

Abstract
The muscleblind-like (MBNL) genes encode alternative splicing factors that are essential for the postnatal development of multiple tissues, and the inhibition of MBNL activity by toxic C(C)UG repeat RNAs is a major pathogenic feature of the neuromuscular disease myotonic dystrophy. While MBNL1 controls fetal-to-adult splicing transitions in muscle and MBNL2 serves a similar role in the brain, the function of MBNL3 in vivo is unknown. Here, we report that mouse Mbnl3, which encodes protein isoforms that differ in the number of tandem zinc-finger RNA-binding motifs and subcellular localization, is expressed primarily during embryonic development but also transiently during injury-induced adult skeletal muscle regeneration. Mbnl3 expression is required for normal C2C12 myogenic differentiation and high-throughput sequencing combined with cross-linking/immunoprecipitation analysis indicates that Mbnl3 binds preferentially to the 3′ untranslated regions of genes implicated in cell growth and proliferation. In addition, Mbnl3ΔE2 isoform knockout mice, which fail to express the major Mbnl3 nuclear isoform, show age-dependent delays in injury-induced muscle regeneration and impaired muscle function. These results suggest that Mbnl3 inhibition by toxic RNA expression may be a contributing factor to the progressive skeletal muscle weakness and wasting characteristic of myotonic dystrophy.
INTRODUCTION
Myotonic dystrophy (dystrophia myotonica, DM), one of the most variable human hereditary diseases, is caused by microsatellite expansions in two distinct genes (1). DM type 1 (DM1) results from (CTG)37->3500 expansions in the 3′ untranslated region (UTR) of the DMPK gene, while (CCTG)75-∼11 000 repeats in the first intron of CNBP cause DM2 (2,3). Characteristic clinical features of adult-onset DM1 and DM2 include myotonia (muscle hyperexcitability), cardiac arrhythmia, subcapsular ocular cataracts and progressive muscle weakness/wasting with early stage involvement of hand and forearm muscles leading to functional impairment. In contrast to DM2, DM1 is also a congenital disorder (CDM) when repeats exceed ∼1000 CTGs with characteristic features of neonatal hypotonia, mental retardation and pulmonary insufficiency.
Although several pathogenic models have been proposed to explain how CTG and CCTG, or C(C)TG, repeat expansions in non-coding regions result in a dominantly inherited disorder, current evidence argues that DM disease is primarily RNA-mediated (3,4). Mutant DMPK and CNBP C(C)UG expansion [C(C)UGexp] RNAs fold into stable double-stranded (ds)RNA stem-loop structures which disrupt the activities of alternative splicing factors during postnatal development, including the CUGBP1/ETR3-like factors (CELF) and the muscleblind-like (MBNL) proteins (5–8). Three MBNL genes exist in mammals (MBNL1, MBNL2, MBNL3) and MBNL1 functions as a CELF1 antagonist to promote the skipping of fetal, and inclusion of adult, exons during postnatal development. Loss of MBNL1 splicing activity due to sequestration by C(C)UGexp RNAs leads to the persistence of fetal isoforms in adult muscles (7,9,10) and characteristic features of DM muscle including myotonia, which is caused by aberrant inclusion of the skeletal muscle chloride channel ClC-1/CLCN1 fetal exon 7a in adults (11,12). Support for this toxic RNA model comes from Mbnl1 (Mbnl1ΔE3/ΔE3) and Mbnl2 (Mbnl2ΔE2/ΔE2) isoform knockout mice which recapitulate multiple features of adult-onset DM (13,14). However, these models fail to develop adult-onset muscle wasting, a characteristic DM manifestation or the neonatal features associated with CDM, and additional RNA-binding proteins have also been implicated in DM pathogenesis, including hnRNP H, p68/DDX5 and Staufen1 (15–17).
The RNA-mediated pathogenesis model predicts that all three MBNL (MBNL1– MBNL3) proteins are sequestered by dsCUG RNAs in the nucleus (6,18). While both MBNL1 and MBNL2 are splicing factors, MBNL1 plays a primary role in alternative splicing in skeletal and cardiac muscle and MBNL2 serves a related function in the central nervous system (4,14). Previous studies have indicated that MBNL3 is an unusual member of the MBNL gene family. RNA blot, microarray profiling and reverse transcription polymerase chain reaction (RT–PCR) analyses indicate that mouse Mbnl3 is expressed at a very low level in adult tissues but is detectable by RT–PCR in the lung, spleen and thymus (19–21). Mbnl3 RNA and protein levels decrease during C2C12 myoblast differentiation and constitutive overexpression of human MBNL3, or mouse Mbnl3, inhibits MyoD-dependent gene expression and myotube formation in vitro (20,22,23). These results led to the suggestion that MBNL3 acts as an antagonist of myogenesis possibly by maintaining myoblasts in a proliferative state. Here, we report that the knockdown of mouse Mbnl3 RNA in C2C12 myoblasts delays myotube formation and Mbnl3 isoform knockout mice show age-dependent impairment of adult muscle regeneration. Binding site analysis using high-throughput sequencing combined with cross-linking/immunoprecipitation (HITS-CLIP) indicates that Mbnl3 targets YGCY motifs primarily in non-coding regions. Our results expand the functional repertoire of the Mbnl proteins and suggest that multiple cellular pathways may be adversely affected by the expression of microsatellite expansion RNAs.
RESULTS
Mbnl3 is both a nuclear and cytoplasmic RNA-binding protein
To clarify Mbnl3 protein functions in myogenic differentiation in vitro and muscle function in vivo, we first generated a monospecific anti-Mbnl3 antibody reactive against an epitope represented in the majority of Mbnl3 isoforms but which was absent in Mbnl1 and Mbnl2. To identify this epitope, mouse Mbnl3 alternative splicing patterns in several tissues were determined during embryonic, neonatal and adult periods using RT–PCR followed by cDNA cloning and sequencing (Supplementary Material, Fig. S1A and B). Although this analysis detected multiple Mbnl3 proteins varying in the C-terminal exon 7, the majority of the Mbnl3 mRNAs expressed in all tissues contained exon 7C. This result agrees with prior observations that Mbnl proteins share high overall sequence similarity but tend to be variable at the C terminus, which mediates interactions between Mbnl proteins (24). Because most tissues express Mbnl3 isoforms containing exon 7C spliced directly to exon 8, a rabbit polyclonal antibody (Mb3/7C) was generated using a 15-amino acid C-terminal peptide encoded by these two exons (Supplementary Material, Fig. S1A). By immunoblot analysis, Mb3/7C specifically detected myc-tagged murine Mbnl3 protein (Mbnl3-myc) but not Mbnl1-myc or Mbnl2-myc transiently expressed in COSM6 cells, which do not express endogenous Mbnl3 (Supplementary Material, Fig. S1C). Mbnl3-myc was also specifically immunopurified by Mb3/7C, while an anti-myc antibody purified all three myc-tagged Mbnl proteins (Supplementary Material, Fig. S1D).
When Mb3/7C antibody was used to detect Mbnl3 in C2C12 myoblasts, two proteins (27 and 38 kDa) were detected by immunoblot analysis with the larger 38 kDa protein the more abundant isoform (Fig. 1A and B). Several approaches were used to confirm that both proteins were encoded by the Mbnl3 gene. First, siRNA-mediated knockdowns resulted in an equivalent decrease in both Mbnl3 proteins, as determined by immunoblotting with Mb3/7C, whereas Mbnl1 protein levels were unaffected (Fig. 1A). Second, immunoprecipitation followed by in-gel tryptic digestion and analysis by liquid chromatography followed by mass spectrometry (LCMS), as well as cDNA cloning and sequencing, demonstrated that these two isoforms use translation initiation codons in either exon 2, to generate the 38 kDa protein with two tandem ZnF motifs (hereafter referred to as Mbnl3ZnF1-4), or exon 3, which produces the 27 kDa isoform with a single tandem ZnF (Mbnl3ZnF3/4) (Fig. 1B). Because tandem ZnF motifs are important for high-affinity binding of Mbnl proteins to dsCUG RNAs and the formation of nuclear RNA foci (25), we next determined whether both of these isoforms localized to the nucleus. Surprisingly, immunoblotting of subcellular fractions revealed that the Mbnl3ZnF1-4 isoform was distributed in both nuclear and cytoplasmic fractions in both C2C12 myoblasts (Fig. 1C, left panel) and mouse embryonic forelimbs (Fig. 1C, right panel), while the shorter isoform was predominantly cytoplasmic. Immunocytochemistry confirmed that Mbnl3 localized primarily to the nucleus but was also present in the cytoplasm of C2C12 cells (Supplementary Material, Fig. S1E). Although detectable in cytoplasmic foci, Mbnl3 did not co-localize with the P-body protein GW182.
