

Abstract
Growing evidence for glutamate abnormalities in schizophrenia support the development of novel antipsychotic agents targeting this system. Early studies investigating modulation of the glutamate system using glycine, D-serine and sarcosine in patients with schizophrenia have demonstrated significant effects, particularly on negative symptoms, conventionally thought to be refractory to antipsychotic drug treatment. Drugs targeting the glutamate system also have a completely different side-effect profile to dopamine D2 antagonists, with no propensity to extrapyramidal side effects, prolactinaemia or weight gain. It has been hypothesized that glutamatergic drugs may be of benefit to the 20–30% of individuals with schizophrenia who fail to show any response to dopaminergic agents, and may be particularly useful in the early stages of the illness, where they may be disease-modifying. A number of glutamatergic compounds have been reported as having promising results in phase II drug trials. If these reach the clinic, they will represent the first truly novel approach to pharmacotherapy in schizophrenia for more than 50 years.
Keywords: schizophrenia, psychosis, pharmacology, antipsychotic drugs, glutamate
Introduction
For more than 50 years, the only effective antipsychotic drugs available have been dopamine D2 receptor antagonists [Kapur and Mamo, 2003], with their clinical potency directly corresponding to their affinity at D2 receptors [Seeman and Lee, 1975]. Despite leading to at least a partial clinical response in around two thirds of patients with schizophrenia, the other third of patients will fail to respond to D2 antagonists [Stone et al. 2010b]. Furthermore, although positive symptoms generally show a reasonable response to these drugs, there frequently remains a core of negative symptoms that are refractory to antipsychotic treatment [Javitt, 2001; Buchanan et al. 1998; Tamminga et al. 1998].
All currently available antipsychotic drugs have significant, and sometimes potentially life-threatening, side effects, which may lead to discontinuation of the treatment. Although the second generation of antipsychotic drugs have a lower incidence of extrapyramidal side effects, they are associated with other debilitating effects such as impaired glucose tolerance and weight gain, which can have significant health consequences. Thus, there has been a great deal of interest in developing new targets for pharmacological treatment in schizophrenia: drugs which might have fewer side effects and/or lead to response in patients who do not respond fully to currently available drugs [Stone et al. 2010b]. So far, the only major advance in drug treatments for schizophrenia has been the discovery of clozapine, which has been consistently shown to have superior efficacy in patients unresponsive to other antipsychotic drugs [McEvoy et al. 2006; Kane et al. 1988]. No other agent developed since clozapine has shown equivalent efficacy, and improvements over first-generation antipsychotic drugs have been incremental at best. Part of the reason for this lack of rapid progress may be due to the fact that drug development in schizophrenia has primarily focused on the strategy of developing new drugs that act on the dopamine system rather than developing compounds for other targets.
Glutamatergic neurotransmission
Glutamate is the main excitatory neurotransmitter in the brain. Between 60% and 80% of total brain metabolic activity in the nonstimulated cerebral cortex is utilized by glutamatergic neurones, with the remainder being used by GABAergic neurones and glial cells. The synaptic release of glutamate and recycling to glutamine in astrocytes is a major metabolic pathway (Figure 1), and accounts for between 80% and 100% of total glutamate trafficking, and it is now accepted that no meaningful distinction can be made between metabolic and neurotransmitter glutamate [Rothman et al. 2003].
Figure 1.

Glutamatergic synapse showing glutamate release, binding to AMPA and NMDA receptors on the postsynaptic membrane, and subsequent reuptake by astrocytes where it is metabolized to glutamine by glutamine synthetase (GS).
Glutamate acts at two main subtypes of neuroreceptor, the metabotropic glutamate receptors (mGluR) and the ionotropic glutamate receptors. mGluR are composed of three groups (groups I-III) distinguished by their sequence homology, pharmacology and second messenger systems. Group I receptors (mGluR1 and mGluR5) are predominantly postsynaptic in somatodendritic domains and couple via Gq/G11 to phospholipase C, whereas groups II (mGluR2 and mGluR3) and III (mGluR4, mGluR6, mGluR7 and mGluR8) are coupled via Gi/G0 to inhibition of adenylate cyclase activity and are primarily presynaptic in axonal domains and terminals, where they modulate neurotransmitter release [Kew and Kemp, 2005].
Ionotropic glutamate receptors are also divided into three groups, named after the agonists originally found to selectively activate them: α-amino-3-hydroxy-5-methyl-4-isoazolepropionic acid (AMPA), kainate and N-methyl-D-aspartate (NMDA) receptors. The ionotropic glutamate receptors are all heteromeric ion channels, composed of multiple protein subunits. When activated they lead to an increase in cation conductance with differential permeability to Na+ and Ca2+ depending upon receptor type and subunit composition. NMDA receptors in the adult brain generally show increased Ca2+ conductance on activation, whereas kainate receptors lead to an increase in Na+ conductance. AMPA receptors expressed on GABAergic hippocampal and amygdala interneurons appear to lack an edited GluR2 subunit, and show preferential Ca2+ conductance, whereas those on pyramidal neurons are non-Ca2+ permeable [Kew and Kemp, 2005; Dingledine et al. 1999].
NMDA receptor structure and function
The NMDA receptor is a heteromeric ion channel, formed from a number of subunits (NR1, NR2A-NR2D, NR3A and NR3B). NR1 and NR2(A-D) subunits are obligatory for a functional NMDA receptor, and contain binding sites for glycine (glycineB site) and glutamate respectively. NR2 subunits show distinct regional and developmental distribution, with NR2A and NR2B being expressed primarily in the forebrain, NR2C in cerebellar granule cells and NR2D being expressed during foetal development in the midbrain and diencephalon. NR3 subunits require both NR1 and NR2 subunits to form functional NMDA receptors. NR3A receptors are expressed primarily during development, and NR3B are only found in somatic neurons in brainstem and spinal cord. Activation of the NMDA receptor requires two obligatory co-agonists, binding at the glycine and glutamate sites. Two independent glycine, and two independent glutamate binding sites appear to be required. Therefore, it has been suggested, the minimum requirement for a functional NMDA receptor is two NR1 and two NR2(A-D) subunits [Kew and Kemp, 2005]. At resting potential, NMDA receptors are blocked by extracellular Mg2+, which binds to an intrachannel site of the NMDA receptor complex. In order to allow Ca2+ to enter the cell, in addition to glutamate and glycine binding, the cell must depolarize, removing the Mg2+ block [Dingledine et al. 1999] (see Figure 2). Uncompetitive allosteric antagonists of the NMDA receptor such as ketamine, phencyclidine (PCP) and dizocilpine (MK-801) bind to the inside of the NMDA receptor ion channel when it is in its open state, and prevent Ca2+ influx.
Figure 2.

Simplified diagram of an NMDA receptor with glycine (Gly), glutamate (Glu) and MK-801/PCP/ ketamine (PCP) binding sites displayed. Extracellular calcium entry through the NMDA receptor occurs only when both glutamate and glycine bind to their respective binding sites, and the cell membrane depolarises allowing removal of the voltage-dependent block due to extracellular magnesium ions binding inside the pore of the NMDA receptor.
