. Mutant ECs, however, show a chronic, ligand-insensitive PDGFB deficiency, which is more profound than that induced by the ligand.
We were able to confirm the PDGFB defect detected in vitro, ex vivo in patient tissues endogenously expressing mutant TIE2. Malformed veins had lower levels of PDGFB when compared with normal veins or arteries, which were robustly stained. This is in keeping with the identified drop in serum levels of PDGFB in the circulation and draining veins of lesions from patients with large VMs (but not other malformations) (35). Measures to normalize levels of PDGFB produced by abnormal vessel ECs, including inhibition of chronic TIE2 or AKT activity, may therefore have beneficial effects. While it is not known whether established lesions could be induced to undergo remodeling in response to such interventions, they may be effective in reducing VM regrowth, frequent after incomplete surgical resection.
The effects detailed here are shared between L914F and R849W, but greatly attenuated in the latter, to the extent that none is significantly different from the WT in genome-wide transcriptional profiling. R849W has been shown to require inactivation of the normal allele by a second hit (2). We have previously hypothesized that this may be because the type or magnitude of signaling abnormalities caused by multimers composed exclusively of mutant receptors, may be different from those caused by heteromers including both normal and mutant receptors (2). The presence of WT TIE2 may serve to dilute, or perhaps actively counteract, the effect of the mutant in ECs. The level of overexpression of R849W (when compared with the level of the endogenous WT receptor; Fig. 1A) would argue for the latter. R849W may therefore need to be studied more extensively in cells without any endogenous WT expression, and/or in the presence of ligand-mediated receptor multimerization. Finally, the causes and implications of dysregulated expression of numerous other molecules and pathways of importance in the vasculature remain to be further explored.
MATERIAL AND METHODS
HUVEC samples and cell culture
Full-length WT-TIE2 (WT), R849W-TIE2 (R849W) or L914F-TIE2 (L914F) was cloned into pMXs vector (gift from T. Kitamura, University of Tokyo, Japan) and packaging cell line 293-GPG VSV-G (36) was transfected for retrovirus production with Fugene 6 (Roche). NT and retrovirally transfected HUVEC lines were maintained in EC growth medium (EC basal medium and growth supplement, Cell Applications, San Diego, CA, USA), supplemented with 10% FBS. For microarray experiments, cells from three separate confluent cultures were used per cell type. Each in vitro validation experiment was carried out on triplicate wells of: (i) the same cultures (or supernatants thereof) on which microarrays had been performed, (ii) separate cultures of the same stock of transfected cells, and (iii) cultures from a second, separate batch of retroviral infections, all grown to confluence under similar conditions.
Microarray procedure
Total RNA was extracted from frozen cell pellets using TriPure (Roche Diagnostics, Mannheim, Germany). RNA quantity was determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). RNA quality was assessed using Agilent RNA 6000 NanoChips on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Gene expression profiles were generated using GeneChip® Human Exon 1.0 ST arrays (Affymetrix, Santa Clara, CA, USA). Briefly, 1 µg total RNA was used for initial ribosomal RNA reduction with the RiboMinus Human/Mouse Transcriptome Isolation Kit (Invitrogen, Carlsbad, CA, USA). Successful rRNA reduction was assessed using Agilent RNA 6000 NanoChips on the Agilent 2100 Bioanalyzer (Agilent Technologies). cDNA was synthesized using the GeneChip® WT (Whole Transcript) cDNA Synthesis and Amplification Kit along with the Control Reaction Kit. The cDNA was fragmented with UDG (uracil DNA glycosylase) and APE 1 (apurinic/apyrimidic endonuclease 1) and biotin-labeled with TdT (terminal deoxynucleotidyl transferase) using the GeneChip® WT Terminal Labeling Kit. Five micrograms of fragmented, biotinylated target were hybridized to the Affymetrix GeneChip® Human Exon 1.0 ST Array at 45°C for 16–18 h. Microarrays were washed and stained on the GeneChip® Fluidics Station 450 using the GeneChip® Hybridization Wash and Stain Kit, and scanned using the GeneChip® Scanner 3000 7G. A balanced experimental design was used, with one sample each of the four cell lines (NT, WT, R849W and L914F) included per batch, with a total of three batches processed. The microarray data have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus and are available through GEO series accession number GSE46684.