Figure 1.

Nuclear and cytoplasmic localization of Mbnl3 isoforms. (A) Immunoblot analysis showing siRNA-mediated knockdown of both Mbnl3ZnF1-4 (38 kDa) and Mbnl3ZnF3/4 (27 kDa) isoforms in C2C12 myoblasts. (B) Illustration of the primary structure of Mbnl3 isoforms with one (Mbnl3ZnF3/4) or two (Mbnl3ZnF1-4) tandem ZnF motifs (CCCH ZnF motifs, blue boxes). The linker region between the two pairs of tandem ZnF motifs (red boxes) is also indicated. (C) The Mbnl3ZnF1-4 isoform is both nuclear and cytoplasmic, while Mbnl3ZnF3/4 is only detectable in C2C12 and muscle cytoplasmic, fractions. Nuclear and cytoplasmic fractions were isolated from either C2C12 cells or muscle (quadriceps) tissue and immunoblotted with antibodies against Mbnl3, Mbnl1 (family member control), Celf1 (nuclear marker) and Ldha (cytoplasmic marker). (D) The Mbnl3ZnF1-4 protein associates with polysomes, while Mbnl3ZnF3/4 remains at the top of the gradient.
MBNL proteins have been proposed to function in both nuclear pre-mRNA splicing and cytoplasmic mRNA localization (7,13,26,27). However, both Mbnl1 and Mbnl2 are predominantly nuclear proteins and shorter Mbnl1ZnF3/4 and Mbnl2ZnF3/4 protein isoforms have not been detected immunologically. Since Mbnl3 showed a different localization pattern, the cytoplasmic distribution of Mbnl3 was assessed by polysome analysis. Interestingly, the Mbnl3ZnF1-4 isoform, which localized to both the nucleus and cytoplasm, co-fractionated with polysomes, while the majority of the cytoplasmic Mbnl3ZnF3/4 protein remained at the top of the gradient (Fig. 1D). Polysome disruption with either puromycin or ethylenediamine tetraacetic acid (EDTA) led to a shift of the Mbnl3ZnF1-4 isoform to lower density fractions (Supplementary Material, Fig. S2). Because these results suggested that the larger Mbnl3 isoform might be required for the regulation of specific cytoplasmic mRNAs, we next identified the RNA binding sites for the Mbnl3ZnF1-4 protein.
Preferential binding of Mbnl3 to mRNA 3′ UTRs in C2C12 myoblasts
Mbnl3 RNA-binding targets in C2C12 myoblasts were identified using HITS-CLIP (28). Covalent RNA–protein complexes, generated by exposure to UV light, were treated with RNase A to generate RNA tags for subsequent sequencing. Complexes were identified by protein gel electrophoresis and only RNA–protein complexes ∼12–15 kDa larger than the Mbnl3 38 kDa isoform were excised from the gel. This protocol minimized the sequencing of RNAs cross-linked to the 27 kDa protein. Interestingly, Mbnl3 showed reduced binding to C2C12 myoblast intronic targets (7% of total targets) compared with a recent analysis of Mbnl1 (65.5%) (27) with elevated binding to 3′ UTRs (57% for Mbnl3; 18.5% for Mbnl1) of target mRNAs (Fig. 2A). None of the Mbnl3 target introns identified in C2C12 cells flank known alternatively spliced exons that have been implicated previously in DM. Prior studies have noted that Mbnl1 and Mbnl2 proteins recognize a YGCY (Y = pyrimidine) binding motif (8,14,27,29), and this was also the preferred recognition site for Mbnl3, although YGCY motifs were reiterated in the Mbnl3 38 kDa data set with either −1 (Fig. 2B), 0 (YGCYYGCY) or +1 (YGCYYYGCY) spacing.
Figure 2.

Mbnl3 primarily targets mRNA 3′ UTRs in C2C12 myoblasts. (A) Pie chart showing a unique CLIP tag distribution with the majority in 3′ UTRs. (B) Mbnl3 RNA binding motif. (C) Example of CLIP tag distribution on the top scoring Mbnl3 target, the Canx 3′ UTR. Gene orientation is right to left (5′ to 3′) showing the Canx 3′ exon (ORF, blue rectangle; 3′ UTR, black line) and CLIP tags obtained from three biological replicates (red, green and turquoise rectangles with inset orientation arrows). (D) RNA levels assayed by qRT–PCR for the top scoring Mbnl3 CLIP targets S100a4, Canx, Myh9, Spp1 following siRNA-mediated knockdown of Mbnl3 (siMbnl3) versus control non-targeting siRNA. Data are SEM (n = 3) and significant (*P < 0.05, **P < 0.01, ***P < 0.001). (E) GO analysis highlighting molecular pathways affected by Mbnl3 depletion in C2C12 cells.
Mbnl proteins are important for calcium homeostasis/signaling and alterations in calcium homeostasis results in ER stress (30). Interestingly, the top five Mbnl3 RNA targets included Canx (calnexin) (Fig. 2C), a calcium-binding ER-associated factor up-regulated in DM1 skeletal muscle (31) and Spp1 (secreted phosphoprotein, osteopontin), a secreted glycoprotein/cytokine expressed by myoblasts and up-regulated in dystrophic muscles (32–34). Also included in this RNA target set were S100a4, a member of the dimeric EF-hand calcium-binding S100 protein family that regulates non-muscle myosin II-A dissociation (35,36), and Myh9, the non-muscle myosin IIA heavy chain involved in diverse functions including cell morphology, motility and division. Specific interactions between Mbnl3 and these RNAs were confirmed using RNA immunoprecipitation analysis (Supplementary Material, Fig. S3A). Since Mbnl3 preferentially targeted the 3′ UTRs of these mRNAs (Supplementary Material, Table S1), including Canx (Fig. 2C), we assessed the impact of Mbnl3 depletion on the mRNA levels of several of these top targets by qRT–PCR. We discovered that Mbnl3 knockdown led to an increase in Canx/calnexin, Spp1/osteopontin and Myh9, but a decrease in S100a4, mRNA levels (Fig. 2D). To determine if Mbnl3 bound to RNAs that were functionally linked, gene ontology (GO) analysis was performed on Mbnl3 CLIP targets (Fig. 2E) and cell growth and proliferation was the top scoring category. Overall, HITS-CLIP analysis revealed that Mbnl3 binds to C2C12 mRNA 3′ UTRs via the recognition of a core YGCY motif, a characteristic shared by Mbnl1 and Mbnl2, and loss of Mbnl3 binding to specific mRNAs results in alterations in mRNA levels, some of which have been previously observed in DM1 (Canx) and other dystrophic muscles (Spp1).
Mbnl3 depletion delays C2C12 differentiation
An earlier study reported that ectopic overexpression of either human MBNL3, or mouse Mbnl3, inhibits C2C12 differentiation and myosin heavy chain (MyHC) induction, while an antisense morpholino (ASO) targeting the translation initiation codon in Mbnl3 exon 2 has the opposite effect (22). Because sequences targeted by this ASO are absent in Mbnl3ZnF3/4 mRNA, we tested the effect of depletion of all endogenous mouse Mbnl3 isoforms on C2C12 differentiation. C2C12 cells, grown on Matrigel-coated plates to accelerate myogenic differentiation, were treated with either control or Mbnl3 siRNAs for 48 h followed by replacement with differentiation media. Mbnl3 siRNA-treated cells showed a delay in the appearance of muscle-specific gene expression as monitored by MyHC expression and myogenin levels, which were noticeably reduced following Mbnl3 depletion (Fig. 3A). A more striking effect on myotube formation was observed by microscopy, particularly at 48 h after transfer into the differentiation medium (Fig. 3B). Quantitative analysis of MyHC positive cells with >2 nuclei at the 48 h time point showed a significant reduction in the number of myotubes in Mbnl3 siRNA versus control siRNA-treated C2C12 cells (Fig. 3C). Based on these observations, we concluded that Mbnl3 expression was required for initiating optimal myogenic differentiation in vitro.
