

Abstract
A texaphyrin–oxaliplatin conjugate, oxaliTEX, was designed to test the concept that a platinum analog can overcome defects in drug accumulation and p53-dependent DNA damage response in a tumor model expressing multifactorial mechanisms of cisplatin resistance. Cytotoxic studies resulted in a resistance factor of only 1.2, which essentially indicated complete reversal of resistance in 2780CP cells expressing a factor of 22 with cisplatin. Unlike cisplatin, oxaliTEX accumulated and formed DNA adducts, stabilized and activated p53 at similar levels in both sensitive and resistant cells, and induced apoptosis in both models. The ability and importance of a designer drug, such as oxaliTEX, to overcome cisplatin resistance by targeting two dominant resistance mechanisms is discussed.
Introduction
Cisplatin and carboplatin (Fig. 1) are two of the most important antitumor agents in the clinic. They have demonstrated significant activities against several cancers and are critical components of front line therapeutic regimens in medical oncology.1 These drugs are curative against testicular cancer, but their clinical utility against other cancers remains limited by intrinsic or acquired resistance, which in the case of NSCLC, mesothelioma, and ovarian cancer is reflected in low 5-year survival rates of only 5–20%.2 The biochemical and pharmacologic mechanisms of platinum resistance (e.g., decreased drug uptake, increased glutathione levels, and increased DNA adduct repair) are well established.1 These mechanisms co-exist with molecular mechanisms of resistance and present treatment-limiting barriers to the effective application of these two platinum agents in the clinic.
Fig. 1.

Various Pt(II) complexes, motexafin gadolinium (1, MGd), and texaphyrin–platinum conjugates 2 (cisTex), and 3 (oxaliTEX).
Among molecular mechanisms inducing cisplatin or carboplatin resistance, the most formidable involves loss of function of the tumor suppressor p53, often through missense gene mutation.3,4 However, over a half of advanced cancers (e.g., ovarian cancer, NSCLC and mesothelioma) that retain wild-type p53 are also resistant to cisplatin, and this is ascribed to loss of p53 regulation.3–5 Data from our group has established that Pt analogs, such as DAP [1,2-diaminocyclohexane-diacetato-dichloro-Pt(IV)], can restore stabilization and activation of wild-type p53 and effectively circumvent the molecular mechanism of resistance.5 This has led us to propose that oxaliplatin (Fig. 1), which contains the same diaminocyclohexane (DACH) ligand found in DAP, should also activate p53 function in resistant cells.
Unfortunately, restoring p53 function solves only part of the problem associated with clinical Pt resistance. Among the most important of the biochemical/pharmacologic mechanisms of resistance is reduced drug accumulation. However, finding an effective means to overcome resistance that occurs as a result of diminished Pt drug accumulation has proved elusive. Approaches that have been tested to address this limitation include conjugation to carrier molecules, such as folate, poly-(ethyleneglycol) (PEG), porphyrins, peptides, and nanoparticles.6–13 While in selected cases increased delivery of a Pt drug was achieved, the drug in question did not circumvent other mechanisms of resistance (including diminished p53 function). For an effective strategy, therefore, it is necessary not just to deliver the increased Pt payload to resistant tumor cells, but the Pt drug must have the properties needed to also overcome multifactorial mechanisms of resistance, particularly those associated with reduced p53 function, thereby restoring its dominant apoptotic activity.
Texaphyrins are unique tumor-localizing agents that are ideally suited for creating Pt conjugates.14 Motexafin gadolinium (1, MGd, cf. Fig. 1) has been extensively studied as a water soluble gadolinium(III) texaphyrin complex, and its ability to localize within cancerous lesions in vivo makes it an attractive choice for the design of conjugates to deliver a Pt payload.15–17 Herein, we report the design, synthesis, and biological evaluation of a novel texaphyrin–oxaliplatin like conjugate, and demonstrate as proof-of-principle that this conjugate is capable of overcoming both pharmacologic and molecular mechanisms associated with cisplatin resistance.