Statistical analysis of microarray data
Statistical analysis and visualization of the microarray data were carried out using GeneSpring software (Agilent Technologies). Data were quantile normalized, summarized using robust multichip analysis and adjusted for probe sequence. A log2 conversion and a baseline transformation to the median of all samples were carried out. The lowest 20th percentile of raw intensity values were filtered out, while retaining entities for which all three samples in any one condition had values above the cut-off.
An ANOVA was performed on the four groups (NT, WT, R849W and L914F), applying a threshold of P ≤ 0.05 with no correction for multiple testing, with a subsequent Tukey HSD post hoc test. A Mann–Whitney U test was carried out to compare WT versus R849W and WT versus L914F, applying a threshold of P ≤ 0.05 and a limit of 2-fold, with no correction for multiple testing.
Pathway analyses were performed on genes significantly differently expressed between WT and L914F according to the above criteria. GO enrichment analysis was performed to identify significantly over-represented biological categories, and TFactS analysis (http://www.tfacts.org) (24) to identify transcription factors inferred to be modulated by comparison of up- and downregulated genes, to a curated database containing common transcription factors and known positive and negative target genes.
FOXO1 target genes included in the TFactS database were supplemented with targets from published literature on HUVECs (25,26), and genes with a fold-change of at least 1.5 were hierarchically clustered to generate the FOXO1-signature heat map.
Cell lysis and fractionation
Cells were washed with ice-cold PBS and lysed in ice-cold lysis buffer [50 mm Tris–HCl pH 8, 150 mm NaCl, 1% NP40, 2 mm EDTA, 1 tablet Complete Protease Inhibitor and 2 tablets PhosStop Phosphatase Inhibitor Cocktail (both Roche Diagnostics) per 20 ml]. Lysates were homogenized using a 21G needle and spun down at 12 000g, 4°C, to pellet debris.
Western blot
Protein lysates were resolved on precast polyacrylamide gels (Biorad, Hercules, CA, USA) and immunoblotted with one of the following antibodies: anti-AKT, anti-phospho-AKT (Ser473), anti-p44/42 MAPK (Erk1/2), anti-phospho-p44/42 MAPK (Erk1/2; Thr202/Tyr204), anti-p38, anti-phospho-p38 (Thr180/Tyr182), anti-FOXO1 (C29H4), anti-phospho-FOXO1 (Ser256), anti-STAT1, anti-phospho-STAT1 (Tyr701) (all Cell Signaling Technology, Danvers, MA, USA) at 1:1000, anti-TIE2 (C-20, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:500, anti-phospho-tyrosine (PY99, Santa Cruz Biotechnology) at 1:400 or anti-β-actin (Clone AC-15, Sigma-Aldrich, St Louis, MO, USA) at 1:15 000. Membranes were first blotted with a phospho-antibody, then stripped and reprobed with its corresponding total antibody and/or β-actin. Immunoreactive proteins were visualized using a goat anti-mouse-HRP secondary antibody (Sigma-Aldrich; for TIE2, phospho-tyrosine and β-actin) or a donkey anti-rabbit-HRP secondary antibody (GE Healthcare Europe, Freiburg, Germany; for all others), both at 1:10 000, with a femto-range-sensitive ECL detection system (Thermo Scientific).
Total cell number
For evaluation of total cell numbers, NT, WT, R849W and L914F HUVECs were plated at low density (5000 cells/96 well). Cells (at day 2, 5, 8, 11 and 16 after plating) were fixed in cold methanol, stained with 0.05% crystal violet (Sigma-Aldrich) and lysed in 10 mm HEPES and 1% sodium deoxycholate. The amount of crystal violet was measured using a microplate spectrophotometer for absorbance at 540 nm, and at 405 nm to subtract background.
Proliferation assay
To measure the extent of cell proliferation, NT, WT, R849W and L914F HUVECs were plated at low density (5000 cells/96 well). The extent of cell proliferation was measured with BrdU ELISA (Roche Diagnostics) according to the manufacturer's protocol at days 2, 5, 8, 11 and 16 after plating. Proliferation index was calculated as a ratio of cell proliferation/total cell number.