Figure 3.
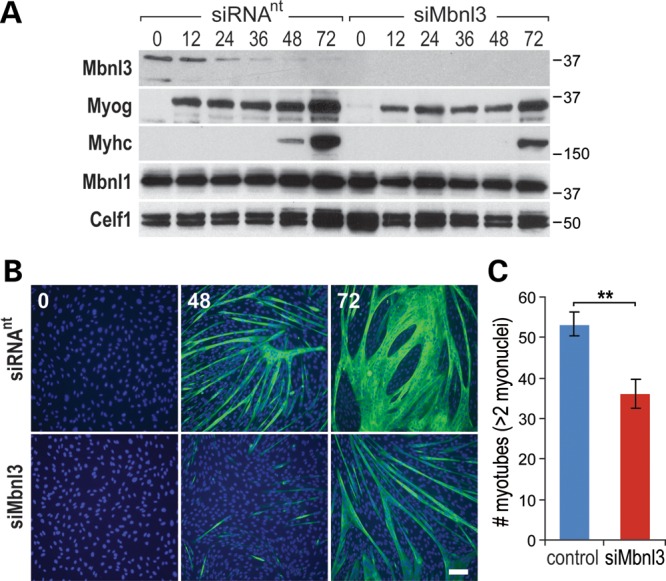
Loss of Mbnl3 expression inhibits C2C12 myogenic differentiation. (A) C2C12 myoblasts were grown on plates supplemented with Matrigel, transfected with either non-targeting (siRNAnt) or Mbnl3 (siMbnl3) siRNA pools, and then induced to differentiate 24 h post-siRNA treatment. Mbnl3, Mbnl1 and Celf1 protein levels were monitored by immunoblot analysis for 0–72 h post-induction and differentiation status was assessed using antibodies against myogenin (Myog) and Myhc. (B) Cell immunofluorescence analysis of C2C12 myotube formation showing impaired myogenic differentiation at 48 and 72 h after transfer to differentiation media following siMbnl3-induced knockdown compared with siRNAnt control (scale bar = 50 μM). (C) Myotube number (>2 myonuclei/myotube per field) decreases following Mbnl3 depletion. Data are SEM and significant (**P < 0.01).
Mbnl3 expression peaks during embryogenesis and adult muscle regeneration
Mbnl1 and Mbnl2 regulate alternative splicing during postnatal development in muscle and the brain, respectively (7,13,14). Mbnl3 is an unusual member of the Mbnl family because the peak expression of this gene occurs early during the embryonic period and Mbnl3 RNA is either absent or detectable only at low levels in adult tissues (19). These expression characteristics argue that, in contrast to Mbnl1 and Mbnl2, Mbnl3 may not be essential in adults but may be required during embryogenesis and its sequestration by toxic C(C)UG RNAs during embryogenesis might influence tissue development in DM1 and DM2. This possibility prompted us to evaluate Mbnl3 spatial and temporal expression patterns during both embryonic and postnatal developmental periods. Total RNA blot analysis confirmed that two major RNAs (1.5, 9.8 kb), which differed in alternative poly(A) site selection and 3′ UTR length (Supplementary Material, Fig. S4A), were expressed at embryonic day (E)15 in placenta but declined by E18 so they were undetectable in most adult tissues examined except for the lung and spleen (Supplementary Material, Figs S1B and S4B). Interestingly, the longer Mbnl3 9.8 kb mRNA was preferentially expressed in both embryonic and adult tissues. To determine Mbnl3 protein expression in specific mouse tissues, immunoblot analysis was performed using the Mb3/7C antibody (Fig. 4A). Placenta (E15) was used as a control, since Mbnl3 RNA levels are relatively high in this tissue (18,19). In contrast to Mbnl3, Mbnl1 protein was readily detectable in adult tissues. This pattern correlated with Mbnl1 and Mbnl3 RNA fetal and adult expression patterns (Supplementary Material, Fig. S1B and S4B) with the exception of a cross-reacting, slightly slower migrating protein that was detected by the Mb3/7C antibody in adult brain (Fig. 4A). However, this protein was identified as glutamine synthetase (Glul), a protein that is abundantly expressed in astroglia. No antibody cross-reaction with other proteins was observed in any other tissues examined including skeletal muscle.
Figure 4.

Mbnl3 expresses two major isoforms during embryonic development. (A) Immunoblot analysis of Mbnl3 protein expression during embryonic (E15, E18), neonatal (P1) and adult periods using the anti-Mbnl3 (Mb3/7) antibody. The band in adult brain (*) migrating at 40 kDa is glutamine synthetase (Glul); Mbnl1 and Gapdh expression patterns are also shown. (B) Transverse sections of TA muscle (mice were 10 weeks of age) either prior to or various (1,3,5,7) days following, notexin injection and stained with H&E (top row) or anti-desmin antibody (bottom row). Note that on day 3, most myofibers are absent and the majority of cells are mononuclear (scale bar = 50 μM). (C) Mbnl3ZnF1-4 protein is only detectable at day 3 PI (Mbnl3ZnF3/4 was not detectable even with longer exposure times). Immunoblot analysis showing expression levels of Mbnl3, myogenin (Myog), Mbnl1 and Gapdh (loading control). Note that expression of Mbnl3 parallels myogenin. (D) RT–PCR analysis showing RNA levels for Mbnl3, myogenin (Myog), Mbnl1, Dmpk (DM1) and Cnbp (DM2). Ppia is the loading control.
Because Mbnl3 is expressed in C2C12 myoblasts, which were originally isolated from regenerating adult skeletal muscle (37), we postulated that this gene might also be expressed during adult muscle regeneration. To test this hypothesis, we injected tibialis anterior (TA) muscles of 10-week-old mice with notexin, a myotoxic phospholipase A2, to induce rapid muscle damage and then followed the time course of muscle regeneration. Notexin-induced injury/regeneration is a rapid process with necrosis, myoblast proliferation, which is prominent by day 3 post-injection (PI), and then muscle regeneration by day 7 PI, although many of the myofibers at this endpoint have centralized nuclei (Fig. 4B). Remarkably, Mbnl3 protein, and only the larger Mbnl3ZnF1-4 isoform, was detectable at day 3 PI (Fig. 4C) which corresponds to the peak of Mbnl3 RNA, as well as myogenin (Myog) RNA and protein expression (Fig. 4C and D). In contrast, Mbnl1 RNA and protein were expressed throughout the regeneration time course, although there was a transient decrease in Mbnl1 RNA and protein during necrosis at day 1 PI. We confirmed that the majority of mononuclear cells at day 3 PI were myoblasts by anti-desmin staining in agreement with the expression of Mbnl3 in C2C12 myoblasts (Fig. 4B). Importantly, Dmpk and Cnbp mRNA expression was seen throughout the 7-day time course (Fig. 4D). Since the Mbnl3ZnF1-4 protein was the predominant isoform detectable during all developmental stages, Mbnl3 isoform knockout mice were generated to determine if the full-length Mbnl3 was essential for normal muscle development and regeneration.
Mbnl3 isoform knockout mice and identification of Mbnl3 RNA targets in vivo
Mbnl3ΔE2/ΔE2 isoform knockout mice were generated using a homologous recombination strategy previously described for Mbnl1ΔE3/ΔE3 and Mbnl2ΔE2/ΔE2 knockouts (Supplementary Material, Fig. S5A). Mbnl3 is an X-linked gene so, for simplicity, we refer to Mbnl3 isoform knockouts as Mbnl3ΔE2 mice, although our analysis included both males and females. Genomic blot analysis confirmed the germline deletion of Mbnl3 exon2 in Mbnl3ΔE2 mice (Fig. 5A). Interestingly, the deletion of exon 2 inclusive isoforms resulted in the up-regulation of Mbnl3 RNA and protein encoding the shorter Mbnl3ZnF3/4 isoform in E15 forelimb (Fig. 5B and C).
Figure 5.