Results
Conjugate design
We have recently reported the synthesis of a texaphyrin–platinum conjugate (2, cisTEX, cf. Fig. 1) that produced equivalent levels of intracellular Pt and DNA–Pt adducts in both sensitive A2780 and isogenic multifactorial-resistant 2780CP cells upon exposure to conjugate 2.18,19 This conjugate was designed to form qualitatively identical adducts to those induced by cisplatin or carboplatin.18 Consequently, it displayed potent cytotoxicity in the A2780 human ovarian tumor model. However, its activity in the ~20-fold resistant cell line was improved by only a modest amount (i.e., a reduction in the resistance factor to ~10-fold).19 Thus, cisTEX is able to overcome the pharmacologic transport-related mechanism of resistance but is unable to modulate the molecular mechanisms of resistance. On the other hand, we have demonstrated that the DACH-containing analog DAP was potently effective at circumventing molecular mechanisms of resistance, although in this latter case residual resistance from defects in drug uptake remained.20 This has led us to consider that combining both approaches might be particularly effective. Specifically, we postulated that incorporation of a DACH–platinum within our conjugate design would provide a texaphyrin–platinum congener (3, oxaliTEX, cf. Fig. 1) that would release the DACH–Pt moiety within the cell and circumvent both pharmacologic and molecular mechanisms of resistance. The present study was designed to test this hypothesis.
Cytotoxicity, comparison of resistance factors, and reactivity with plasma proteins
Proof-of-principle insights into whether a particular conjugate design has the potential to circumvent cisplatin resistance may be garnered from the relative activities of the conjugate in a sensitive/resistant pair of intimately related cell lines. Therefore, we compared the activity of conjugate 3 in the A2780 human ovarian cancer model and its ostensibly isogenic cisplatin-resistant 2780CP model.
Cytotoxic studies were conducted following exposure of tumor cells to the Pt agents, and the activity of conjugate 3 (oxaliTEX) in inhibiting cell proliferation was compared to that of conjugate 2 (cisTEX), oxaliplatin, and cisplatin. Typical dose–response curves are shown for conjugates 2 and 3 in Fig. 2. The two curves for conjugate 2 in A2780 and 2780CP models are substantially separated, with the curve for 2780CP cells shifted to higher concentrations, which is an indication of resistance to this particular conjugate in the case of these resistant tumor cells. In contrast, the dose–response curves for conjugate 3 in the two models are almost superimposable. This finding is taken as evidence that this newly designed conjugate is equi-effective against both sensitive and resistant tumor cells and a strong indication that it can fully circumvent resistance in 2780CP cells.
Fig. 2.

Cytotoxicity profiles of (A) cisTEX (2) and (B) oxaliTEX (3) with cisplatin sensitive A2780 and cisplatin resistant 2780CP. The complex was made up as a stock solution (for which the Pt concentration was confirmed by FAAS) and serially diluted before adding to cells, which were then incubated for 5 days at 37 °C in 5% CO2. Error bars represent the standard deviation.
The IC50 values generated from these curves are presented in Table 1. This table also includes values for oxaliplatin and cisplatin for the sake of comparison, together with associated resistance factors in the 2780CP cell line relative to parent A2780 cell line. Conjugate 3 (IC50 = 0.55 ± 0.06 μM) provided a dose potency in the A2780 cell line that was nearly 3-fold greater than conjugate 2 (IC50 = 1.63 ± 0.2 μM). The FDA-approved platinum complexes oxaliplatin and cisplatin provided IC50 values of 0.15 ± 0.05 and 0.33 ± 0.02, respectively. Against 2780CP cells, oxaliTEX (3) and oxaliplatin maintained their potent activities, with IC50 values of 0.65 ± 0.09 and 0.30 ± 0.05 μM, respectively. In contrast, our first generation conjugate 2 and cisplatin produced significantly greater IC50 values of 17.0 ± 1.5 and 7.3 ± 0.2 μM, respectively, values that reflect a 11- to 26-fold lower potency relative to conjugate 3.
Table 1.
IC50 values of platinum complexes in cisplatin sensitive A2780 ovarian and its isogenic cisplatin resistant (2780CP) cell lines
| Complex | IC50 (μM) A2780 | IC50 (μM) 2780CP | Resistance factor |
|---|---|---|---|
| 2 (cisTEX) | 1.63 ± 0.2 | 17.0 ± 1.5a | 10.4 ± 0.71 |
| 3 (oxaliTEX) | 0.55 ± 0.06 | 0.65 ± 0.09 | 1.2 ± 0.18 |
| Oxaliplatin | 0.15 ± 0.05 | 0.30 ± 0.05a | 2.0 ± 0.29 |
| Cisplatin | 0.33 ± 0.02 | 7.3 ± 0.2a | 22.1 ± 0.76 |
p < 0.05 by Student’s t-test vs. IC50 of A2780.