Glutamate and schizophrenia
There is growing evidence that changes in glutamatergic neurotransmission may occur in schizophrenia, and it has been hypothesized that glutamatergic changes may precede, or give rise to, alterations in other downstream neurotransmitter systems such as dopamine [Stone et al. 2007]. The glutamate hypothesis of schizophrenia was founded on a number of observations. Drugs that act as uncompetitive antagonists at NMDA receptors such as PCP and ketamine reliably and instantly induce a drug-induced state that closely resembles the symptoms of schizophrenia, including thought disorder, odd ideas and delusions, cognitive impairment and, most notably, an emotional withdrawal that has been likened to the negative symptoms of schizophrenia [Javitt, 2007; Morgan and Curran, 2006; Krystal et al. 1994]. In contrast, drugs that increase brain dopamine transmission, such as amphetamine, do not induce cognitive or negative symptoms [Krystal et al. 2005]. Blockade of NMDA receptors by ketamine has been shown to be most closely related to negative, rather than positive symptoms [Stone et al. 2008], suggesting that dopamine and glutamatergic changes may give rise to different symptoms of the illness [Stone et al. 2008; Krystal et al. 2005]. Second, candidate risk genes for schizophrenia are not related to the dopamine system, but rather converge on molecules involved in glutamatergic neurotransmission [Harrison and Weinberger, 2005].
These findings suggest, as hypothesized previously, that negative and cognitive symptoms may be at the core of schizophrenia [Andreasen, 1999]. Cognitive symptoms have been found to be closely associated with negative symptoms in patients with schizophrenia [Ventura et al. 2009; Addington et al. 1991], and negative symptoms are most closely associated with functional outcome [Ventura et al. 2009]. Early and subtle cognitive decline is one of the first (usually undetected) symptoms of schizophrenia [Bilder et al. 2006], and it is interesting to speculate that these changes might arise secondary to abnormalities in NMDA receptor function or glutamatergic transmission.
Studies of the effect of NMDA receptor antagonists on brain structure and function are supportive of the hypothesis of abnormalities of glutamatergic transmission in schizophrenia. Seminal work by Olney and Farber showed that exposure of rats to systemic injections of NMDA receptor antagonists led to neurotoxic changes in cortical brain regions, which they suggested closely resembled the reductions in grey matter volume seen in patients with schizophrenia [Olney and Farber, 1995]. They showed that antagonists for the AMPA subtype of glutamate receptor blocked this toxicity, and hypothesized that the cortical toxicity was caused, somewhat counterintuitively, by excess cortical glutamate release. They put forward the theory that NMDA receptors expressed on GABAergic interneurons were particularly sensitive to NMDA receptor antagonists and that the resultant reduction in inhibitory tone led to disinhibition of glutamatergic projection neurons leading to glutamate release and excitotoxicity [Olney and Farber, 1995] (Figure 3).
Figure 3.

Hypothesized mechanism whereby NMDA receptor antagonists lead to increased cortical glutamate release: inhibition of NMDA receptors expressed on GABAergic interneurons (A) leads disinhibition of glutamatergic projection neurons (B).
This hypothesis was supported by later microdialysis studies showing increased glutamate in prefrontal cortex following systemic administration of ketamine [Lorrain et al. 2003a; Moghaddam et al. 1997]. Interestingly, injection of the NMDA receptor antagonist MK-801 into cortical regions did not lead to any evidence of neurodegenerative changes, whereas injection into anterior thalamus led to the same cortical changes as seen with systemic administration [Sharp et al. 2001], suggesting that thalamus might be a primary site of NMDA receptor blockade in the generation of downstream effects by NMDA receptor antagonists and, by extension, may also be a site of NMDA receptor dysfunction in schizophrenia [Stone et al. 2007; Olney et al. 1999].
Studies of patients with schizophrenia and first-episode psychosis, and in individuals with prodromal symptoms of schizophrenia (‘at risk mental state’ [ARMS]), who are at high risk of developing schizophrenia [Phillips et al. 2000], have generally been supportive of the hypothesis of NMDA receptor dysfunction and altered glutamate transmission in the illness [Stone, 2009]. A study using a single photon emission tomography (SPET) ligand for the NMDA receptor revealed that individuals with schizophrenia who were not currently medicated had lower NMDA receptor binding in the left hippocampus compared with healthy volunteers [Pilowsky et al. 2006] (Figure 4). This finding was in close agreement with an earlier postmortem study of mRNA of the NMDA receptor subunit NMDAR1, which revealed that patients with schizophrenia showed reduced levels in left dentate gyrus [Law and Deakin, 2001]. Furthermore, in patients treated with typical antipsychotic drugs, levels of NMDA receptor binding in the left hippocampus were inversely correlated with negative symptoms [Pilowsky et al. 2006].
Figure 4.

Reduced NMDA receptor binding in left hippocampus in drug free patients with schizophrenia and matched healthy controls [Pilowsky et al. 2006].
Studies employing proton magnetic resonance spectroscopy (1H-MRS) have measured cortical glutamate and glutamine levels in individuals at high risk of psychosis, as well as patients with first-episode psychosis and chronic schizophrenia. Glutamine is produced in astrocytes following synaptic release of glutamate, and so has been suggested to be a marker of glutamatergic transmission. Studies in individuals at risk of psychosis, and patients with first-episode psychosis have found evidence of increased glutamatergic transmission in anterior cingulate and frontal cortex [Stone et al. 2009; Tibbo et al. 2004; Théberge et al. 2002; Bartha et al. 1997]. In contrast, studies of patients with chronic schizophrenia have generally found normal or reduced cortical glutamate levels [Ongur et al. 2009; Rowland et al. 2009; Tayoshi et al. 2009; Lutkenhoff et al. 2010; Ohrmann et al. 2005; Théberge et al. 2003; Block et al. 2000; Kegeles et al. 2000].
Subcortical measures of glutamate are less consistent. Individuals at risk of psychosis have been shown to have reduced thalamic glutamate levels [Stone, 2009], whereas patients with first-episode and chronic schizophrenia have been reported to have increased thalamic glutamine [Théberge et al. 2003, 2002]. The reason for this discrepancy is not clear, but it may represent differences in imaging methodology, or possibly differences in illness subtype as increases in cortical glutamate release could be driven by reduced subcortical glutamatergic transmission on GABA interneurons or by dysfunctional NMDA receptors expressed on the same population of neurons [Olney and Farber, 1995].
There has been much speculation about how glutamate and dopamine may be related in schizophrenia, and whether glutamate or dopamine might be more ‘upstream’ in the illness. Olney and Farber hypothesized that glutamatergic changes might be related to a primary dopaminergic abnormality [Olney and Farber, 1995], whereas others have suggested that abnormalities in glutamate transmission or NMDA receptor function could drive changes in dopamine [Stone et al. 2007; Coyle, 2006; Harrison and Weinberger, 2005]. A plausible animal model of schizophrenia suggests that increased glutamate efferents from hippocampus may drive increased dopamine neuron responsivity [Lodge and Grace, 2006] (Figure 5). In support of this hypothesis, lower levels of hippocampal glutamate (potentially driving increased hippocampal outputs through reduced stimulation of GABA interneurons) are associated with increased striatal [18F]DOPA uptake (a marker of presynaptic dopamine function) in individuals with an ARMS but not in healthy volunteers [Stone et al. 2010a]. The fact that this relationship was found in ARMS subjects and not in controls suggests GABA intereneurons in individuals with an ARMS may be more sensitive to presynaptic glutamate levels, possibly due to lower hippocampal NMDA receptor expression [Pilowsky et al. 2006] (Figure 5). Drugs targeting glutamatergic transmission might help to normalize these deficits and have additional downstream effects on normalising dopamine neuron activity.