Mouse Mbnl3 knockout characterization and Mbnl3 muscle RNA targets in vivo. (A) Genomic DNA blot of wild-type (Mbnl3+) and Mbnl3 isoform knockout (Mbnl3ΔE2), showing the reduction in KpnI fragment length in Mbnl3ΔE2 knockouts. (B) RT–PCR using primers in Mbnl3 exons 1 and 8 (top panel) or exons 2 and 8 (middle) and wild-type (Mbnl3+) and Mbnl3 isoform knockout (Mbnl3ΔE2) skeletal muscle RNA. Ppia was included as a loading control. (C) Immunoblot analysis of Mbnl3, Mbnl1 and Gapdh (loading control) protein in Mbnl3+ versus Mbnl3ΔE2 knockout skeletal muscle. Note the up-regulation of Mbnl3ZnF3/4 following loss of Mbnl3ZnF1-4. (D) Mbnl3ZnF3/4 relocalizes to the nucleus in Mbnl3ΔE2 isoform knockouts. Immunoblots were performed with anti-Mbnl3 or anti-Ldha (cytoplasmic marker). In comparison with Figure 1C, Mbnl3ZnF3/4 is equivalently distributed in the nucleus and cytoplasm in Mbnl3ΔE2 mutants. (E) Mbnl3 RNA binding motifs in embryonic (E15) forelimb determined by HITS-CLIP and MEME analysis of wild-type (Mbnl3+) versus Mbnl3ΔE2 knockouts. (F) Pie chart summaries of CLIP tag distribution of wild-type mouse embryonic day (E)15 forelimb for Mbnl3ZnF1-4 (top) versus Mbnl3ZnF3/4 in Mbnl3ΔE2 (bottom) mice. (G) Wiggle plots of Mbnl3 binding sites on Igf2 pre-mRNA in Mbnl3+ wild-type (blue, Mbnl3ZnF1-4), and Mbnl3ΔE2 knockout (red, Mbnl3ZnF3/4), E15 forelimbs. Gene orientation is right to left (5′ to 3′) with coding (large boxes) and non-coding (small boxes) regions and introns (lines with orientation arrows).
Mbnl3ΔE2 mice were viable and did not display fetal or early postnatal muscle, or other, overt phenotypes. The lack of DM-associated manifestations was surprising, although DM-relevant phenotypes are generally adult-onset in Mbnl1ΔE3/ΔE3 and Mbnl2ΔE2/ΔE2 knockouts. However, Mbnl1 and Mbnl2 knockouts do not show up-regulation of shorter Mbnl1 or Mbnl2 ZnF3/4 isoforms, so we tested the possibility that the Mbnl3ZnF3/4 protein might relocalize into the nucleus and compensate for the loss of Mbnl3ZnF1-4 nuclear function. In support of this idea, the Mbnl3ZnF3/4 protein localized primarily to the cytoplasm in wild-type muscle (Fig. 1C) but loss of the full-length isoform in Mbnl3ΔE2 mutants resulted in equivalent levels of the shorter Mbnl3ZnF3/4 protein in the nucleus and cytoplasm (Fig. 5D). Interestingly, HITS-CLIP analysis (Fig. 5E) of E15 Mbnl3 wild-type forelimb for the major Mbnl3ZnF1-4 isoform showed an altered genomic distribution pattern compared with C2C12 myoblasts with the number of intron tags increased from 7 to 38%, while 3′ UTR tags decreased (57 to 38%) in the embryonic tissue (compare Fig. 2A and Fig. 5F, top pie chart). A similar pattern was observed for the Mbnl3ZnF3/4 protein in Mbnl3ΔE2 E15 forelimb (Fig. 5F, bottom) and the binding sites for these two isoforms generally overlapped as illustrated for insulin-like growth factor 2 (Igf2, Fig. 5G). IGF2/IGF-II is an important regulator of skeletal myogenesis during embryonic development in vivo and myogenic differentiation in vitro (38,39), and the distribution of HITS-CLIP tags indicated that Mbnl3 might influence both Igf2 5′ exon use (Fig. 5G, isoform 2) and 3′ end formation (isoforms 1–3). To determine if loss of Mbnl3ZnF1-4 led to significant mis-splicing during fetal development, RNA-seq was performed on Mbnl3ΔE2 E15 forelimb. In striking contrast to Mbnl1 and Mbnl2 isoform knockouts, Mbnl3ΔE2 showed relatively modest changes in splicing patterns with some variation among samples of the same genotype but none of these events were validated by RT–PCR (Supplementary Material, Table S2 and data not shown). Additionally, gene expression changes were also modest compared with C2C12 myoblasts following siRNA-mediated knockdown of both Mbnl3 isoforms (Fig. 2D and data not shown), suggesting functional compensation by the up-regulation of the shorter Mbnl3ZnF3/4 isoform. Since Mbnl3 was transiently expressed during muscle regeneration (Fig. 4C and D), and Igf2 was a direct binding target (Supplementary Material, Table S1 and Fig. S3B), we next tested if the loss of Mbnl3 influenced the regenerative process in vivo.
Progressive impairment of muscle regeneration in Mbnl3 knockout mice
Injury-induced muscle regeneration was normal in young (10 weeks of age) Mbnl3ΔE2 knockouts (Supplementary Material, Fig. S5B). However, the muscle satellite cell pool decreases with age and regenerative defects become more conspicuous during aging (40,41). Therefore, Mbnl3ΔE2 mice were also evaluated at a later stage (>32 weeks of age) for muscle function and regeneration defects. These older mice showed delayed muscle regeneration, as monitored by H&E staining (Fig. 6A) and the number of MyHC positive myofibers (Fig. 6B), but Mbnl3 was still transiently expressed at day 3 in wild-type muscles from older animals (Fig. 6C). The myofiber cross-sectional area and number (day 5 after notexin injection) (Fig. 6D) and grip strength (Fig. 6E) were significantly reduced in Mbnl3ΔE3 knockout mice. Given the important role of the MBNL gene family in myotonic dystrophy, these results suggest that Mbnl3ΔE3 mice provide a novel model for age-associated muscle atrophy in this disease.
Figure 6.

Impaired muscle regeneration in Mbnl3ΔE2 knockouts. (A) H&E (scale bar = 50 μM) and (B) MyHC (scale bar = 100 μM) immunofluorescence of transverse TA muscle sections from Mbnl3+ and Mbnl3ΔE2 8-month-old mice before (0) or 3, 5 and 7 days post-notexin injection to induce muscle regeneration. (C) Immunoblot confirming that the Mbnl3ZnF1-4 protein is detectable at day 3 PI during TA regeneration of older (8-month old) Mbnl3+ wild-type mice. (D) Bar graphs showing that the fiber cross-sectional area (left) and fiber number (right) in Mbnl3ΔE2 knockouts (red) are significantly reduced compared to wild-type (blue) following notexin treatment (+notexin) but are not affected in muscles in the absence of notexin (-notexin). (E) Grip strength assay indicating average force exerted by forelimb muscles in wild-type (Mbnl3+) versus Mbnl3ΔE2 mutant mice. Data in (C) and (D) are SEM (n = 3) and significant (***P < 0.001).
DISCUSSION
The MBNL loss-of-function model for DM proposes that this multi-systemic disease is caused by sequestration of the MBNL proteins by C(C)UG repeat expansion RNAs (2,6,42). While Mbnl knockout mice have highlighted primary roles for MBNL1 and MBNL2 in adult-onset DM and alternative splicing regulation during postnatal development of skeletal muscle and the brain, respectively, nothing is known about the function(s) of MBNL3 in vivo, although this protein is also sequestered by toxic CUGexp RNAs (18). Here, we characterize the temporal and spatial expression patterns of mouse Mbnl3 RNA and protein, generate and characterize novel Mbnl3 isoform knockout mice and demonstrate an unanticipated role for Mbnl3 in muscle regeneration during aging. Our results support the MBNL combinatorial loss-of-function model for DM (6,14) and suggest that studies on Mbnl3 functions in vivo will provide unique insights into additional cellular pathways influenced by microsatellite expansion mutations.