The above results lead us to suggest that conjugate 3 may have a role to play in overcoming the resistance observed in cisplatin resistant wild type p53 ovarian cancer. Although conjugate 3 displays lower potency (higher IC50) than oxaliplatin in the A2780 and 2780CP cell lines, this does not negate its clinical potential. This key point is underscored by carboplatin, which has substantially lower potency than cisplatin (1.6 vs. 0.31 μM in A2780 human ovarian cancer cell line),19 but nevertheless enjoys widespread clinical use.21 The fact that conjugate 3 also overcomes resistance pathways makes it attractive for further study.
A better understanding of relative activity of complexes against resistant cells can be garnered from the resistance factor, which is a ratio of IC50 of an agent in sensitive and resistant cells (Table 1). As expected, 2780CP cells demonstrated 22.1-fold resistance to cisplatin22 and a lower 10.4-fold cross-resistance to conjugate 2. In contrast, 2780CP cells were only 2-fold resistant to oxaliplatin, but were almost devoid of cross-resistance to conjugate 3 (cross-resistance factor, 1.2). This is consistent with essentially complete circumvention of resistance.
The high potencies of conjugate 3 in the A2780 and 2780CP cell lines, as shown in Table 1, is consistent with the design expectation that the conjugate is labile and rapidly converts to a reactive product that is able to interact covalently with cellular macromolecules, including DNA. Based on the underlying chemistry this conversion was expected to involve release of a free DACH–Pt aquo species. To determine the activation rate of conjugate 3 relative to cisplatin and oxaliplatin, samples of fetal bovine serum (FBS) were incubated with the Pt-based agents and the amount of free (unbound) Pt was determined over time. It is presumed that the irreversible reaction of platinum with FBS results in inactivation of the platinum drug. The half-life (t1/2) of the initial logarithmic decay phase was determined by linear regression analyses (Fig. 3). Cisplatin and oxaliplatin reacted rapidly with serum proteins, and this corresponded to an accelerated decrease in free Pt (t1/2 = 5.1–5.3 hours). Conjugate 3 did become activated but its binding to proteins was comparatively slower (t1/2 = 8.3 hours). However, conjugate was found to react more quickly than the clinically approved agent carboplatin (t1/2 = 36–45 hours) and conjugate 2 (t1/2 = 16 hours).19,23
Fig. 3.

Platination of FBS proteins with various platinum agents. Half-lives were determined by fitting the data to an exponential decay curve between 0 and 13 h during which time the logarithmic decay phase was linear.
Intracellular platinum uptake and DNA–Pt lesion quantification
Cisplatin resistance is multifactorial and is reflective of a variety of biochemical pharmacologic mechanisms, including reductions in cellular uptake and retention, decreased DNA adduct formation and a greater tolerance for DNA damage.24–27 The minimal resistance factor obtained for conjugate 3 reflects an ability to overcome most forms of resistance present in the 2780CP model. To gain insights into the basis of this high activity against resistant cells, the intracellular uptake of 3 was analyzed, along with that of the control complexes oxaliplatin and cisplatin. The decrease (>50%) in cisplatin uptake in 2780CP cells relative to A2780 cells was readily apparent and statistically significant, as was the reduced accumulation of oxaliplatin in resistant cells (Fig. 4). Such findings are consistent with the reported 2-fold reduction in uptake seen in resistant cells exposed to platinum anticancer agents.28 Interestingly, no difference in uptake was observed for conjugate 3 between A2780 and 2780CP tumor cells. Relative to oxaliplatin, conjugate 3 demonstrated a ≥2- and ≥4-fold increase in platinum uptake for the A2780 and 2780CP cell lies, respectively. Additionally, it is notable that conjugate 3 also demonstrated a ≥2-fold higher uptake than cisplatin in 2780CP cells.
Fig. 4.
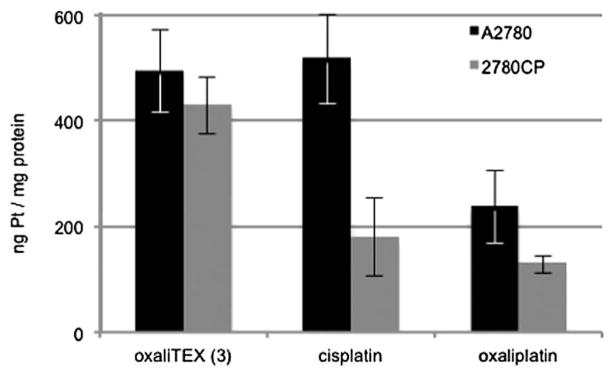
Cellular uptake of platinum drugs. Levels of intracellular platinum in A2780 and 2780CP were determined by FAAS after a 4-hour incubation with 200 μM of the respective complex (concentrations confirmed by FAAS). p < 0.05 by Student’s t-test for platinum uptake of cisplatin and oxaliplatin in 2780CP vs. A2780.