Figure 5.
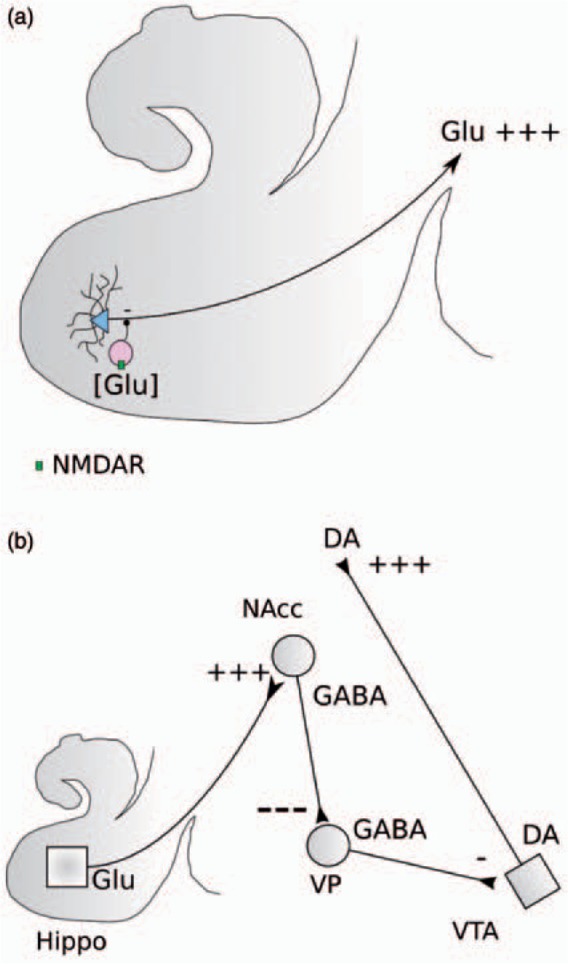
Reduced NMDA receptor (NMDAR) expression on hippocampal GABA interneurons leads to increased sensitivity to reductions in presynaptic glutamate levels and disinhibition of glutamate efferents from hippocampus (A) leading to enhanced stimulation of GABA neurons projecting from the nucleus accumbens (NAcc) to ventral pallidum (VP) resulting in enhanced inhibition of GABA neurons projecting from the VP to the ventral tegmental area (VTA) and disinhibition of dopamine (DA) neurons projecting to striatum (after Lodge et al. [2006]).
Drugs targeting glutamate abnormalities in schizophrenia: Studies in patients
A number of different potential targets have been suggested to reverse the hypothesized abnormality of glutamatergic transmission in schizophrenia [Stone, 2009] (see Figure 6). These include i) enhancement of the function of NMDA receptors expressed on GABAergic interneurons: by increasing synaptic glycine levels, through direct action at the glycineB site, or by mGlu5 receptor agonism; ii) enhancement of GABAergic inter-neuron function (through agonism of Trk1 receptors, alpha-7 nicotinic receptors or M1 receptors); iii) enhancement of GABA tone on glutamatergic projection neurons (through drugs with preferential effects at alpha-2 containing GABA receptors); and iv) reduction of the effect of excess downstream glutamate release by antagonism of AMPA glutamate receptors, or by reducing glutamate release through agonism of mGlu2/3 autoreceptors (Figure 6).
Figure 6.

Potential targets for drugs to reduce excess cortical glutamate release: Enhancement of NMDA receptor function by direct action at glycineB site (2), or by increasing synaptic glycine concentrations through the block of glycine transporters (1). Enhancement of NMDA receptor function through agonism of mGluR5 metabotropic glutamate receptors (3). Enhancement of GABA interneuron function through M1 or nicotinic alpha-7 agonism (4), or enhancement of BDNF function (5). Enhancement of GABA action on alpha-2 containing GABA-A receptors (6). Reduction of the effects of cortical glutamate release by blocking AMPA receptors (7), or by reducing glutamate release through mGlu2/3 autoreceptor agonism (8) (An earlier version of this figure appeared in Stone [2009]), reproduced with permission from Bentham Science Publishers Ltd.
Enhancement of NMDA receptor function
Several studies have investigated the effect of drugs which increase NMDA receptor function via the NMDA receptor glycine site: either increasing synaptic glycine levels by inhibiting type 1 glycine transporters (GlyT1) and preventing reuptake of synaptic glycine (sarcosine), or by acting as agonists at the glycineB modulatory site (glycine and D-serine). As these compounds were not developed as CNS agents, relatively high doses are required for a clinical response (30–60 g glycine per day has commonly been used). Nonetheless, a number of studies have employed these agents as adjunctive treatments in patients with schizophrenia. These have recently been reviewed in a meta-analysis, which showed a modest effect on both positive and negative psychotic symptoms [Tsai and Lin, 2010]. No trials of glycineB agonists as monotherapy have been published to date, and it is possible that the effect size would be more marked when used as first-line treatment. A single study of sarcosine as monotherapy showed efficacy, but patients were randomized to low-dose (1 g) or high-does (2 g) sarcosine and so a direct comparison against dopaminergic agents has not yet been made [Lane et al. 2008]. It is interesting to note that glycine, D-serine and sarcosine did not have any additional effect when added to clozapine [Tsai and Lin, 2010], possibly because part of the superior efficacy of clozapine may be due to intrinsic agonist action at the glycineB modulatory site [Schwieler et al. 2008]. It must be noted that other currently available antipsychotic drugs (including haloperidol, thioridazine, chlorpromazine and clozapine) appear to interact with GlyT1 as noncompetitive antagonists at therapeutic doses [Williams et al. 2004].
Reduction of downstream glutamate release and its effects
Drugs enhancing the function of alpha-2 subunit containing GABA-A receptors should, theoretically, lead to reduced downstream glutamate release (Figure 6) [Lewis et al. 2005]. One study of MK-0777, a benzodiazepine-like drug with selectivity as a partial agonist at alpha-2 and alpha-3 GABA-A receptor subunits, reported improved cognition in patients with schizophrenia, but no effect on psychotic symptoms [Lewis et al. 2008].
Lamotrigine, a drug which inhibits glutamate release, has been investigated as an adjunctive treatment in schizophrenia. Lamotrigine has been shown to reverse positive, negative and cognitive symptoms associated with ketamine administration in healthy volunteers [Hosak and Libiger, 2002], and to reverse ketamine-associated changes in brain function measured using fMRI [Deakin et al. 2008]. A recent meta-analysis suggests that lamotrigine, in contrast to drugs acting through glycine enhancement of NMDA receptor function, is effective as an add-on medication for patients who are only partially responsive to clozapine, although effects were relatively modest [Tiihonen et al. 2009].