MBNL loss-of-function model for DM: roles for MBNL3
The MBNL proteins were first identified as 40–45 kDa factors that bind and photocross-link with UV light to expanded ds, but not primarily single-stranded, CUG repeat RNAs under in vitro splicing/polyadenylation conditions where MBNL proteins must compete with many other RNA-binding factors (6). RNA recognition by MBNL proteins is mediated by one or two pairs of tandem CCCH ZnF motifs (ZnF1/2, ZnF3/4) and MBNL isoforms with a single tandem ZnF pair have a reduced affinity for C(C)UG repeat RNAs (25,43). A recent study that evaluated the effects of various MBNL1 ZnF mutations using cell-based splicing assays concluded that these ZnF pairs possess different RNA-binding affinities with the N-terminal ZnF1/2 having a higher affinity (44). Importantly, the ZnF1/2 and ZnF3/4 pairs do not possess equivalent splicing functions and a single MBNL1 tandem ZnF pair maintains ∼80% of wild-type splicing activity for some RNA targets. MBNL3 splicing activation and repression activity requires a 74-amino acid region located between the ZnF1/2 and ZnF3/4 pairs (45). In contrast to our prior reports on Mbnl1 and Mbnl2 isoform knockouts (13,14), in which the exon encoding the first translation initiation codon was deleted, Mbnl3ΔE2 mice showed elevated expression of the Mbnl3ZnF3/4 isoform that re-localized to the nucleus in skeletal muscle. This Mbnl3 isoform is missing ZnF1/2 but contains most of the splicing regulatory domain, although the N-terminal 14 amino acids of this 74-amino acid region are absent. Therefore, this truncated Mbnl3 isoform or the Mbnl1 and/or Mbnl2 proteins, which are also expressed in embryonic muscle and during regeneration, may compensate for the loss of the Mbnl3ZnF1-4 full-length protein.
Overexpression of human or mouse Mbnl3 in C2C12 cells promotes myoblast proliferation while antagonizing myogenic differentiation via inhibition of MyoD-dependent transcription (22,23,46). Constitutive overexpression of Mbnl3 in C2C12 cells causes a delay in differentiation and blocking Mbnl3 expression, using an ASO to inhibit Mbnl3ZnF1-4 translation, accelerated MyHC expression and C2C12 differentiation (22). These results led to the current model that MBNL3 functions as an inhibitor of muscle differentiation (46). To test this model and clarify Mbnl3 functions in vivo, we first analyzed mouse Mbnl3 RNA and protein expression in both embryonic and adult skeletal muscle. Mbnl3 was expressed during embryonic muscle development but also transiently in adult skeletal muscle following injury-induced regeneration. Surprisingly, Mbnl3 siRNA-mediated knockdown delayed C2C12 myotube formation. Since experimental differences (ASO translation inhibition versus knockdowns) might account for the discrepancy between our results and earlier studies (22,23,46), we analyzed the effects of Mbnl3 loss in vivo using an isoform knockout strategy previously successful for analyzing Mbnl1 and Mbnl2 functions (13,14). In contrast to Mbnl1 and Mbnl2 isoform knockouts, Mbnl3ΔE2 mutants fail to develop overt mutant muscle or CNS phenotypes during postnatal development but instead show a late-onset and age-associated impairment of muscle regeneration following injury. Progressive muscle weakness and wasting is a debilitating manifestation of DM disease (1,2), and our findings suggest that MBNL3 expression is important for normal adult muscle satellite cell activation and/or myoblast function, particularly in older muscle when regenerative capacity declines. Significantly, we demonstrate that the Dmpk and Cnbp genes are both expressed during adult muscle regeneration (Fig. 4D), so MBNL3 sequestration by toxic C(C)UGexp RNAs may be an important factor in age-associated muscle wasting in DM muscle.
Although Mbnl3ΔE2 mice show a progressive delay in muscle regeneration, abnormalities in embryonic muscle differentiation leading to neonatal hypotonia in CDM was not recapitulated in either Mbnl3ΔE2 single or Mbnl1ΔE3/ΔE3; Mbnl3ΔE2 double knockouts (data not shown). Since overexpression of the Mbnl3ZnF3/4 protein in Mbnl3ΔE2 mice could mask the essential roles for Mbnl3 in embryonic muscle development, it will be important to generate Mbnl3−/− null, as well as Mbnl1ΔE3/ΔE3; Mbnl3−/− double, knockout mice to determine if Mbnl loss of function leads to a congenital DM-associated phenotype.
Nuclear and cytoplasmic functions for MBNL proteins
RNA-binding proteins function at multiple steps in the expression of genes from transcription to RNA turnover (47–49). While MBNL proteins are alternative splicing factors, Mbnl1 and Mbnl2 are also required for the localization of specific mRNAs, including α3 integrin and multiple RNAs encoding rough endoplasmic reticulum signal sequences (26,27). In this study, HITS-CLIP identified Mbnl3 targets in C2C12 myoblasts in vitro as well as mouse E15 forelimbs in vivo (Supplementary Material, Fig. S6). Surprisingly, RNA-seq analysis combined with RT–PCR validation failed to detect significant changes in the alternative splicing of these targets in Mbnl3ΔE2 knockouts. However, these results do not exclude the possibility that Mbnl3 functions as a splicing factor particularly, since loss of the full-length Mbnl3ZnF1-4 protein led to up-regulation, together with increased nuclear trafficking, of the Mbnl3ZnF3/4 isoform. Interestingly, the Mbnl3ZnF1-4 full-length protein co-fractionated with polysomes in C2C12 myoblasts, while Mbnl3ZnF3/4 did not so it will be interesting to determine the cytoplasmic function(s) of this shorter isoform. We anticipate that future studies on the Mbnl protein family will uncover additional cellular pathways impacted by these multifunctional RNA-binding proteins and provide novel molecular targets for therapeutic development.
MATERIALS AND METHODS
Mbnl plasmids
Mbnl1, Mbnl2 and Mbnl3 coding sequences were amplified using primers MSS3580-MSS3581, MSS3582-MSS3583, and MSS3584-MSS3585 (see Supplementary Material, Table S3 for primers) from mouse embryonic fibroblast (MEF) cDNA using the PCR Extender System (5') according to the manufacturer's protocol. PCR conditions were 96°C for 2 min, 96°C for 30 s, 58 °C for 30 s, 72°C for 90 s (30 cycles) followed by 72°C for 5 min (see Supplementary Material, Table S3 for primers). PCR products were extracted, precipitated and cut with BamHI and XbaI. Following digestion, amplicons were resolved on a 1.0% agarose gel, gel extracted (Qiagen) and resuspended in 50 μl of water. Five micrograms of pcDNA3.1/myc-His MCS A (Invitrogen) was digested under the same conditions and Mbnl inserts were ligated with T4 DNA ligase buffer (NEB) followed by transformation into Top10 cells (Invitrogen) and plating. Individual colonies were picked, grown in 150 ml of LBamp overnight and the plasmids were purified (Qiagen) and sequenced (University of Florida Biotech).
Antibody generation and immunoblotting
Anti-Mbnl3 polyclonal antibodies were generated in rabbits (Genscript, Piscataway, NJ, USA). Briefly, the Mbnl3 C-terminal peptide NVPYVPTTTGNQLKY was conjugated to KLH through an N-terminal Cys residue, and rabbits (n = 4) were injected followed by two additional booster injections. Test bleeds were screened by immunoblot analysis using 50 μg of E15 placenta lysate and anti-sera were selected for affinity purification using the immunizing peptide.
For immunoblotting, whole cell lysates were prepared from staged C57BL6/J embryos, postnatal day 1 (P1) and adult (22 weeks) tissues, and cells by disruption with a sterile pestle in lysis buffer (20 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-KOH, pH 8.0, 100 mm KCl, 0.1% Igepal, 0.5 mm phenylmethylsulfonyl fluoride, 5 μg/ml pepstatin A, 1 μg/ml chymostatin, 1 mm ɛ-aminocaproic acid, 1 mm p-aminobenzamidine, 1 μg/ml leupeptin, 1 μg/ml aprotinin) on ice. Protein samples were sonicated and spun at 16 100g for 20 min at 4°C. Protein concentrations were determined using the DC Protein Assay (Bio-Rad) and total proteins (50 μg/lane) were resolved on 12.5% SDS-acrylamide gels followed by electroblotting to nitrocellulose. Blots were blocked for 45 min in 5% non-fat dry milk in phosphate buffered saline (PBS) (pH 7.4) and immunoblotted in 5% non-fat dry milk in PBS (pH 7.4) with 0.05% Igepal for 1 h at room temperature (RT) with a primary antibody [α-Mbnl3 Mb3/7C, 1:5000 dilution; α-Mbnl1 A2764 antisera, 1:1000; α-Gapdh 6C5, 1:25 000 (Abcam)]. Blots were washed 3× in PBS (pH 7.4) with 0.05% Igepal for 10 min and incubated for 1 h at RT in blotting milk with α-rabbit IgG, or α-mouse IgG, horseradish peroxidase (1:5000; GE Healthcare) secondary antibody. Membranes were washed as described above, followed by a PBS (pH 7.4) wash, developed in Amersham ECL detection reagents (GE Healthcare) and exposed to Biomax Film (Kodak).