The reduction in platinum uptake in resistant cells generally translates into reduced Pt-adducts formed with DNA.29 This, however, is not always the case and is not seen with every platinum drug. Because this correlation is not certain, the Pt–DNA adducts formed by conjugate 3, as well as the Pt control complexes oxaliplatin and cisplatin, were quantified in both the A2780 and 2780CP cell lines. With cisplatin and oxaliplatin, a statistically significant 50% reduction in Pt–DNA adducts was seen in the 2780CP cells (Fig. 5). This is considered to reflect the corresponding differences in drug uptake noted in Fig. 4, and is fully consistent with what was expected for these known agents. Also in accord with expectations with oxaliplatin,30 the observed adduct levels of conjugate 3 in A2780 cells were lower than those seen for cisplatin, even though the uptake of these two platinum species is comparable (see Fig. 4). This is attributed to the differences in Pt coordination provided by the two species in question. Finally, and in contrast to cisplatin and oxaliplatin, the adduct levels provided by conjugate 3 were not reduced in resistant cells (Fig. 5).
Fig. 5.
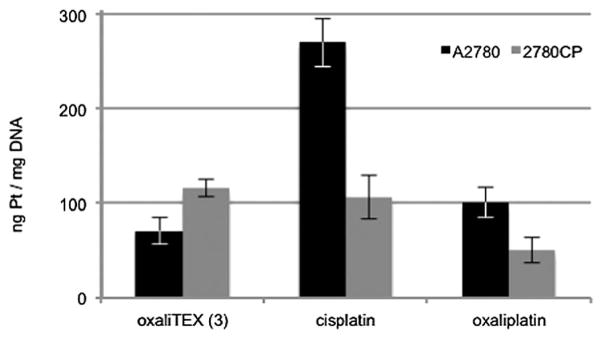
Cellular platination of DNA by oxaliTEX (3), cisplatin, and oxaliplatin. DNA adducts were determined by FAAS after a 4-hour incubation with 200 μM of the respective complex (concentrations confirmed by FAAS). p < 0.05 by Student’s t-test for Pt–DNA lesions by oxaliTEX, cisplatin, and oxaliplatin in 2780CP vs. A2780.
It is evident from an inspection of Fig. 5 and Table 1 that adduct levels may not reflect the potency of the molecule. This is clearly demonstrated by the fact that oxaliplatin adducts are formed at a lower level than those of cisplatin in sensitive A2780 cells, and yet, oxaliplatin is the more potent drug. To generalize this finding and expand it to conjugate 3 in both sensitive and resistant cells, the DNA damage tolerance ratio was calculated. This tolerance is determined from the level of adducts in Fig. 5 and extrapolating to the IC50 concentration; that is, the level of adducts required to kill 50% of tumor cells. The DNA damage tolerance data in Fig. 6 demonstrate that resistant 2780CP cells require 10-fold greater adduct levels as compared to A2780 cells for equivalent antiproliferative activity. As reported prevously,19 cisplatin resistant 2780CP cells were capable of similarly tolerating ca. 10-fold the number of Pt–DNA lesions formed by conjugate 2. Conjugate 3 or oxaliplatin, on the other hand, induced cytotoxicity in the 2780CP model at substantially lower adduct levels.
Fig. 6.

DNA Damage tolerance of oxaliTEX, oxaliplatin, and cisplatin. These calculated values were determined from the ratio of DNA–Pt adducts/([complex] × IC50), with the primary data obtained from three separate experiments.
FACS analysis
Apoptosis is a desirable endpoint for antitumor agents in cancer chemotherapy. To determine whether such a feature was inherent to conjugate 3, and also to ascertain the underlying basis for its antiproliferative activity in Table 1, we conducted apoptotic studies using FACS analysis to detect Annexin V-positive cells. We included the parent drug oxaliplatin as a positive control and MGd (1) as a model for the Pt-free conjugating texaphyrin moiety present in conjugate 3. A2780 and 2780CP cells were exposed to these agents for 24, 72, and 120 hours and the fraction of apoptotic cells determined. The findings are presented in Fig. 7. The data in Fig. 7A demonstrate that the 14–16% of drug-induced apoptotic A2780 cells at 24 hours were not significantly different from controls (12%). No gross changes were observed at 72 and 120 hours in Annexin V-positive control cells or those treated with MGd (1). In contrast, the level of apoptotic cells following exposure to conjugate 3 and oxaliplatin increased 2- to 4-fold with time at 72 (~40%) and 120 (55–60%) hours. Similar temporal profiles for apoptosis were observed with the cisplatin-resistant 2780CP model. Such findings are consistent with the notion that 3 is capable of inducing a similar degree of apoptosis in both A2780 and 2780CP cell lines. Moreover, conjugate 3 is capable of inducing apoptosis to the same extent as oxaliplatin. Finally, the observed apoptosis is attributable to the Pt-bearing conjugate as a whole and not to the texaphyrin carrier.