Cell viability assays
For evaluation of cell viability, NT, WT, R849W and L914F HUVECs were plated on cover slips (40 000 cells/200 mm2) in complete growth medium (EC basal medium ECBM and growth supplement, Cell Applications, supplemented with 10% FBS, HyClone, Logan, UT, USA) for 30 h, then incubated for 15 min in the presence of a blue-fluorescence dye Hoechst (stains all nuclei, Sigma-Aldrich) and a red-fluorescence propidium iodide (excluded from viable cells, BD Pharmingen, San Diego, CA, USA) and fixed. Cell death was measured based on nuclear morphology (pyknotic/fragmented nuclei in Hoechst stain) and cell membrane leakage (positive nuclei in propidium-iodide stain) using Olympus FluoView FV1000 confocal microscope. 1905–2365 cells were included in each group calculated from 30 microscopic fields.
To evaluate cell death after serum starvation, HUVECs were plated on cover slips (40 000 cells/200 mm2) in complete growth medium for 12 h, washed four times with ECBM basal media (no FBS or growth supplements) and subsequently grown in full growth medium (10% FBS and growth supplements) or in serum-free conditions (ECBM basal media) for 18 h, stained with Hoechst for 15 min and then fixed. Numbers of adherent cells (Hoechst positive nuclei) were calculated from fixed specimens (30 microscopic fields from three cover slips in each group, 1512–2567 cells were included) and cell numbers were compared between complete and serum-free media.
For caspase assay, HUVECs were plated in complete growth medium, washed and subsequently grown in complete medium or starved in ECBM basal medium (no FBS or growth supplements) for 12 h followed by luminometric caspase 3/7 activity measurement according to the manufacturer's protocol (Promega, Fitchburg, WI, USA).
ELISAs for secreted proteins
For ELISA, conditioned media from confluent cultures were used. Culture media were changed, the supernatants collected 24 h later and spun at 2500 g for 15 min to remove cellular particles. ELISA was carried out for human PDGFB (R&D Systems, Minneapolis, MN, USA) following the manufacturer's instructions. For ELISA of ANGPT1-stimulated cells, confluent cells were starved over night in serum-low medium (2% FBS), and supernatants were collected as above after 6 h of stimulation with 500 ng/ml ANGPT1 in serum-low medium.
AKT inhibition
AKT inhibition experiments were carried out on confluent cells. A dose curve was initially performed to identify the inhibitor concentration most effective in reducing AKT phosphorylation without causing significant cell death. This dose was used in all subsequent experiments shown. Culture medium was replaced by medium containing MK2206 (Selleck Chemicals, Houston, USA) or vehicle control (DMSO). After 24 h, supernatants were collected for PDGFB ELISA and cells were lysed for protein extraction. Western blot was used to assess for levels of AKT and FOXO1 phosphorylation. Quantification of intensities in western blotting was carried out using the GeneTools software on images acquired using GeneSnap software (both Syngene, Cambridge, UK). Intensities of phospho-bands were normalized to corresponding β-actin signals.
Immunofluorescence on tissues
Informed consent was obtained from each patient prior to enrollment, as approved by the ethical committee of the Medical Faculty at the Université catholique de Louvain, Brussels, Belgium. Paraffin blocks of seven surgically resected VMs with an L914F mutation and six GVMs were used. Five-micrometer sections were deparaffinized with xylene (VWR, Leuven, Belgium) and rehydrated through a degraded ethanol series, into water. For hematoxylin–eosin staining, sections were incubated in hematoxylin for 1 min and 30 s, followed by an acid wash. They were then incubated in eosin for 20 s. For immunofluorescence, tissue sections underwent heat-induced antigen retrieval in 0.1 m citrate buffer (pH 6.0) in a microwave, for 2 min at full power followed by 10 min at 30% power. Sections were then blocked in 1% bovine serum albumin in 1X PBS before incubation in α-PDGFB (1:50) for overnight at 4°C, then in α-SMA (1:200) for 2 h at room temperature. Fluorescent secondary antibodies used were goat-α-rabbit AlexaFluor-594 against α-PDGFB, and donkey-α-mouse AlexaFluor-488 against α-SMA (Life Technologies). Slides were mounted with VectaShield HardSet mounting medium containing DAPI (Vector Labs, Peterborough, UK). Images were visualized with an Axiovert 200M wide-field fluorescent microscope (Zeiss, Jena, Germany). Quantification of PDGFB, was done using AxioVision software (Zeiss), on at least 10 fields per vessel type (GVM, VM, normal artery, normal vein). Values were normalized to lumen length visible within captured images. A non-parametric test (Kruskal–Wallis) was used to compare the four groups (normal artery, normal vein, GVM, VM); the Mann–Whitney U-test was used to compare pairs of groups.