Glutamate mGlu 2/3 receptors are presynaptic autoreceptors [Kew and Kemp, 2005]. Agonists inhibit synaptic glutamate release (Figure 6), and have been shown to reduce the effects of NMDA receptor antagonists, and amphetamine in both animal and human studies [Javitt, 2004; Moghaddam, 2004]. A recent phase II trial of an mGlu2/3 receptor agonist (LY2140023, an oral prodrug of LY404039), in a sample of patients with chronic schizophrenia, reported significant improvement in positive and negative symptoms compared with placebo [Patil et al. 2007]. Olanzapine (15 mg daily) was used as an active control group in this study, and although not planned, a post hoc comparison of olanzapine versus LY2140023 revealed no statistically significant difference in terms of response to positive and negative symptoms. LY2140023 showed no propensity to elevated prolactin, weight gain or extrapyramidal side effects, however. Its main reported side effects were affective lability (although this was reported to be beneficial in some patients who had severe affective flattening leading to increased emotional response and spontaneous emotional fluctuations), and a mild reduction in body weight and body mass index. One curious feature of the study was that the group of patients on placebo showed no improvement during the trial duration, whereas it is usual for improvement in symptomatology to be seen in patients in clinical trials on both placebo and active drug. A subsequent phase II trial of LY2140023 was reported by Lilly to be ‘inconclusive’ due to a large placebo response, with neither LY2140023 nor olanzapine showing a significant improvement over placebo. They also reported that convulsions occurred in 3 out of the 669 patients recruited [Kinon et al. 2010]. The study has not been published in full, and it is not clear whether Lilly plan to pursue further trials using this prodrug.
It has been suggested that mGlu2/3 agonists may work primarily through dopaminergic mechanisms [Seeman and Guan, 2009]. However, recent work reveals that the efficacy of mGlu2/3 agonists to block the effects of amphetamine, ketamine and PCP are lost in mGlu2/3 knockout mice [Fell et al. 2009]. It is possible that mGlu2/3 agonists may have downstream effects reducing D2High expression [Seeman et al. 2009]. Further studies of LY404039, LY2140023 and related compounds are awaited.
Topiramate, an antiepileptic drug with AMPA antagonist properties has been found to be effective as an adjunctive therapy in treatment-resistant patients with schizophrenia [Tiihonen et al. 2005], and to reduce the effects of MK-801 in rats [Deutsch et al. 2002], although it is possible that these effects of topiramate may occur through enhancement of GABA transmission, as AMPA antagonism only occurs at higher concentrations [Gibbs et al. 2000].
Other mechanisms
The antibiotic minocycline has, somewhat unexpectedly, been shown to inhibit the effects of NMDA receptor antagonism by MK-801 on rats [Levkovitz et al. 2007; Zhang et al. 2007], and to reverse PCP-induced cognitive deficits [Fujita et al. 2008]. A double-blind, randomized controlled trial of minocycline as add-on treatment in patients with early phase schizophrenia (within the first 5 years of diagnosis) revealed a significant effect on negative and cognitive symptoms [Levkovitz et al. 2010]. Although the exact mechanism of action for minocycline in schizophrenia has still to be ascertained, it is possible that its effect arises through the inhibition of glutamate excitotoxicity (mediated via nitric oxide) by blocking p38 MAP kinase and c-jun N-terminal kinase (mitogen-activated protein kinases responsive to stress stimuli that regulate cellular functions including neurodegeneration, apoptosis, cell differentiation and proliferation) [Pi et al. 2004; Wilkins et al. 2004].
Cannabidiol (CBD), a constituent of cannabis, may also have a modulatory effect on glutamatergic transmission, as it has been shown to inhibit ketamine and MK-801-induced effects in animal models [Long et al. 2006; Moreira and Guimaraes, 2005], and in humans [Hallak et al. 2011]. Cannabis users with detectable levels of both CBD and delta-9 tetrahydrocannabinol (THC) in hair samples reported a lower incidence of schizophrenia-like symptoms than those in whom THC alone was detected [Morgan and Curran, 2008]. Furthermore, acute intoxication with cannabis containing low CBD led to impairments in recall, whereas high CBD cannabis did not induce any cognitive deficits [Morgan et al. 2010]. CBD has been shown to have the opposite effect to THC on neural activation measured using fMRI during an emotional processing task and a verbal memory task [Bhattacharyya et al. 2010; Fusar-Poli et al. 2009], and pretreatment with CBD significantly attenuates the psychotogenic effects of THC [Bhattacharyya et al. 2010; Karniol et al. 1974]. Preliminary work suggests that CBD is effective as an antipsychotic in patients with schizophrenia [Zuardi et al. 2006], although it had no additional beneficial effect in a small open-label study of clozapine-resistant patients [Zuardi et al. 2006].
The mechanism of action of CBD has not yet been elucidated completely. It has been demonstrated that CBD antagonizes the inhibitory effect of endocannabinoids and THC on GABA and glutamate transmission, mediated via CB1 receptors [Godino Mdel et al. 2007; Neu et al. 2007]. Given the hypothesized mechanism of ketamine action on GABA and glutamate systems, it is possible that the enhancement of GABA-A function is its primary mode of action in reducing ketamine-induced effects (Figure 6). However, a CB1 antagonist was not found to be effective in patients with schizophrenia [Meltzer et al. 2004], and there is growing evidence that some of the beneficial effects of CBD, like minocycline, may be mediated via inhibition of p38 MAP kinase [El-Remessy et al. 2008; Esposito et al. 2006].
Drugs targeting glutamate in schizophrenia: Drugs in development
GlyT1 inhibitors
Several pharmaceutical companies have published data on GlyT1 receptor inhibitors (see Table 1). Roche reported in a press release that their GlyT1 inhibitor, RG1678, was successful in treating negative symptoms in a phase II drug trial, but they have not published any further data on this compound at present [Pinard et al. 2010]. Johnson and Johnson have reported that the GlyT1 inhibitor, R231857, improved scopolamine-induced cognitive impairments in healthy volunteers [Liem-Moolenaar et al. 2010]. Schering-Plough report that they are investigating the effects of Org 25935 on negative symptoms, but no data have yet been released to the public domain. One concerning potential side effect of glycine transporter inhibitors is respiratory depression, although it is not clear whether this affects all compounds in this class [Perry et al. 2008]. Another issue is that the effect of GlyT1 inhibitors appears to occur only within a particular dose range, increasing NMDA receptor currents and long-term potentiation (LTP) within this range, but leading to reductions in NMDA receptor currents at higher doses [Martina et al. 2004].
Table 1.
Glutamatergic drugs currently in development for the treatment of schizophrenia.
| Company name | Drug name | Mechanism | Phase | Results |
|---|---|---|---|---|
| Roche | RG1678 | GlyT1 inhibitor | Phase II | Effective vs. negative symptoms in patients with schizophrenia |
| Johnson & Johnson | R231857 | GlyT1 inhibitor | Phase I | Improved scopolamine-induced cognitive impairments in healthy volunteers |
| Schering-Plough | Org 25935 | GlyT1 inhibitor | ?Phase II | No results in public domain |
| Lilly | LY379268 | Allosteric mGlu2 potentiator | ? | Preclinical effectiveness vs. ketamine and MK-801 models |
| Merck | CDPPB | Allosteric mGlu5 potentiator | ? | Preclinical effectiveness vs. amphetamine, MK-801 and sucrose-preference (negative symptoms) models |
Metabotropic glutamatergic receptors: Allosteric potentiators
There has been some interest in developing allosteric potentiators of metabotropic glutamate receptors [Johnson et al. 2004], and two pharmaceutical companies have published data on these compounds (see Table 1). There are several theoretical advantages of allosteric potentiation in targeting the glutamatergic system. As endogenous ligand is required for their action, they should have a lower propensity to side effects; they may also be less prone to desensitization which occurs with drugs targeting the active site. LY379268 is an allosteric drug potentiating glutamate signalling at the mGlu2 receptor currently in development by Lilly. It has been shown to block ketamine-induced glutamate release, as well as ketamine-induced dopamine and histamine release in the prefrontal cortex, and norepinephrine release in hippocampus [Fell et al. 2010; Lorrain et al. 2003a, 2003b]. It has also been shown to inhibit MK-801-induced retrosplenial cortex damage when injected into thalamus or cortex [Carter et al. 2004], suggesting that it may be neuroprotective in the early stages of psychosis.