Fig. 7.

Percent positive Annexin V-FITC signal of (A) A2780 and (B) 2780CP cells after exposure to oxaliTEX (3), oxaliplatin, and MGd (1).
Western blot
Facile apoptosis following DNA damage, as observed with conjugate 3 and oxaliplatin in Fig. 7, can occur in a p53-dependent or an independent manner. Both A2780 and 2780CP models have wild-type p53 function, but only in A2780 cells can p53 be induced and activated by cisplatin through post-translational modification events via, e.g., Ser-15 phosphorylation.22,31 To investigate the effect of conjugate 3 and oxaliplatin on p53 induction and Ser-15 phosphorylation, A2780 and 2780CP cells were exposed to these agents at concentrations equal to 2.5× and/or 5× the IC50 values for 6-, 12-, and 24 hours. At these time points, cells were collected, washed with cold PBS, and cell lysates prepared for subsequent analysis by Western blotting. Equal protein loading was confirmed by β-actin immunoblots. The protein bands in immunoblots were quantified by densitometry and the data presented in Fig. 8. Induction of p53 in A2780 cells increased 2-fold (relative to basal p53 levels) upon exposure to 3 and oxaliplatin and peaked after 12 hours of exposure (Fig. 8A and C). A corresponding increase in Ser-15-p53 over the time course was also evident. The exposure of cisplatin resistant 2780CP cells to these two Pt-complexes led to similar increases in p53 and Ser-15-p53, with total p53 increasing progressively to over 2-fold at 24 hours (Fig. 8B and D).
Fig. 8.

Induction of total and Ser-15 phosphorylated p53 protein following incubation of (A) A2780 and (B) 2780CP human ovarian cancer cells with conjugate 3 and oxaliplatin. Quantification of fold increase in total p53 protein in (C) A2780 and (D) 2780CP.
Induction of p53 and its functional activation are separate events. It was therefore necessary to assess p53 function by monitoring the transcriptional activation of p21 as a downstream target of p53. Both conjugate 3 and oxaliplaitin induced p21, which increased in a time-dependent manner to 3- to 5-fold above controls by 24 hours (Fig. 9). Such a finding supports the conclusion that the p53–p21 pathway is similarly upregulated by conjugate 3 and oxaliplatin and that this occurs in both the Pt-sensitive A2780 and Pt-resistant 2780CP cell lines.
Fig. 9.

Transactivation of p21 protein resulting from incubation of (A) A2780 and (B) 2780CP human ovarian cancer cells with oxaliplatin and oxaliTEX. Fold p21 protein increase in (C) A2780 and (D) 2780CP.
Discussion
While active in several cancer types and included in front line therapy by oncologists, platinum anticancer agents display acquired resistance in many cancers, which limits their clinical utility. The cause of this resistance is multifactorial and includes both pharmacologic mechanisms (e.g., decreased drug uptake, increased glutathione, and increased DNA adduct repair) and molecular mechanisms of resistance (e.g., loss of p53 function, increase in survivin, and increase in Bcl2).1,3,4 In this study, we have focused on two major cisplatin resistance mechanisms, reduced drug uptake and attenuated wild-type p53 function. Specifically, we sought to target these mechanisms via a novel platinum drug design. With this goal in mind, we designed conjugate 3 (oxaliTEX). As detailed below, this new agent not only circumvents these two mechanisms, it also demonstrates antiproliferative activity in a multifactorial-resistant in vitro tumor model (the Pt-resistant 2780CP ovarian cancer cell line) that makes the cells in question as responsive as the corresponding Pt-sensitive parental (A2780) cell line to this DNA-damaging agent.