Statistical analysis
All graphed data (with the exception of Fig. 7) are expressed as mean ± standard deviation (SD) of at least n = 3 separate samples per group (mutant type, and/or treatment condition). Statistical analyses were carried out using one-way ANOVA followed by Tukey's post hoc test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Interuniversity Attraction Poles initiated by the Belgian Federal Science Policy (networks 6/05, 7/43); the National Institutes of Health (program project P01 AR048564-01A1) and the Fonds national de la recherche scientifique (all to M.V.); the Academy of Finland (136880) and the Finnish Center of Excellence Program 2012-2017 (both to L.E.). N.L. is a ‘chercheur qualifié’ of the F.R.S.-FNRS, Belgium. M.U. was supported by the Fonds spécial de la recherche (FSR) de l'UCL, Belgium, a F.R.S.-FNRS-Télévie fellowship, Belgium and the Patrimoine de la Faculté de Médecine de l'UCL, Belgium.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Bernard Lauwerys for kindly providing access to the GeneSpring software, Ms. Liliana Niculescu for her superb secretarial help, Jaana Träskelin and Elisabeth Verhamme for their excellent technical assistance and Dr Deepti Narasimhaiah for assistance accessing patient tissues.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Calvert J.T., Riney T.J., Kontos C.D., Cha E.H., Prieto V.G., Shea C.R., Berg J.N., Nevin N.C., Simpson S.A., Pasyk K.A., et al. Allelic and locus heterogeneity in inherited venous malformations. Hum. Mol. Genet. 1999;8:1279–1289. doi: 10.1093/hmg/8.7.1279. doi:10.1093/hmg/8.7.1279. [DOI] [PubMed] [Google Scholar]
- 2.Limaye N., Wouters V., Uebelhoer M., Tuominen M., Wirkkala R., Mulliken J.B., Eklund L., Boon L.M., Vikkula M. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat. Genet. 2009;41:118–124. doi: 10.1038/ng.272. doi:10.1038/ng.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nobuhara Y., Onoda N., Fukai K., Hosomi N., Ishii M., Wakasa K., Nishihara T., Ishikawa T., Hirakawa K. TIE2 gain-of-function mutation in a patient with pancreatic lymphangioma associated with blue rubber-bleb nevus syndrome: report of a case. Surg. Today. 2006;36:283–286. doi: 10.1007/s00595-005-3138-9. doi:10.1007/s00595-005-3138-9. [DOI] [PubMed] [Google Scholar]
- 4.Soblet J., Limaye N., Uebelhoer M., Dompmartin A., Vanwijck R., Boon L.M., Vikkula M. Variable somatic TIE2 mutations in half of sporadic venous malformations. J. Mol. Syndromol. doi: 10.1159/000348327. in press doi 10.1159/000348327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vikkula M., Boon L.M., Carraway K.L., III, Calvert J.T., Diamonti A.J., Goumnerov B., Pasyk K.A., Marchuk D.A., Warman M.L., Cantley L.C., et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. doi:10.1016/S0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 6.Wouters V., Limaye N., Uebelhoer M., Irrthum A., Boon L.M., Mulliken J.B., Enjolras O., Baselga E., Berg J., Dompmartin A., et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur. J. Hum. Genet. 2010;18:414–420. doi: 10.1038/ejhg.2009.193. doi:10.1038/ejhg.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris P.N., Dunmore B.J., Tadros A., Marchuk D.A., Darland D.C., D'Amore P.A., Brindle N.P. Functional analysis of a mutant form of the receptor tyrosine kinase Tie2 causing venous malformations. J. Mol. Med. (Berl.) 2005;83:58–63. doi: 10.1007/s00109-004-0601-9. doi:10.1007/s00109-004-0601-9. [DOI] [PubMed] [Google Scholar]
