

Abstract
Angiotensin (ANG)-converting enzyme (ACE)2 in brain regions such as the paraventricular nucleus (PVN) controlling cardiovascular function may be involved in the regulation of sympathetic outflow in chronic heart failure (CHF). The purpose of this study was to determine if ACE2 plays a role in the central regulation of sympathetic outflow by regulating neuronal nitric oxide (NO) synthase (nNOS) in the PVN. We investigated ACE2 and nNOS expression within the PVN of rats with CHF. We then determined the effects of ACE2 gene transfer in the PVN on the contribution of NO-mediated sympathoinhibition in rats with CHF. The results showed that there were decreased expressions for ACE2, the ANG-(1–7) receptor, and nNOS within the PVN of rats with CHF. After the application of adenovirus vectors encoding ACE2 (AdACE2) into the PVN, the increased expression of ACE2 in the PVN was confirmed by Western blot analysis. AdACE2 transfection significantly increased nNOS protein levels (change of 50 ± 5%) in the PVN of CHF rats. In anesthetized rats, AdACE2 treatment attenuated the responses of renal sympathetic nerve activity (RSNA), mean arterial pressure, and heart rate to the NOS inhibitor N-monomethyl-l-arginine in rats with CHF (RSNA: 28 ± 3% vs. 16 ± 3%, P < 0.05) compared with CHF + AdEGFP group. Furthermore, neuronal NG-108 cells incubated with increasing doses of AdACE2 showed a dose-dependent increase in nNOS protein expression (60% at the highest dose). Taken together, our data highlight the importance of increased expression and subsequent interaction of ACE2 and nNOS within the PVN, leading to a reduction in sympathetic outflow in the CHF condition.
Keywords: paraventricular nucleus, nitric oxide and sympathetic nerve activity
impaired cardiovascular reflexes and increased neurohumoral drive are characteristic manifestations of chronic heart failure (CHF) (19, 21). An altered central mechanism(s) may be responsible for the elevated neurohumoral drive in CHF (22, 48). Studies have shown the increased neuronal activity in the paraventricular nucleus (PVN) of rats with CHF, as gauged by hexokinase activity (24), Fra-like activity (36), or direct recording (43) is related to the increased sympathetic activity. The CHF condition produces attenuated vasodilator responses to agonist known to act via a nitric oxide (NO)-mediated mechanism (8). Concomitantly, levels of endogenous endothelial NO synthase (NOS) protein and mRNA in peripheral tissues are reduced in the CHF state (34); however, there are few studies examining the NO system within the central nervous system in CHF, including studies from our laboratory (14, 23, 40, 41), which have indicated that NO-mediated mechanisms [neuronal NOS (nNOS) activity, mRNA, and protein] in the PVN are decreased in CHF.
Angiotensin (ANG) II may act via the PVN to influence the regulation of sympathetic activity by either affecting the circumventricular organs (CVOs) or by an intrinsic angiotensinergic system in the PVN (11). The forebrain areas contain both ANG-converting enzyme (ACE) as well as ANG II type 1 receptors in high concentrations. A new member of the ACE family has been identified and named ACE2 (35). ACE2 acts on ANG II to produce ANG-(1–7) (29). Although the exact function of ANG-(1–7) in the brain is unclear, there is considerable evidence for a role of ANG-(1–7) in the brain (5). ACE2 mRNA has been demonstrated in the medulla oblongata (26), and ACE2 activity has been measured in the mouse brain (9). Recently, Lazartigues and colleagues (7) have shown the presence of ACE2 protein and mRNA in various central brain regions, such as the PVN, subfornical organ, area postrema, nucleus tractus solitarii (NTS), and rostral ventrolateral medulla (RVLM), involved in centrally cardiovascular regulation (7).
ANG-(1–7) has a hypotensive effect in hypertensive but not normotensive rats (2). ANG-(1–7) acts centrally to increase the sensitivity of the cardiac baroreflex (1). When injected directly into the NTS and RVLM, it induced depressor and pressor responses, respectively (28). ANG-(1–7) has been reported to increase vasopressin (30) and NO release (27). The direct acute action of ANG-(1–7) in the RVLM appears to be sympathoexcitatory (46). Bilateral microinjections of the Mas receptor antagonist A-779 into the PVN decrease renal sympathetic nerve activity (RSNA) (31). However, there are other studies showing that the chronic effect of ANG-(1–7) maybe different from its acute direct action. Overexpression of ACE2 in the RVLM of spontaneously hypertensive rats caused a long-lasting hypotensive effect (38). The action of ACE2 on nNOS may be more long term and generally lead to sympathoinhibition via NO, so the long-term effect may be more important in a disease state, such as CHF or hypertension, where there is a decrease in the levels of nNOS. The details of these long-term effects on nNOS/NO remain to be elucidated.