The design of oxaliTEX was based on literature data and our earlier studies involving conjugate 2. The focus on targeting the tumor suppressor p53 derived from an appreciation that cisplatin has a greater curative rate in ovarian cancer when p53 is present in its wild-type state than in the mutant form.3,4 Paradoxically, about a half of advanced ovarian cancers that harbor wild-type p53 are resistant, primarily as a result of failure of upstream DNA damage signaling to stabilize and activate the p53. Furthermore, in these resistant cancers, the presence of wild-type p53 can lead to a “gain-of-resistance” phenotype, where the resistance is greater than those with mutant p53.3,4 Thus, loss of function of wild-type p53 is one of the most formidable molecular mechanism of resistance. However, we have reported that a panel of resistant ovarian tumor models respond to DACH-based platinum drug (e.g., DAP) through distinctly different DNA damage signaling processes that serve to restore p53 function and cellular apoptotic activity.20,32,33 Such a restoration of activity was considered likely to hold in the case of DACH-based oxaliplatin, and was specifically confirmed in the present study using the resistant 2780CP cell line.
Unfortunately, circumvention of resistance by DAP or oxaliplatin is not complete and at least a 2-fold level of resistance remains in the test 2780CP cell line, as seen with oxaliplatin in the present study. This residual resistance is ascribed to resistant cells displaying reduced accumulation of DAP or oxaliplatin.20,34 On the other hand, we have previously demonstrated in an independent study with conjugate 2, which has a cisplatin-like diammine-Pt configuration around the Pt(II) center, that Pt accumulation in 2780CP cells was not diminished, and that this translated into a concomitant 2-fold reduction in cross-resistance.19 Collectively, these previous investigations provided support for the hypothesis that conjugating texaphyrin with a DACH–Pt structure, such as that present in DAP and oxaliplatin, would serve to circumvent both pharmacologic and molecular mechanisms of resistance. Our present study, involving the new conjugate 3 (oxaliTEX) provides proof-of-principle support that this goal can be accomplished in vitro.
That activation of wild-type p53 is sufficient to overcome multifactorial molecular mechanisms of resistance is intriguing. Normally, wild-type p53 plays a critical role in drug-induced apoptosis. However, this activity becomes compromised when p53 is mutated, which leads to cisplatin/carboplatin resistance and, in the specific case of advanced ovarian cancer for which statistics are available, a 4- to 5-fold reduction in the 5-year survival rate compared to the wild-type p53 cancer sub-group.3,4 Advanced cancers other than ovarian cancer (e.g., NSCLC and mesothelioma) that retain wild-type p53 also demonstrate resistance to cisplatin,4 an observation ascribed to a number of mechanisms, including the critical post-translational modifications of p53 to release p53 from its inhibitory interaction with Mdm2.35,36 Based on reports from molecularly engineered mouse models,37 it appears that activation of wild-type p53 for apoptosis is a dominant DNA-damaging effect, and is sufficient to override the potential negative influence of other molecular defects that may co-exist in multifactorial resistant tumor cells. The 2780CP tumor cells used as a model for Pt resistance in ovarian cancer have been characterized as having a multifactorial cisplatin-resistant phenotype.20 The ability of oxaliTEX to restore platinum sensitivity in this model highlights the potential of this agent to enhance in due course both the initial response and the long-term cure rate in patients with cancers harboring wild-type p53.3,4
In addition to circumventing molecular mechanisms of resistance, we have demonstrated that oxaliTEX is capable of delivering the DACH–Pt payload at similar levels in both sensitive and resistant tumor cells. This was ascertained in the present study from examinations of both total intracellular Pt and DNA adduct levels. The similar delivery of Pt is likely due to the of the expanded porphyrin, texaphyrin, which has been shown to localize selectively within tumors.14–17 That the effective delivery of Pt is due to the conjugating texaphyrin carrier and not the DACH–Pt moiety can be inferred from the knowledge that uptake and DNA adduct data with oxaliTEX (conjugate 3) mirror those reported by us for cisTEX (conjugate 2), which has an alternate diamine-Pt coordination environment.19
It is further apparent from apoptotic investigations using Annexin V as a biomarker in the present study that the texaphyrin control, MGd (1), is devoid of antiproliferative effects at concentrations that were equivalent to those employed in the studies of oxaliTEX. This is taken as evidence that the cellular effects are predominantly due to the Pt moiety and that the texaphyrin is functioning primarily as a delivery vehicle. Support for this conclusion comes from noting the close similarities between oxaliTEX and oxaliplatin in the level of adducts formed, the phosphorylation of p53, the induction of the p53/p21 pathway, and the extent and temporal profile of apoptosis in both sensitive and resistant tumor cells. However, there is an apparent difference in drug potency between oxaliTEX and oxaliplatin in both the A2780 and 2780CP cell lines. This difference likely reflects the slow release of the DACH–Pt moiety from the conjugate, which serves to increase the time for Pt activation by ~50%.