3-Cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB) is an allosteric potentiator of the mGlu5 receptor developed by Merck [Lindsley et al. 2004]. It increases the affinity of glutamate for the receptor, leading to an enhancement of NMDA receptor activity (Figure 6). It has been shown to attenuate amphetamine-induced PPI deficits [Kinney et al. 2005], and to reverse MK-801-induced elevation in pyramidal cell activity [Lecourtier et al. 2007]. It was also effective in an animal model of negative symptoms (MK-801-induced impairment of sucrose preference) [Vardigan et al. 2010], and showed superior efficacy compared with mGlu2/3 agonists in reversing MK-801-induced cognitive impairment [Vales et al. 2010; Stefani and Moghaddam, 2010]. CDPPB was found to have a U-shaped dose—response curve on cognitive function and on GluR1 phosphorylation, however, suggesting a fairly tight therapeutic window [Uslaner et al. 2009].
Glutamate and illness progression in schizophrenia: A critical window of opportunity?
Elevated cortical glutamate activity in schizophrenia appears to be most marked in the early phases of the illness [Théberge et al. 2002] and in the prodrome [Stone et al. 2009]. Thus, it makes intuitive sense that drugs targeting excess glutamate release may be of most benefit when given during these stages. Indeed, as the illness progresses, it appears that, rather than being overactive, that cortical glutamate system may be reduced in function compared with healthy volunteers [Théberge et al. 2003]. It is tempting to speculate that administration of drugs that reduce cortical glutamate release (such as mGlu2/3 agonists), or inhibit its effects (such as AMPA antagonists and possibly minocycline) may be disease-modifying if given early enough, preventing transition to psychosis in individuals with prodromal symptoms and improving outcome in individuals with first-episode schizophrenia.
The evidence for a phase-specific deficit in NMDA receptor function is less clear, however. In one small SPECT study with the NMDA receptor ligand [123I]CNS-1261, unmedicated individuals with chronic schizophrenia were shown to have reduced NMDA receptor binding in left hippocampus [Pilowsky et al. 2006]. Thus, drugs targeting NMDA receptor enhancement (glycineB agonists, GlyT1 antagonists and mGlu5 receptor agonists) may be of benefit at other phases of the illness including the prodrome and first episode.
Clearly further work is required to investigate these hypotheses as trials to date have generally targeted individuals with chronic schizophrenia.
Conclusions
Novel drugs targeting glutamate transmission have shown considerable promise in the treatment of schizophrenia. Current evidence supports their use as adjunctive agents in individuals who fail to respond to conventional dopaminergic antipsychotic drugs, and preliminary data suggests that they are also efficacious as monotherapy. There are currently a large number of glutamatergic compounds in development, with a great deal of excitement about their potential as novel therapeutic agents in schizophrenia. It seems likely that the next wave of drugs for schizophrenia will target this system.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Dr Stone was employed on a non-restricted academic fellowship by GlaxoSmithKline 2008–2009. He has received travel expenses for conference attendance from Janssen. He has received honoraria from Berenberg Bank and from Pfizer.
References
- Addington J., Addington D., Maticka-Tyndale E. (1991) Cognitive functioning and positive and negative symptoms in schizophrenia. Schizophrenia Res 5: 123–134 [DOI] [PubMed] [Google Scholar]
- Andreasen N.C. (1999) A unitary model of schizophrenia: Bleuler's “fragmented phrene” as schizencephaly. Arch Gen Psychiatry 56: 781–787 [DOI] [PubMed] [Google Scholar]
- Bartha R., Williamson P.C., Drost D.J., Malla A., Carr T.J., Cortese L., et al. (1997) Measurement of glutamate and glutamine in the medial prefrontal cortex of never-treated schizophrenic patients and healthy controls by proton magnetic resonance spectroscopy. Arch Gen Psychiatry 54: 959–965 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S., Morrison P.D., Fusar-Poli P., Martin-Santos R., Borgwardt S., Winton-Brown T., et al. (2010) Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology 35: 764–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder R.M., Reiter G., Bates J., Lencz T., Szeszko P., Goldman R.S., et al. (2006) Cognitive development in schizophrenia: Follow-back from the first episode. J Clin Exp Neuropsychol 28: 270–282 [DOI] [PubMed] [Google Scholar]
- Block W., Bayer T.A., Tepest R., Traber F., Rietschel M., Muller D.J., et al. (2000) Decreased frontal lobe ratio of N-acetyl aspartate to choline in familial schizophrenia: A proton magnetic resonance spectroscopy study. Neurosci Lett 289: 147–151 [DOI] [PubMed] [Google Scholar]
- Buchanan R.W., Breier A., Kirkpatrick B., Ball P., Carpenter W.T., Jr (1998) Positive and negative symptom response to clozapine in schizophrenic patients with and without the deficit syndrome. Am J Psychiatry 155: 751–760 [DOI] [PubMed] [Google Scholar]
- Carter K., Dickerson J., Schoepp D.D., Reilly M., Herring N., Williams J., et al. (2004) The mGlu2/3 receptor agonist LY379268 injected into cortex or thalamus decreases neuronal injury in retrosplenial cortex produced by NMDA receptor antagonist MK-801: Possible implications for psychosis. Neuropharmacology 47: 1135–1145 [DOI] [PubMed] [Google Scholar]
- Coyle J.T. (2006) Glutamate and schizophrenia: Beyond the dopamine hypothesis. Cellular Mol Neurobiol 26: 365–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin J.F., Lees J., McKie S., Hallak J.E., Williams S.R., Dursun S.M. (2008) Glutamate and the neural basis of the subjective effects of ketamine: A pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry 65: 154–164 [DOI] [PubMed] [Google Scholar]
- Deutsch S.I., Rosse R.B., Billingslea E.N., Bellack A.S., Mastropaolo J. (2002) Topiramate antagonizes MK-801 in an animal model of schizophrenia. Eur J Pharmacol 449: 121–125 [DOI] [PubMed] [Google Scholar]
- Dingledine R., Borges K., Bowie D., Traynelis S.F. (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7–61 [PubMed] [Google Scholar]
- El-Remessy A.B., Tang Y., Zhu G., Matragoon S., Khalifa Y., Liu E.K., et al. (2008) Neuroprotective effects of cannabidiol in endotoxin-induced uveitis: Critical role of p38 MAPK activation. Mol Vision 14: 2190–2203 [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Esposito G., De Filippis D., Maiuri M.C., De Stefano D., Carnuccio R., Iuvone T. (2006) Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci Lett 399: 91–95 [DOI] [PubMed] [Google Scholar]