- 8.Korpelainen E.I., Karkkainen M., Gunji Y., Vikkula M., Alitalo K. Endothelial receptor tyrosine kinases activate the STAT signaling pathway: mutant Tie-2 causing venous malformations signals a distinct STAT activation response. Oncogene. 1999;18:1–8. doi: 10.1038/sj.onc.1202288. doi:10.1038/sj.onc.1202288. [DOI] [PubMed] [Google Scholar]
- 9.Hu H.T., Huang Y.H., Chang Y.A., Lee C.K., Jiang M.J., Wu L.W. Tie2-R849W mutant in venous malformations chronically activates a functional STAT1 to modulate gene expression. J. Invest. Dermatol. 2008;128:2325–2333. doi: 10.1038/jid.2008.89. doi:10.1038/jid.2008.89. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y.H., Wu M.P., Pan S.C., Su W.C., Chen Y.W., Wu L.W. STAT1 activation by venous malformations mutant Tie2-R849W antagonizes VEGF-A-mediated angiogenic response partly via reduced bFGF production. Angiogenesis. 2013;16:207–222. doi: 10.1007/s10456-012-9313-x. doi:10.1007/s10456-012-9313-x. [DOI] [PubMed] [Google Scholar]
- 11.Fukuhara S., Sako K., Minami T., Noda K., Kim H.Z., Kodama T., Shibuya M., Takakura N., Koh G.Y., Mochizuki N. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat. Cell Biol. 2008;10:513–526. doi: 10.1038/ncb1714. doi:10.1038/ncb1714. [DOI] [PubMed] [Google Scholar]
- 12.Saharinen P., Eklund L., Miettinen J., Wirkkala R., Anisimov A., Winderlich M., Nottebaum A., Vestweber D., Deutsch U., Koh G.Y., et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat. Cell Biol. 2008;10:527–537. doi: 10.1038/ncb1715. doi:10.1038/ncb1715. [DOI] [PubMed] [Google Scholar]
- 13.Kim I., Kim H.G., So J.N., Kim J.H., Kwak H.J., Koh G.Y. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3’-Kinase/Akt signal transduction pathway. Circ. Res. 2000;86:24–29. doi: 10.1161/01.res.86.1.24. doi:10.1161/01.RES.86.1.24. [DOI] [PubMed] [Google Scholar]
- 14.Papapetropoulos A., Fulton D., Mahboubi K., Kalb R.G., O'Connor D.S., Li F., Altieri D.C., Sessa W.C. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J. Biol. Chem. 2000;275:9102–9105. doi: 10.1074/jbc.275.13.9102. doi:10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- 15.Yoon M.J., Cho C.H., Lee C.S., Jang I.H., Ryu S.H., Koh G.Y. Localization of Tie2 and phospholipase D in endothelial caveolae is involved in angiopoietin-1-induced MEK/ERK phosphorylation and migration in endothelial cells. Biochem. Biophys. Res. Commun. 2003;308:101–105. doi: 10.1016/s0006-291x(03)01341-x. doi:10.1016/S0006-291X(03)01341-X. [DOI] [PubMed] [Google Scholar]
- 16.Brigstock D.R. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61) Angiogenesis. 2002;5:153–165. doi: 10.1023/a:1023823803510. doi:10.1023/A:1023823803510. [DOI] [PubMed] [Google Scholar]
- 17.Fischer C., Mazzone M., Jonckx B., Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat. Rev. Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki Y., Montagne K., Nishihara A., Watabe T., Miyazono K. BMPs promote proliferation and migration of endothelial cells via stimulation of VEGF-A/VEGFR2 and angiopoietin-1/Tie2 signalling. J. Biochem. 2008;143:199–206. doi: 10.1093/jb/mvm215. doi:10.1093/jb/mvm215. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Q., Heinke J., Vargas A., Winnik S., Krauss T., Bode C., Patterson C., Moser M. ERK signaling is a central regulator for BMP-4 dependent capillary sprouting. Cardiovasc. Res. 2007;76:390–399. doi: 10.1016/j.cardiores.2007.08.003. doi:10.1016/j.cardiores.