Conclusion
The ability of oxaliTEX and oxaliplatin in the present study, and of DAP in an earlier study,20 to activate p53 stands in direct contrast to our previous finding that cisplatin fails to activate the p53/p21 pathway in 2780CP cells.20,31 The ability to upregulate p53 function in resistant cells augurs well for the eventual application of 3 in treating refractory cancers that harbor wild-type p53. The ability to enhance the uptake of a DACH–Pt payload into resistant 2780CP ovarian cancer cells distinguishes oxaliTEX (3) from other Pt delivery strategies, and identifies this conjugate as the first small molecule platinum complex capable of overcoming both pharmacologic and molecular mechanisms of resistance in vitro.
It is normally presumed that success of chemotherapy in resistant disease may require addressing each of the many mechanisms of resistance. However, it is evident from the present study that targeting only a few critical cellular impediments with a designer drug may be sufficient to achieve a markedly improved therapeutic response. The fact that texaphyrin is known to localize selectively to tumors, leads us to predict that suitably designed conjugates, such as 3, may prove effective in treating resistant cancers. Further tests of this paradigm, including in vivo studies to determine the ability of 3 to selectively localize to tumor, are currently in progress.
Acknowledgments
The work described herein was supported in part by the NIH (grant CA-68682 to J.L.S.; grants CA-127263 and CA-160687 to Z.H.S.), CPRIT (grant RP120393 to J.L.S.) and the Robert A. Welch Foundation (grant F-1018 to J.L.S.). Support under the Korean WCU program (grant R32-20080-000-10217-0) is acknowledged. Collaborative grant support from UT Austin TI-3D (Robert A. Welch Foundation Grant H-F-0032) and UT MD Anderson Cancer Center CCD (Grant 1003020-2100) is also acknowledged. JFA is supported by a postdoctoral fellowship (Grant PF-11-015-01-CDD) by the American Cancer Society. The authors thank Dr Guangan He for his expert guidance in the implementation of experimental protocols. Fruitful discussions with Dr Darren Magda are also acknowledged.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c2md20206a
Author contributions and conflicts of interest: all authors contributed equally and there are no conflicts of interest.
Contributor Information
Jonathan L. Sessler, Email: sessler@mail.utexas.edu.
Zahid H. Siddik, Email: zsiddik@mdanderson.org.
Notes and references
- 1.Siddik ZH. Oncogene. 2003;22:7265. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society. Cancer Facts and Figures 2012. ACS; Atlanta, GA: 2012. [Google Scholar]
- 3.Siddik ZH. Drug Resistance and the Tumor Suppressor p53: The Paradox of Wild-Type Genotype in Chemorefractory Cancers. In: Mehta K, Siddik ZH, editors. Drug Resistance in Cancer Cells. Springer Science; 2009. [Google Scholar]
- 4.Martinez-Rivera M, Siddik ZH. Biochem Pharmacol. 2012;83:1049. doi: 10.1016/j.bcp.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siddik ZH, Hagopian GS, Thai G, Tomisaki S, Toyomasu T, Khokhar AR. J Inorg Biochem. 1999;77:65. doi: 10.1016/s0162-0134(99)00144-0. [DOI] [PubMed] [Google Scholar]
- 6.Aronov O, Horowitz AT, Gabizon A, Fuertes MA, Perez JM, Gibson D. Bioconjugate Chem. 2004;15:814. doi: 10.1021/bc0499331. [DOI] [PubMed] [Google Scholar]
- 7.Aronov O, Horowitz AT, Gabizon A, Gibson D. Bioconjugate Chem. 2003;14:563. doi: 10.1021/bc025642l. [DOI] [PubMed] [Google Scholar]
- 8.Lottner C, Knuechel R, Bernhardt G, Brunner H. Cancer Lett. 2004;215:167. doi: 10.1016/j.canlet.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 9.Barnes KR, Kutikov A, Lippard SJ. Chem Biol. 2008;11:557. doi: 10.1016/j.chembiol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 10.Galanski M, Keppler BK. Anti-Cancer Agents Med Chem. 2007;7:55. doi: 10.2174/187152007779314017. [DOI] [PubMed] [Google Scholar]