- Fell M.J., Katner J.S., Johnson B.G., Khilevich A., Schkeryantz J.M., Perry K.W., et al. (2010) Activation of metabotropic glutamate (mGlu)2 receptors suppresses histamine release in limbic brain regions following acute ketamine challenge. Neuropharmacology 58: 632–639 [DOI] [PubMed] [Google Scholar]
- Fell M.J., Perry K.W., Falcone J.F., Johnson B.G., Barth V.N., Rash K.S., et al. (2009) In vitro and in vivo evidence for a lack of interaction with dopamine D2 receptors by the metabotropic glutamate 2/3 receptor agonists. [DOI] [PubMed]
- 1S,2S,5R,6S-2-aminobicyclo[3.1.0]hexane-2,6-bicaroxylate monohydrate (LY354740) and (-)-2-oxa-4-aminobicyclo[3.1.0] Hexane-4,6-dicarboxylic acid (LY379268). J Pharmacol Exp Therapeutics 331: 1126–1136 [DOI] [PubMed] [Google Scholar]
- Fujita Y., Ishima T., Kunitachi S., Hagiwara H., Zhang L., Iyo M., et al. (2008) Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the antibiotic drug minocycline. Progress Neuro-psychopharmacol Biol Psychiatry 32: 336–339 [DOI] [PubMed] [Google Scholar]
- Fusar-Poli P., Crippa J.A., Bhattacharyya S., Borgwardt S.J., Allen P., Martin-Santos R., et al. (2009) Distinct effects of {delta}9-tetrahydrocannabinol and cannabidiol on neural activation during emotional processing. Arch Gen Psychiatry 66: 95–105 [DOI] [PubMed] [Google Scholar]
- Gibbs J.W., III, Sombati S., DeLorenzo R.J., Coulter D.A. (2000) Cellular actions of topiramate: Blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia 41(Suppl. 1): S10–S16 [DOI] [PubMed] [Google Scholar]
- Godino Mdel C., Torres M., Sanchez-Prieto J. (2007) CB1 receptors diminish both Ca(2+) influx and glutamate release through two different mechanisms active in distinct populations of cerebrocortical nerve terminals. J Neurochem 101: 1471–1482 [DOI] [PubMed] [Google Scholar]
- Hallak J.E., Dursun S.M., Bosi D.C., de Macedo L.R., Machado-De-Sousa J.P., Abrao J., et al. (2011) The interplay of cannabinoid and NMDA glutamate receptor systems in humans: Preliminary evidence of interactive effects of cannabidiol and ketamine in healthy human subjects. Progress Neuro-psychopharmacol Biol psychiatry 35(1): 198–202 [DOI] [PubMed] [Google Scholar]
- Harrison P.J., Weinberger D.R. (2005) Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol Psychiatry 10: 40–68 [DOI] [PubMed] [Google Scholar]
- Hosak L., Libiger J. (2002) Antiepileptic drugs in schizophrenia: A review. Eur Psychiatry 17: 371–378 [DOI] [PubMed] [Google Scholar]
- Javitt D.C. (2001) Management of negative symptoms of schizophrenia. Curr Psychiatry Rep 3: 413–417 [DOI] [PubMed] [Google Scholar]
- Javitt D.C. (2004) Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry 9: 984–997, 979 [DOI] [PubMed] [Google Scholar]
- Javitt D.C. (2007) Glutamate and schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine—glutamate Interactions. Int Rev Neurobiol 78: 69–108 [DOI] [PubMed] [Google Scholar]
- Johnson M.P., Nisenbaum E.S., Large T.H., Emkey R., Baez M., Kingston A.E. (2004) Allosteric modulators of metabotropic glutamate receptors: Lessons learnt from mGlu1, mGlu2 and mGlu5 potentiators and antagonists. Biochem Soc Trans 32: 881–887 [DOI] [PubMed] [Google Scholar]
- Kane J., Honigfeld G., Singer J., Meltzer H. (1988) Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry 45: 789–796 [DOI] [PubMed] [Google Scholar]
- Kapur S., Mamo D. (2003) Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry 27(7): 1081–1090 [DOI] [PubMed] [Google Scholar]
- Karniol I.G., Shirakawa I., Kasinski N., Pfeferman A., Carlini E.A. (1974) Cannabidiol interferes with the effects of delta 9 — tetrahydrocannabinol in man. Eur J Pharmacol 28: 172–177 [DOI] [PubMed] [Google Scholar]
- Kegeles L.S., Shungu D.C., Anjilvel S., Chan S., Ellis S.P., Xanthopoulos E., et al. (2000) Hippocampal pathology in schizophrenia: Magnetic resonance imaging and spectroscopy studies. Psychiatry Res 98: 163–175 [DOI] [PubMed] [Google Scholar]
- Kew J.N., Kemp J.A. (2005) Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 179: 4–29 [DOI] [PubMed] [Google Scholar]
- Kinney G.G., O'Brien J.A., Lemaire W., Burno M., Bickel D.J., Clements M.K., et al. (2005) A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Therapeutics 313: 199–206 [DOI] [PubMed] [Google Scholar]
- Kinon B.J., Zhang L., Williams J.E., Osuntokun O.O., Millen B.A., Kollack-Walker S. (2010) Ly2140023 monohydrate: An agonist at the Mglu2/3 receptor for the treatment of schizophrenia. Schizophrenia Res 117: 379–379 [Google Scholar]
- Krystal J.H., Perry E.B., Jr, Gueorguieva R., Belger A., Madonick S.H., Abi-Dargham A., et al. (2005) “Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: Implications for glutamatergic and dopaminergic model psychoses and cognitive function”. Arch Gen Psychiatry 62(9): 985–94 [DOI] [PubMed] [Google Scholar]
- Krystal J.H., Karper L.P., Seibyl J.P., Freeman G.K., Delaney R., Bremner J.D., et al. (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199–214 [DOI] [PubMed] [Google Scholar]
- Lane H.Y., Liu Y.C., Huang C.L., Chang Y.C., Liau C.H., Perng C.H., et al. (2008) Sarcosine (N-methylglycine) treatment for acute schizophrenia: A randomized, double-blind study. Biol Psychiatry 63: 9–12 [DOI] [PubMed] [Google Scholar]
- Law A.J., Deakin J.F. (2001) Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport 12: 2971–2974 [DOI] [PubMed] [Google Scholar]
- Lecourtier L., Homayoun H., Tamagnan G., Moghaddam B. (2007) Positive allosteric modulation of metabotropic glutamate 5 (mGlu5) receptors reverses N-Methyl-D-aspartate antagonist-induced alteration of neuronal firing in prefrontal cortex. Biol Psychiatry 62: 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkovitz Y., Levi U., Braw Y., Cohen H. (2007) Minocycline, a second-generation tetracycline, as a neuroprotective agent in an animal model of schizophrenia. Brain Res 1154: 154–162 [DOI] [PubMed] [Google Scholar]
- Levkovitz Y., Mendlovich S., Riwkes S., Braw Y., Levkovitch-Verbin H., Gal G., et al. (2010) A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry 71: 138–149 [DOI] [PubMed] [Google Scholar]
- Lewis D.A., Cho R.Y., Carter C.S., Eklund K., Forster S., Kelly M.A., et al. (2008) Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am J Psychiatry 165: 1585–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D.A., Hashimoto T., Volk D.W. (2005) Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6: 312–24 [DOI] [PubMed] [Google Scholar]