Immunoprecipitation and mass spectrometry
Protein-A dynabeads, pre-bound with 20 μg purified Mb3/7C antibody, or other primary antibodies, and incubated for 30 min at RT, were washed two times with 0.2 m triethanolamine (pH 8.2), resuspended and incubated in 1 ml of 20 mm dimethyl pimelimidate (Sigma) in 0.2 m triethanolamine (pH 8.2) for 30 min at RT for antibody-Protein A cross-linking. This reaction was quenched using 1 ml of 50 mm Tris (pH 7.5) followed by incubation for 15 min at RT. For immunoprecipitation, protein lysates were prepared from cells or tissues in IPP150 (50 mm Tris–Cl, pH 7.4, 150 mm NaCl, 0.1% Igepal, 0.5 mm phenylmethylsulfonyl fluoride, 5 μg/ml pepstatin A, 1 μg/ml chymostatin, 1 mm ɛ-aminocaproic acid, 1 mm p-aminobenzamidine, 1 μg/ml leupeptin, 1 μg/ml aprotinin) and added to Protein-A:Mb3/7C beads followed by incubation at 4°C for 2 h. Beads were collected by magnetic capture, washed extensively, resuspended in 35 μl of sodium dodecyl sulfate - polyacrylamide gel electrophoresis (SDS-PAGE) loading cocktail buffer, incubated at 95°C for 5 min at 1100 rpm (Eppendorf Thermomixer) followed by bead removal. Immunopurified proteins were resolved on a 12.5% SDS-acrylamide gel, which was then stained with colloidal Coomassie blue (Invitrogen) and bands of interest were excised from the gel and submitted for LCMS analysis (University of Florida Biotech Proteomic Core).
Polysome analysis
Polysome analysis was performed according a previously published protocol (50) with several modifications. C2C12 cells were washed with ice-cold PBS containing 0.1 mg/ml cycloheximide (CHX) and homogenized in 1 ml of polyribosome lysis buffer [10 mm HEPES-KOH, pH 7.4, 150 mm NaCl, 5 mm MgCl2, 0.5 mm dithiothreitol, 0.1% Igepal, 0.1 mg/ml CHX, 1× protease inhibitor cocktail (Roche) and 40 U/ml rRNAsin (Promega)]. The lysate was spun at 12 000g for 10 min at 4°C. The supernatant was loaded on 15–45% linear sucrose gradients, centrifuged at 40 000 rpm (∼18 000g) in a Beckman SW41 rotor for 2 h and 10–12 fractions (0.75 ml each) were collected with constant monitoring at 254 nm (ISCO UA-6 detector). Protein was trichloroacetic acid precipitated from each fraction. For puromycin treatment, cells were treated with puromycin (100 µg/ml) for 2 h prior to harvesting and CHX was omitted from the gradient. For EDTA-mediated polysome release, a final concentration of 25 mm EDTA was used in the polysome lysis buffer.
Mbnl3 splicing and RNA blot analysis
Total RNA was isolated from C57BL6/J tissues (embryonic tissues pooled) or MEFs with Tri Reagent (Sigma) according to the manufacturer's protocol. For northern blot analysis, ∼15 μg of total RNA was denatured in glyoxal/dimethylsulfoxide (DMSO) buffer (8% deionized glyoxal, 60% DMSO, 12 mm 1,4 piperazine diethane sulfonic acid (PIPES), 36 mm Bis–Tris, 1.2 mm EDTA, pH ∼6.5) at 55°C for 1 h, immediately transferred to ice and resolved on a 1.2% agarose gel in 1× PIPES + Bis-Tris + EDTA (BPTE) buffer (10 mm PIPES, 30 mm Bis–Tris, 1 mm EDTA, pH ∼6.5) for 2 h at 100 V. Following electrophoresis, the gel was rinsed in water, treated with 50 mm NaOH for 20 min and neutralized in 20× saline sodium citrate (SSC) (pH 7) for 40 min. RNAs were transferred to Hybond-N+ nylon membranes (GE Healthcare) in 10× SSC using neutral capillary transfer. Blots were UV cross-linked (Stratalinker 1800, Stratagene) and prehybridized for 2 h in ExpressHyb (Clontech) at 68°C. Hybridization probes were generated by [α-32P]dCTP-labeling using 50 ng of DNA template (Mbnl1, Mbnl3, Ppia) with DNA Labeling Beads-dCTP (GE Healthcare) and purified with Illustra ProbeQuant G-50 Micro Columns (GE Healthcare). DNA probes and 2 mg sheared salmon sperm DNA (Invitrogen) were denatured and added to hybridization solution and blots were hybridized overnight at 68°C. Following hybridization, blots were washed once in 1× SSC, 0.1% SDS at RT on a shaker for 10 min followed by three washes in 0.5× SSC, 0.1% SDS at 65°C in a heated/shaking water bath for 10 min each. Blots were exposed to Biomax Film and X-Omatic Intensifying Screens (Kodak) at −80°C. For RT–PCR analysis, ∼25 μg of total RNA + 2.5 μg oligo-d(T)12-18 primers (Invitrogen) were denatured at 65°C for 5 min, transferred to ice for 1 min and then added to a final volume of 50 μl in 0.2 mm dNTPs, 1× 1st Strand Buffer (Invitrogen) + 8 mm dithiothreitol, 100 U RNasin (Promega) and 500 U Superscript III RT (Invitrogen). Reverse transcription reaction conditions were performed in a thermocycler at 25°C for 5 min, 50°C for 60 min and 70°C for 15 min. After reverse transcription, RNase H (2 U, Invitrogen) was added to the RT reaction which was incubated at 37°C for 20 min. Mbnl3 isoform-specific PCR was carried out with forward primers in exon 1 (MSS3648), exon 2 (MSS3649), exon 3 (MSS3655) and reverse primers in exon 7a (MSS3759), exon 7b (MSS3760), exon 7c (MSS3763) and exon 8 (MSS3652). Ppia PCR was carried out with a forward primer in exon 4 (MSS3586) and a reverse primer spanning exon 4/exon 5 (MSS3587). For PCRs, 2 μl of cDNA was used as template in a 50 μl reaction containing 1× High Fidelity buffer (5'), 0.4 mm dNTPs, 30 pmol forward primer, 30 pmol reverse primer and 2.5 U Triplemaster Taq (5'). PCRs were performed at 96°C for 30 s, 58°C for 30 s and 72°C for 1 min (35 cycles, MSS3648–MSS3652, MSS3649–MSS3652, MSS3655–MSS3652; 32 cycles, MSS3655–MSS3759, MSS3655– MSS3760; 30 cycles, MSS3655–MSS3763) for Mbnl3 and 96°C for 30 s, 57°C for 30 s and 72°C for 30 s (30 cycles, MSS3586–MSS3587) for Ppia. PCR products were resolved on a 1–1.5% agarose gel, stained with ethidium bromide and images collected (ImageQuant 400, GE Healthcare).
siRNA and plasmid transfections
For siRNA treatment, C2C12 cells, which were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen), 20% fetal bovine serum (FBS) (Invitrogen), 1% penicillin/streptomycin (P/S) (Invitrogen) were seeded in 6-well plates (1.5 × 105 cells/well) in 2 ml antibiotic-free media. Cells were either mock-treated or treated with siMbnl3 (ON-TARGET plus SMARTpool, Dharamcon) or a scrambled siRNA control 6 h post-seeding following the manufacturer's protocol and media was replaced by fresh C2C12 antibiotic-free growth media 24 h post-transfection.
For COSM6 transfections, cells were grown in DMEM (Invitrogen), 10% FBS (Invitrogen), 1% l-glutamine (Invitrogen) and 1% P/S (Invitrogen) and 6 h prior to transfection 2.0 × 105 cells were seeded per well of a 6-well plate in antibiotic-free growth media. In a microfuge tube, 6 μl Fugene 6 (Roche) was diluted in 180 μl OptiMEM I (Invitrogen), vortexed and incubated for 5 min at RT. After incubation, 2 μg of plasmid (either myc-tagged Mbnl1, Mbnl2, Mbnl3 or empty vector pSP72) was added to the tube containing Fugene 6:OptiMEM I, vortexed and incubated for 15 min at RT. The contents of each transfection were placed in individual wells of a 6-well plate followed by overnight incubation. Cells were washed with PBS (pH 7.4), antibiotic-free growth media was replaced and cell lysates were prepared 48 h post-transfection.