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Maisonpierre P.C., Suri C., Jones P.F., Bartunkova S., Wiegand S.J., Radziejewski C., Compton D., McClain J., Aldrich T.H., Papadopoulos N., et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. doi:10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong L.C., Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2. doi:10.1016/S0945-053X(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 22.Lin E.A., Liu C.J. The role of ADAMTSs in arthritis. Protein Cell. 2010;1:33–47. doi: 10.1007/s13238-010-0002-5. doi:10.1007/s13238-010-0002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter S., Clark I.M., Kevorkian L., Edwards D.R. The ADAMTS metalloproteinases. Biochem. J. 2005;386:15–27. doi: 10.1042/BJ20040424. doi:10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Essaghir A., Toffalini F., Knoops L., Kallin A., van Helden J., Demoulin J.B. Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucleic Acids Res. 2010;38:e120. doi: 10.1093/nar/gkq149. doi:10.1093/nar/gkq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daly C., Wong V., Burova E., Wei Y., Zabski S., Griffiths J., Lai K.M., Lin H.C., Ioffe E., Yancopoulos G.D., et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1) Genes Dev. 2004;18:1060–1071. doi: 10.1101/gad.1189704. doi:10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Potente M., Urbich C., Sasaki K., Hofmann W.K., Heeschen C., Aicher A., Kollipara R., DePinho R.A., Zeiher A.M., Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Invest. 2005;115:2382–2392. doi: 10.1172/JCI23126. doi:10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brouillard P., Boon L.M., Mulliken J.B., Enjolras O., Ghassibe M., Warman M.L., Tan O.T., Olsen B.R., Vikkula M. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (‘glomangiomas’) Am. J. Hum. Genet. 2002;70:866–874. doi: 10.1086/339492. doi:10.1086/339492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hosaka T., Biggs W.H., III, Tieu D., Boyer A.D., Varki N.M., Cavenee W.K., Arden K.C. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc. Natl Acad. Sci. USA. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. doi:10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hellstrom M., Kalen M., Lindahl P., Abramsson A., Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 30.Lindahl P., Johansson B.R., Leveen P., Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. doi:10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 31.Hellstrom M., Gerhardt H., Kalen M., Li X., Eriksson U., Wolburg H., Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. doi:10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888. doi:10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- 33.Li L., Blumenthal D.K., Masaki T., Terry C.M., Cheung A.K. Differential effects of imatinib on PDGF-induced proliferation and PDGF receptor signaling in human arterial and venous smooth muscle cells. J. Cell Biochem. 2006;99:1553–1563. doi: 10.1002/jcb.20993. doi:10.1002/jcb.20993. [DOI] [PubMed] [Google Scholar]
- 34.Li L., Blumenthal D.K., Terry C.M., He Y., Carlson M.L., Cheung A.K. PDGF-induced proliferation in human arterial and venous smooth muscle cells: molecular basis for differential effects of PDGF isoforms. J. Cell Biochem. 2011;112:289–298. doi: 10.1002/jcb.22924. doi:10.1002/jcb.22924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redondo P., Aguado L., Marquina M., Paramo J.A., Sierra A., Sanchez-Ibarrola A., Martinez-Cuesta A., Cabrera J. Angiogenic and prothrombotic markers in extensive slow-flow vascular malformations: implications for antiangiogenic/antithrombotic strategies. Br. J. Dermatol. 2010;162:350–356. doi: 10.1111/j.1365-2133.2009.09513.x. doi:10.1111/j.1365-2133.2009.09513.x. [DOI] [PubMed] [Google Scholar]
- 36.Ory D.S., Neugeboren B.A., Mulligan R.C. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl Acad. Sci. USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. doi:10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.