- Liem-Moolenaar M., Zoethout R.W., de Boer P., Schmidt M., de Kam M.L., Cohen A.F., et al. (2010) The effects of a glycine reuptake inhibitor R231857 on the central nervous system and on scopolamine-induced impairments in cognitive and psychomotor function in healthy subjects. J Psychopharmacol 24: 1681–1687 [DOI] [PubMed] [Google Scholar]
- Lindsley C.W., Wisnoski D.D., Leister W.H., O'brien J.A., Lemaire W., Williams D.L., Jr, et al. (2004) Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-diphenyl-1H- pyrazol-5-yl)benzamides that potentiate receptor function in vivo. J Med Chem 47: 5825–5828 [DOI] [PubMed] [Google Scholar]
- Lodge D.J., Grace A.A. (2006) The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology 31: 1356–61 [DOI] [PubMed] [Google Scholar]
- Long L.E., Malone D.T., Taylor D.A. (2006) Cannabidiol reverses MK-801-induced disruption of prepulse inhibition in mice. Neuropsychopharmacology 31: 795–803 [DOI] [PubMed] [Google Scholar]
- Lorrain D.S., Baccei C.S., Bristow L.J., Anderson J.J., Varney M.A. (2003a) Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 117: 697–706 [DOI] [PubMed] [Google Scholar]
- Lorrain D.S., Schaffhauser H., Campbell U.C., Baccei C.S., Correa L.D., Rowe B., et al. (2003b) Group II mGlu receptor activation suppresses norepinephrine release in the ventral hippocampus and locomotor responses to acute ketamine challenge. Neuropsychopharmacology 28: 1622–1632 [DOI] [PubMed] [Google Scholar]
- Lutkenhoff E.S., van Erp T.G., Thomas M.A., Therman S., Manninen M., et al. (2010) Proton MRS in twin pairs discordant for schizophrenia. Mol Psychiatry 15(3): 308–318 [DOI] [PubMed] [Google Scholar]
- Martina M., Gorfinkel Y., Halman S., Lowe J.A., Periyalwar P., Schmidt C.J., et al. (2004) Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol 557: 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy J.P., Lieberman J.A., Stroup T.S., Davis S.M., Meltzer H.Y. Rosenheck R.A. et al for the CATIE Investigators. (2006) Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 163: 600–610 [DOI] [PubMed] [Google Scholar]
- Meltzer H.Y., Arvanitis L., Bauer D. Rein, W. for the Meta-Trial Study Group. (2004) Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry 161: 975–984 [DOI] [PubMed] [Google Scholar]
- Moghaddam B. (2004) Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacology (Berl) 174: 39–44 [DOI] [PubMed] [Google Scholar]
- Moghaddam B., Adams B., Verma A., Daly D. (1997) Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17: 2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira F.A., Guimaraes F.S. (2005) Cannabidiol inhibits the hyperlocomotion induced by psychotomimetic drugs in mice. Eur J Pharmacol 512: 199–205 [DOI] [PubMed] [Google Scholar]
- Morgan C.J., Curran H.V. (2006) Acute and chronic effects of ketamine upon human memory: A review. Psychopharmacology 188: 408–424 [DOI] [PubMed] [Google Scholar]
- Morgan C.J., Curran H.V. (2008) Effects of cannabidiol on schizophrenia-like symptoms in people who use cannabis. Br J Psychiatry 192: 306–307 [DOI] [PubMed] [Google Scholar]
- Morgan C.J., Schafer G., Freeman T.P., Curran H.V. (2010) Impact of cannabidiol on the acute memory and psychotomimetic effects of smoked cannabis: Naturalistic study. Br J Psychiatry 197: 285–290 [DOI] [PubMed] [Google Scholar]
- Neu A., Foldy C., Soltesz I. (2007) Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol 578: 233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrmann P., Siegmund A., Suslow T., Spitzberg K., Kersting A., Arolt V., et al. (2005) Evidence for glutamatergic neuronal dysfunction in the prefrontal cortex in chronic but not in first-episode patients with schizophrenia: A proton magnetic resonance spectroscopy study. Schizophr Res 73: 153–157 [DOI] [PubMed] [Google Scholar]
- Olney J.W., Farber N.B. (1995) Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 52: 998–1007 [DOI] [PubMed] [Google Scholar]
- Olney J.W., Newcomer J.W., Farber N.B. (1999) NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res 33: 523–533 [DOI] [PubMed] [Google Scholar]
- Ongur D., Prescot A.P., Jensen J.E., Cohen B.M., Renshaw P.F. (2009) Creatine abnormalities in schizophrenia and bipolar disorder. Psychiatry Res 172: 44–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S.T., Zhang L., Martenyi F., Lowe S.L., Jackson K.A., Andreev B.V., et al. (2007) Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized Phase 2 clinical trial. Nat Med 13: 1102–1107 [DOI] [PubMed] [Google Scholar]
- Perry K.W., Falcone J.F., Fell M.J., Ryder J.W., Yu H., Love P.L., et al. (2008) Neurochemical and behavioral profiling of the selective GlyT1 inhibitors ALX5407 and LY2365109 indicate a preferential action in caudal vs. cortical brain areas. Neuropharmacology 55: 743–754 [DOI] [PubMed] [Google Scholar]
- Phillips L.J., Yung A.R., McGorry P.D. (2000) Identification of young people at risk of psychosis: Validation of Personal Assessment and Crisis Evaluation Clinic intake criteria. Aust N Z J Psychiatry 34(Suppl): S164–S169 [DOI] [PubMed] [Google Scholar]
- Pi R., Li W., Lee N.T., Chan H.H., Pu Y., Chan L.N., et al. (2004) Minocycline prevents glutamate-induced apoptosis of cerebellar granule neurons by differential regulation of p38 and Akt pathways. J Neurochem 91: 1219–1230 [DOI] [PubMed] [Google Scholar]
- Pilowsky L.S., Bressan R.A., Stone J.M., Erlandsson K., Mulligan R.S., Krystal J.H., et al. (2006) First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry 11: 118–119 [DOI] [PubMed] [Google Scholar]
- Pinard E., Alanine A., Alberati D., Bender M., Borroni E., Bourdeaux P., et al. (2010) Selective GlyT1 inhibitors: Discovery of [4-(3-fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfon yl-2-((S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat schizophrenia. J Medicinal Chem 53: 4603–4614 [DOI] [PubMed] [Google Scholar]
- Rothman D.L., Behar K.L., Hyder F., Shulman R.G. (2003) In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: Implications for brain function. Annu Rev Physiol 65: 401–427 [DOI] [PubMed] [Google Scholar]
- Rowland L.M., Spieker E.A., Francis A., Barker P.B., Carpenter W.T., Buchanan R.W. (2009) White matter alterations in deficit schizophrenia. Neuropsychopharmacology 34: 1514–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieler L., Linderholm K.R., Nilsson-Todd L.K., Erhardt S., Engberg G. (2008) Clozapine interacts with the glycine site of the NMDA receptor: Electrophysiological studies of dopamine neurons in the rat ventral tegmental area. Life Sci 83: 170–175 [DOI] [PubMed] [Google Scholar]