- 11.van Zutphen S, Reedijk J. Coord Chem Rev. 2005;249:2845. [Google Scholar]
- 12.Mukhopadhyay S, Barnes CM, Haskel A, Short SM, Barnes KR, Lippard SJ. Bioconjugate Chem. 2008;19:39. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arambula JF, Preihs C, Borthwick D, Magda D, Sessler JL. Anti-Cancer Agents Med Chem. 2011;11:222. doi: 10.2174/187152011795255894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magda D, et al. Gadolium(III) Texaphyrin (Xcytrin®): A New Class of Redox Active Drug Leads. In: Sessler JL, Doctrow S, McMurry T, Lippard SJ, editors. Medicinal Inorganic Chemistry. Oxford University Press; 2005. (American Chemical Society Symposium Series 903). [Google Scholar]
- 16.Miller RA, Woodburn K, Fan Q, Renschler M, Sessler JL, Koutcher JA. Int J Radiat Oncol, Biol, Phys. 1999;45:981. doi: 10.1016/s0360-3016(99)00274-6. [DOI] [PubMed] [Google Scholar]
- 17.Mehta MP. Oncol News Int. 2006:12. [Google Scholar]
- 18.Arambula JF, Sessler JL, Fountain ME, Wei W, Magda D, Siddik ZH. Dalton Trans. 2009;48:10834. doi: 10.1039/b912089k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arambula JF, Sessler JL, Siddik ZH. Bioorg Med Chem Lett. 2011;21:1701. doi: 10.1016/j.bmcl.2011.01.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siddik ZH, Hagopian GS, Thai G, Tomisaki S, Toyomasu T, Khokhar AR. J Inorg Biochem. 1999;77:65. doi: 10.1016/s0162-0134(99)00144-0. [DOI] [PubMed] [Google Scholar]
- 21.Ozols RF, Behrens BC, Ostchega Y, Young RC. Cancer Treat Rev. 1985;12(suppl A):59. doi: 10.1016/0305-7372(85)90019-2. [DOI] [PubMed] [Google Scholar]
- 22.Mujoo K, Watanabe M, Nakamura J, Khokhar AR, Siddik ZH. J Cancer Res Clin Oncol. 2003;129:709. doi: 10.1007/s00432-003-0480-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siddik ZH, Jones M, Boxall FE, Harrap KR. Cancer Chemother Pharmacol. 1988;21:19. doi: 10.1007/BF00262732. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Lippard SJ. Nat Rev Drug Discovery. 2005;4:307. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 25.Heiger-Bernays WJ, Essigmann JM, Lippard SJ. Biochemistry. 1990;29:8461. doi: 10.1021/bi00488a037. [DOI] [PubMed] [Google Scholar]
- 26.Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, Hamilton TC, Chaney SG. Cancer Res. 1998;58:3579. [PubMed] [Google Scholar]
- 27.Johnson SW, Laub PB, Beesley JS, Ozols RF, Hamilton TC. Cancer Res. 1997;57:850. [PubMed] [Google Scholar]
- 28.Kelland LR. Crit Rev Oncol Hematol. 1993;15:191. doi: 10.1016/1040-8428(93)90042-3. [DOI] [PubMed] [Google Scholar]
- 29.Kelland LR. Nat Rev Cancer. 1997;7:573. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 30.Raymond E, Faivre S, Channey S, Woynarowski J, Cvitkovic E. Mol Cancer Ther. 2002;1:227. [PubMed] [Google Scholar]
- 31.Siddik ZH, Mims B, Lozano G, Thai G. Cancer Res. 1998;58:698. [PubMed] [Google Scholar]
- 32.Hagopian GS, Mills GB, Khokhar AR, Bast RC, Jr, Siddik ZH. Clin Cancer Res. 1999;5:655. [PubMed] [Google Scholar]
- 33.He G, Kuang J, Khokhar AR, Siddik ZH. Gynecol Oncol. 2011;122:402. doi: 10.1016/j.ygyno.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kido Y, Khokhar A, Al-Baker S, Siddik ZH. Cancer Res. 1993;53:4567. [PubMed] [Google Scholar]
- 35.Sionov RV, Haupt Y. Oncogene. 1999;18:6145. doi: 10.1038/sj.onc.1203130. [DOI] [PubMed] [Google Scholar]
- 36.Shieh SY, Ikeda M, Taya Y, Prives C. Cell. 1997;91:325. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 37.Kastan MB. Cell. 2007;128:837. doi: 10.1016/j.cell.2007.02.022. [DOI] [PubMed] [Google Scholar]