C2C12 differentiation
For C2C12 differentiation, proliferating cells were seeded at a density of 1.5 × 105 cells per well in a 6-well plate coated with Matrigel (BD Biosciences) in C2C12 growth media (DMEM, 20% FBS, 1% P/S). At ∼90% confluency, growth media was aspirated, cells were washed twice with PBS and C2C12 differentiation media (DMEM, 2% horse serum, 1% P/S) was added. Cells were allowed to differentiate for 5 days and media was replaced daily. For siMbnl3 experiments, C2C12 cells were differentiated 24 h post-siRNA treatment.
Muscle injury/regeneration
Lyophilized Notexin (Latoxan, Valence France) was reconstituted in sterile 154 mm NaCl to a final concentration of 100 μg/ml and 10 μl aliquots (1 μg) were stored at −80°C. For injections, 1 × 10 μl aliquot was thawed on ice, diluted to a final working concentration of 1 μg/ml (in sterile 154 mm NaCl) and C57/BL6 anesthetized mice (10–12 or 32–36 weeks old) were injected in the TA muscle with 100 μl (100 ng Notexin) using a 29 G needle. TA muscles were analyzed at days 1, 3, 5 and 7 PI. Each TA muscle was divided in lengthwise sections for protein, RNA analysis and histological analyses.
HITS-CLIP and RNA-Seq
CLIP was performed as reported previously (14,51) with the following modifications. Forelimbs were dissected from wild-type mice embryos (E15), snap frozen in liquid nitrogen, ground to a fine powder and cross-linked with UV light in a Stratalinker 1800 (Stratagene). Three biological replicates were analyzed, each consisting of four forelimbs from two embryos. For C2C12 cells, five 10 cm plates per biological replicate were UV cross-linked followed by scraping cells in ice-cold PBS, centrifugation and freezing of cell pellets. For immunoprecipitation, protein lysates were denatured by adding SDS to a final concentration of 1% followed by heating (100°C/5 min) and subsequent dilution to a final SDS concentration of 0.1% (in lysis buffer). The Mb3/7 anti-Mbnl3 antibody (5 µg) and RNase A (concentrations of 55 and 0.55 U/ml for high and low RNase, respectively) were used and cDNA libraries were generated using RNA linkers and primers described for Ago CLIP (52). For the identification of RNAs bound to full-length Mbnl3 in either C2C12 myoblasts or E15 forelimb, only RNA–protein complexes that migrated 12–15 kDa above the major Mbnl3 38 kDa isoform were excised from the gel and then 90–130 nt RNAs were isolated and used for subsequent sequencing analysis. This procedure minimized the contribution of the Mbnl3 27 kDa isoform in the binding site analysis. All sequencing was performed using an Illumina Genome Analyzer IIx. Raw reads obtained from the Illumina pipeline included a 4 nt barcode (three random positions and a G at the 4th position) at the 5′ end followed by the CLIP tag and the 3′ adaptor sequence. Results were filtered to remove low-quality reads by requiring a minimum score of 20 in barcode positions and an average score of 20 in the following 25 nucleotides. The filtered reads were then aligned to the mouse reference genome (mm9) by novoalign (http://www.novocraft.com) allowing iterative trimming at the 3′ end (53). Potential PCR duplicates, as judged from the starting position of genomic mapping and the barcode sequences, were removed to identify unique CLIP tags as described (14). For motif enrichment, de novo motif searches were performed using the MEME suite (54).
For RNA-seq, total RNA was isolated from staged Mbnl3+/+ and Mbnl3ΔE2 E15 embryos with Tri Reagent (Sigma) according to the manufacturer's protocol. RNA was purified using the RNeasy kit (Qiagen) and RNA quality tested using a Bioanalyzer 2100 (Agilent Technologies). Total RNA (5 μg) was used to generate a 250 bp cDNA library using the Illumina mRNA sequencing preparation kit and cDNA libraries were further analyzed for size and quality using the Agilent DNA-1000 chip for the Bioanalyzer 2100. The libraries were subjected to paired-end sequencing (Illumina Genome IIX). Sequence data were analyzed for global alternative splicing and quantitative changes as described previously (14).
Mbnl3ΔE2isoform knockout generation
The Mbnl3 conditional targeting vector was constructed using standard recombineering bacterial strains and techniques (reagents and protocols can be found at http://recombineering.ncifcrf.gov/). Briefly, an ∼11 kb fragment, containing Mbnl3 exon 2 together with 6.8 kb (upstream) and 4.2 kb (downstream) flanking sequences, was retrieved from CHORI clone RP23-459O21 into a high copy plasmid backbone so that Mbnl3 exon 2 was flanked by an upstream loxP site and downstream NeoR cassette/loxP and HSV-TK cassettes. The Mbnl3 targeting vector was linearized with NotI and electroporated into CMTI-2 BL/6 ESCs (Millipore) followed by positive (neomycin) and negative (HSV-TK) selection, and positive Mbnl3cond euploid ESCs were injected into B6(Cg) -Tyrc-2J/J blastocysts, which were then transferred to pseudo-pregnant C57BL/6 females. To obtain germline transmission of the Mbnl3cond allele, Mbnl3cond chimeric males were mated with female B6(Cg) -Tyrc-2J/J females. To delete Mbnl3 exon 2, Mbnl3cond/+ females were first crossed to B6.C-Tg(CMV-cre)1Cgn/J males (JAX), to obtain F2 Mbnl3ΔE2/+; CMV-cre females (CMV-Cre is also X-linked), and Mbnl3ΔE2/+; CMV-cre F2 females were crossed to wild-type C57BL6/J males to generate Mbnl3ΔE2 male, or Mbnl3ΔE2/ΔE2 female, knockouts. All animal procedures were approved by the University of Florida IACUC.
ACCESSION NUMBERS
HITS-CLIP data have been deposited in GEO under accession number GSE46207.