- Seeman P., Battaglia G., Corti C., Corsi M., Bruno V. (2009) Glutamate receptor mGlu2 and mGlu3 knockout striata are dopamine supersensitive, with elevated D2(High) receptors and marked super-sensitivity to the dopamine agonist (+)PHNO. Synapse 63: 247–251 [DOI] [PubMed] [Google Scholar]
- Seeman P., Guan H.C. (2009) Glutamate agonist LY404,039 for treating schizophrenia has affinity for the dopamine D2(High) receptor. Synapse 63: 935–939 [DOI] [PubMed] [Google Scholar]
- Seeman P., Lee T. (1975) Antipsychotic drugs: Direct correlation between clinical potency and pre-synaptic action on dopamine neurons. Science 188: 1217–1219 [DOI] [PubMed] [Google Scholar]
- Sharp F.R., Tomitaka M., Bernaudin M., Tomitaka S. (2001) Psychosis: Pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia?. Trends Neurosci 24: 330–334 [DOI] [PubMed] [Google Scholar]
- Stefani M.R., Moghaddam B. (2010) Activation of type 5 metabotropic glutamate receptors attenuates deficits in cognitive flexibility induced by NMDA receptor blockade. Eur J Pharmacol 639: 26–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J.M. (2009) Imaging the glutamate system in humans: Relevance to drug discovery for schizophrenia. Curr Pharmaceut Design 15: 2594–2602 [DOI] [PubMed] [Google Scholar]
- Stone J.M., Day F., Tsagaraki H., Valli I., McLean M.A., Lythgoe D.J., et al. (2009) Glutamate dysfunction in people with prodromal symptoms of psychosis: Relationship to gray matter volume. Biol Psychiatry 66: 533–539 [DOI] [PubMed] [Google Scholar]
- Stone J.M., Erlandsson K., Arstad E., Squassante L., Teneggi V., Bressan R.A., et al. (2008) Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: A [(123)I]CNS-1261 SPET study. Psychopharmacology (Berl) 197: 401–408 [DOI] [PubMed] [Google Scholar]
- Stone J.M., Howes O.D., Egerton A., Kambeitz J., Allen P., Lythgoe D.J., et al. (2010a) Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biol Psychiatry 68: 599–602 [DOI] [PubMed] [Google Scholar]
- Stone J.M., Morrison P.D., Pilowsky L.S. (2007) Glutamate and dopamine dysregulation in schizophrenia—a synthesis and selective review. J Psychopharmacol 21: 440–452 [DOI] [PubMed] [Google Scholar]
- Stone J.M., Raffin M., Morrison P., McGuire P.K. (2010b) Review: The biological basis of antipsychotic response in schizophrenia. J Psychopharmacol 24: 953–964 [DOI] [PubMed] [Google Scholar]
- Tamminga C.A., Buchanan R.W., Gold J.M. (1998) The role of negative symptoms and cognitive dysfunction in schizophrenia outcome. Int Clin Psychopharmacol 13(Suppl. 3): S21–S26 [DOI] [PubMed] [Google Scholar]
- Tayoshi S., Sumitani S., Taniguchi K., Shibuya-Tayoshi S., Numata S., Iga J., et al. (2009) Metabolite changes and gender differences in schizophrenia using 3-Tesla proton magnetic resonance spectroscopy (1H-MRS). Schizophrenia Res 108: 69–77 [DOI] [PubMed] [Google Scholar]
- Théberge J., Al-Semaan Y., Williamson P.C., Menon R.S., Neufeld R.W.J., Rajakumar N., et al. (2003) Glutamate and glutamine in the anterior cingulate and thalamus of medicated patients with chronic schizophrenia and healthy comparison subjects measured with 4.0-T proton MRS. Am J Psychiatry 160: 2231–2233 [DOI] [PubMed] [Google Scholar]
- Théberge J., Bartha R., Drost D.J., Menon R.S., Malla A., Takhar J., et al. (2002) Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry 159: 1944–1946 [DOI] [PubMed] [Google Scholar]
- Tibbo P., Hanstock C., Valiakalayil A., Allen P. (2004) 3-T proton MRS investigation of glutamate and glutamine in adolescents at high genetic risk for schizophrenia. Am J Psychiatry 161: 1116–1118 [DOI] [PubMed] [Google Scholar]
- Tiihonen J., Halonen P., Wahlbeck K., Repo-Tiihonen E., Hyvarinen S., Eronen M., et al. (2005) Topiramate add-on in treatment-resistant schizophrenia: A randomized, double-blind, placebo-controlled, crossover trial. J Clin Psychiatry 66: 1012–1015 [DOI] [PubMed] [Google Scholar]
- Tiihonen J., Wahlbeck K., Kiviniemi V. (2009) The efficacy of lamotrigine in clozapine-resistant schizophrenia: A systematic review and meta-analysis. Schizophrenia Res 109: 10–14 [DOI] [PubMed] [Google Scholar]
- Tsai G.E., Lin P.Y. (2010) Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des 16(5): 522–537 [DOI] [PubMed] [Google Scholar]
- Uslaner J.M., Parmentier-Batteur S., Flick R.B., Surles N.O., Lam J.S., McNaughton C.H., et al. (2009) Dose-dependent effect of CDPPB, the mGluR5 positive allosteric modulator, on recognition memory is associated with GluR1 and CREB phosphorylation in the prefrontal cortex and hippocampus. Neuropharmacology 57: 531–538 [DOI] [PubMed] [Google Scholar]
- Vales K., Svoboda J., Benkovicova K., Bubenikova-Valesova V., Stuchlik A. (2010) The difference in effect of mGlu2/3 and mGlu5 receptor agonists on cognitive impairment induced by MK-801. Eur J Pharmacol 639: 91–98 [DOI] [PubMed] [Google Scholar]
- Vardigan J.D., Huszar S.L., McNaughton C.H., Hutson P.H., Uslaner J.M. (2010) MK-801 produces a deficit in sucrose preference that is reversed by clozapine, D-serine, and the metabotropic glutamate 5 receptor positive allosteric modulator CDPPB: Relevance to negative symptoms associated with schizophrenia?. Pharmacol Biochem Behavior 95: 223–229 [DOI] [PubMed] [Google Scholar]
- Ventura J., Hellemann G.S., Thames A.D., Koellner V., Nuechterlein K.H. (2009) Symptoms as mediators of the relationship between neurocognition and functional outcome in schizophrenia: A meta-analysis. Schizophrenia Res 113: 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins A., Nikodemova M., Compston A., Duncan I. (2004) Minocycline attenuates nitric oxide-mediated neuronal and axonal destruction in vitro. Neuron Glia Biol 1: 297–305 [DOI] [PubMed] [Google Scholar]
- Williams J.B., Mallorga P.J., Jeffrey Conn P., Pettibone D.J., Sur C. (2004) Effects of typical and atypical antipsychotics on human glycine transporters. Schizophr Res 71(1): 103–112 [DOI] [PubMed] [Google Scholar]
- Zhang L., Shirayama Y., Iyo M., Hashimoto K. (2007) Minocycline attenuates hyperlocomotion and prepulse inhibition deficits in mice after administration of the NMDA receptor antagonist dizocilpine. Neuropsychopharmacology 32: 2004–2010 [DOI] [PubMed] [Google Scholar]
- Zuardi A.W., Crippa J.A., Hallak J.E., Moreira F.A., Guimaraes F.S. (2006) Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Med Biol Res 39: 421–429 [DOI] [PubMed] [Google Scholar]