SUPPLEMENTARY MATERIAL
FUNDING
This study was supported by grants from the National Institutes of Health (AR046799 and NS058901 to M.S.S.; K99GM95713 to C.Z. and NS34389 to R.B.D.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank E. Chan and C. Thornton for anti-GW182 and Mbnl1 antibodies, respectively, A. Ravel-Chapuis and B. Jasmin for the Matrigel C2C12 protocol, C. Lopez for Illumina sequencing and T. Ashizawa and L. Ranum for comments on the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Harper P.S. Myotonic Dystrophy. London: W.B. Saunders; 2001. [Google Scholar]
- 2.Ranum L.P., Cooper T.A. RNA-mediated neuromuscular disorders. Ann. Rev. Neurosc. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 3.Cooper T.A., Wan L., Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Rourke J.R., Swanson M.S. Mechanisms of RNA-mediated disease. J. Biol. Chem. 2009;284:7419–7423. doi: 10.1074/jbc.R800025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michalowski S., Miller J.W., Urbinati C.R., Paliouras M., Swanson M.S., Griffith J. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Res. 1999;27:3534–3542. doi: 10.1093/nar/27.17.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho T.H., Charlet B.N., Poulos M.G., Singh G., Swanson M.S., Cooper T.A. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du H., Cline M.S., Osborne R.J., Tuttle D.L., Clark T.A., Donohue J.P., Hall M.P., Shiue L., Swanson M.S., Thornton C.A., et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Timchenko L.T., Miller J.W., Timchenko N.A., DeVore D.R., Datar K.V., Lin L., Roberts R., Caskey C.T., Swanson M.S. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philips A.V., Timchenko L.T., Cooper T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 11.Charlet B.N., Savkur R.S., Singh G., Philips A.V., Grice E.A., Cooper T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 12.Mankodi A., Takahashi M.P., Jiang H., Beck C.L., Bowers W.J., Moxley R.T., Cannon S.C., Thornton C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 13.Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 14.Charizanis K., Lee K.Y., Batra R., Goodwin M., Zhang C., Yuan Y., Shiue L., Cline M., Scotti M.M., Xia G., et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–450. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravel-Chapuis A., Belanger G., Yadava R.S., Mahadevan M.S., DesGroseillers L., Cote J., Jasmin B.J. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J. Cell. Biol. 2012;196:699–712. doi: 10.1083/jcb.201108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laurent F.X., Sureau A., Klein A.F., Trouslard F., Gasnier E., Furling D., Marie J. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res. 2012;40:3159–3171. doi: 10.1093/nar/gkr1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paul S., Dansithong W., Kim D., Rossi J., Webster N.J., Comai L., Reddy S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006;25:4271–4283. doi: 10.1038/sj.emboj.7601296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fardaei M., Rogers M.T., Thorpe H.M., Larkin K., Hamshere M.G., Harper P.S., Brook J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 19.Kanadia R.N., Urbinati C.R., Crusselle V.J., Luo D., Lee Y.J., Harrison J.K., Oh S.P., Swanson M.S. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr. Patterns. 2003;3:459–462. doi: 10.1016/s1567-133x(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 20.Lee K.S., Squillace R.M., Wang E.H. Expression pattern of muscleblind-like proteins differs in differentiating myoblasts. Biochem. Biophys. Res. Commun. 2007;361:151–155. doi: 10.1016/j.bbrc.2007.06.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang J., Song W., Tromp G., Kolattukudy P.E., Fu M. Genome-wide survey and expression profiling of CCCH-zinc finger family reveals a functional module in macrophage activation. PLoS One. 2008;3:e2880. doi: 10.1371/journal.pone.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Squillace R.M., Chenault D.M., Wang E.H. Inhibition of muscle differentiation by the novel muscleblind-related protein CHCR. Dev. Biol. 2002;250:218–230. doi: 10.1006/dbio.2002.0798. [DOI] [PubMed] [Google Scholar]
- 23.Lee K.S., Smith K., Amieux P.S., Wang E.H. MBNL3/CHCR prevents myogenic differentiation by inhibiting MyoD-dependent gene transcription. Differentiation. 2008;76:299–309. doi: 10.1111/j.1432-0436.2007.00209.x. [DOI] [PubMed] [Google Scholar]
- 24.Yuan Y., Compton S.A., Sobczak K., Stenberg M.G., Thornton C.A., Griffith J.D., Swanson M.S. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35:5474–5486. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kino Y., Mori D., Oma Y., Takeshita Y., Sasagawa N., Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum. Mol. Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 26.Adereth Y., Dammai V., Kose N., Li R., Hsu T. RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat. Cell Biol. 2005;7:1240–1247. doi: 10.1038/ncb1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S., et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darnell R.B. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA. 2010;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goers E.S., Purcell J., Voelker R.B., Gates D.P., Berglund J.A. MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res. 2010;38:2467–2484. doi: 10.1093/nar/gkp1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osborne R.J., Lin X., Welle S., Sobczak K., O'Rourke J.R., Swanson M.S., Thornton C.A. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 2009;18:1471–1481. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikezoe K., Nakamori M., Furuya H., Arahata H., Kanemoto S., Kimura T., Imaizumi K., Takahashi M.P., Sakoda S., Fujii N., et al. Endoplasmic reticulum stress in myotonic dystrophy type 1 muscle. Acta Neuropath. 2007;114:527–535. doi: 10.1007/s00401-007-0267-9. [DOI] [PubMed] [Google Scholar]
- 32.Uaesoontrachoon K., Wasgewatte Wijesinghe D.K., Mackie E.J., Pagel C.N. Osteopontin deficiency delays inflammatory infiltration and the onset of muscle regeneration in a model of muscle injury. Dis. Model. Mech. 2012;6:197–205. doi: 10.1242/dmm.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uaesoontrachoon K., Yoo H.J., Tudor E.M., Pike R.N., Mackie E.J., Pagel C.N. Osteopontin and skeletal muscle myoblasts: association with muscle regeneration and regulation of myoblast function in vitro. Int. J. Biochem. Cell. Biol. 2008;40:2303–2314. doi: 10.1016/j.biocel.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 34.Zanotti S., Gibertini S., Di Blasi C., Cappelletti C., Bernasconi P., Mantegazza R., Morandi L., Mora M. Osteopontin is highly expressed in severely dystrophic muscle and seems to play a role in muscle regeneration and fibrosis. Histopathology. 2011;59:1215–1228. doi: 10.1111/j.1365-2559.2011.04051.x. [DOI] [PubMed] [Google Scholar]
- 35.Badyal S.K., Basran J., Bhanji N., Kim J.H., Chavda A.P., Jung H.S., Craig R., Elliott P.R., Irvine A.F., Barsukov I.L., et al. Mechanism of the Ca(2)+-dependent interaction between S100A4 and tail fragments of nonmuscle myosin heavy chain IIA. J. Mol. Biol. 2011;405:1004–1026. doi: 10.1016/j.jmb.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elliott P.R., Irvine A.F., Jung H.S., Tozawa K., Pastok M.W., Picone R., Badyal S.K., Basran J., Rudland P.S., Barraclough R., et al. Asymmetric mode of Ca(2)(+)-S100A4 interaction with nonmuscle myosin IIA generates nanomolar affinity required for filament remodeling. Structure. 2012;20:654–666. doi: 10.1016/j.str.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yaffe D., Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- 38.Florini J.R., Magri K.A., Ewton D.Z., James P.L., Grindstaff K., Rotwein P.S. “Spontaneous” differentiation of skeletal myoblasts is dependent upon autocrine secretion of insulin-like growth factor-II. J. Biol. Chem. 1991;266:15917–15923. [PubMed] [Google Scholar]
- 39.Ge Y., Sun Y., Chen J. IGF-II is regulated by microRNA-125b in skeletal myogenesis. J. Cell Biol. 2011;192:69–81. doi: 10.1083/jcb.201007165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlson M.E., Conboy I.M. Loss of stem cell regenerative capacity within aged niches. Aging Cell. 2007;6:371–382. doi: 10.1111/j.1474-9726.2007.00286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conboy I.M., Conboy M.J., Smythe G.M., Rando T.A. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 42.Poulos M.G., Batra R., Charizanis K., Swanson M.S. Developments in RNA splicing and disease. Cold Spring Harb. Perspect. Biol. 2011;3:a000778. doi: 10.1101/cshperspect.a000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teplova M., Patel D.J. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat. Struct. Mol. Biol. 2008;15:1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purcell J., Oddo J.C., Wang E.T., Berglund J.A. Combinatorial mutagenesis of MBNL1 zinc fingers elucidates distinct classes of regulatory events. Mol. Cell. Biol. 2012;32:4155–4167. doi: 10.1128/MCB.00274-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grammatikakis I., Goo Y.H., Echeverria G.V., Cooper T.A. Identification of MBNL1 and MBNL3 domains required for splicing activation and repression. Nucleic Acids Res. 2011;39:2769–2780. doi: 10.1093/nar/gkq1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee K.S., Cao Y., Witwicka H.E., Tom S., Tapscott S.J., Wang E.H. RNA-binding protein muscleblind-like 3 (MBNL3) disrupts myocyte enhancer factor 2 (Mef2) {beta}-exon splicing. J. Biol. Chem. 2010;285:33779–33787. doi: 10.1074/jbc.M110.124255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luco R.F., Allo M., Schor I.E., Kornblihtt A.R., Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ciais D., Cherradi N., Feige J.J. Multiple functions of tristetraprolin/TIS11 RNA-binding proteins in the regulation of mRNA biogenesis and degradation. Cell. Mol. Life Sci. 2012 doi: 10.1007/s00018-012-1150-y. doi:10.1007/s00018-012-1150-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kapeli K., Yeo G.W. Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Front. Neurosci. 2012;6:144. doi: 10.3389/fnins.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savant-Bhonsale S., Cleveland D.W. Evidence for instability of mRNAs containing AUUUA motifs mediated through translation-dependent assembly of a >20S degradation complex. Genes Dev. 1992;6:1927–1939. doi: 10.1101/gad.6.10.1927. [DOI] [PubMed] [Google Scholar]
- 51.Jensen K.B., Darnell R.B. CLIP: crosslinking and immunoprecipitation of in vivo RNA targets of RNA-binding proteins. Methods Mol. Biol. 2008;488:85–98. doi: 10.1007/978-1-60327-475-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chi S.W., Zang J.B., Mele A., Darnell R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang C., Darnell R.B. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011;29:607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.