Although there are a fair amount of data regarding ANG-(1–7) in medullary sites, its role in the forebrain, particularly in the PVN, is not as clear. The present study was designed to examine if there was a concomitant decrease in ACE2, the ANG-(1–7) receptor (Mas receptor), and nNOS within the PVN of rats with CHF. Second, it was designed to determine if upregulation of ACE2 within the PVN would restore the endogenous nNOS within the PVN of rats with CHF both in terms of the levels of protein as well as the functional response in RSNA, mean arterial pressure (MAP), and heart rate (HR) to inhibition of NOS. Finally, the relationship between ACE2 and nNOS was examined in a neuronal cell line (NG-108) that endogenously exhibits the presence of both ACE2 and nNOS.
METHODS
Induction of heart failure.
All procedures on animals in this study were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. Experiments were conducted according to the American Physiological Society's “Guiding Principles for Research Involving Animals and Human Beings” and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Male Sprague-Dawley rats weighing 220–240 g (Sasco Breeding Laboratories, Omaha, NE) were randomly assigned to either the sham-operated control (sham) group or the CHF group. CHF was induced by ligation of the left coronary artery as previously described (24). Left ventricular (LV) dysfunction was assessed using hemodynamic and anatomic criteria (17). To measure infarct size, the heart was dissected free of adjacent tissues, and the atria were removed. The right ventricle (RV) was opened with a lengthwise incision such that the heart was flattened with the LV lying in the middle and the RV on either side of it. The RV was removed, and the remaining LV was laid flat. A digital image of the LV was captured with a digital camera, and the infarct area and total LV area were quantified with SigmaScan Pro. Infarct size (in %) was found by dividing the size of the infarct area by the total size of the LV. Echocardiograms were performed after 6–7 wk of ligation surgery. LV end-diastolic pressure (LVEDP) was measured after the ExT period with a Micro-Tip catheter (Millar Instruments, Houston, TX) inserted into the LV via the right carotid artery at the time of the terminal experiment. Rats with elevated LVEDP (>15 mmHg), an infarct size of >30% of the total LV wall, significant reductions in dP/dtmax, and ejection fraction (<40%) were considered to be in CHF.
In vivo adenovirus injection.
After 8 wk of ligation surgery, adenoviral vectors carrying human (h)ACE2 (AdACE2; kindly supplied from the core facilities at the University of Iowa) or enhanced green fluorescent protein (AdEGFP; as a control vector) were delivered into the PVN by microinjection (1 × 108 plaque-forming units/ml, 100 nl) bilaterally. The following four groups of rats were used in this study: sham + AdEGFP, sham + AdACE2, CHF + AdEGFP, and CHF + AdACE2. Three days after injection, rats were used for functional experiments. Transfection efficiency (ACE2 expression) was determined using Western blot analysis.
RSNA, MAP, and HR recording.
Experiments were performed 8 wk after CHF surgery. Rats were anesthetized with urethane (0.75g/kg ip) and α-chloralose (70 mg/kg ip). The left femoral vein was cannulated with polyethylene tubing for the injection of supplemental anesthesia. The left femoral artery was cannulated and connected via a pressure transducer (Gould P23 1D) to a computer-based data recording and analyzing program (PowerLab) to record MAP and HR.
The left kidney was exposed through a retroperitoneal flank incision. A branch of the renal nerve was isolated from fat and connective tissue. The central end of the nerve was placed on thin bipolar platinum electrodes. The electrical signal was amplified with a Grass amplifier with high- and low-frequency cutoffs of 1,000 and 100 Hz, respectively. The rectified output from the amplifier was displayed using the PowerLab system to record and integrate the raw nerve discharge. Basal nerve activity was determined by efferent RSNA at the beginning of the experiment, and background noise was determined by nerve activity recorded at the end of the experiment [after the rat had been injected with hexamethonium (30 mg/kg iv)]. The nerve activity during the experiment was calculated by subtracting the background noise from the recorded value. The RSNA response to the injection of drugs into the PVN was expressed as the percent change from the basal value.
For the placement of microinjection cannulas into the PVN, the anesthetized rat was placed in a stereotaxic apparatus (David Kopf Instruments, Tujanga, CA). A longitudinal incision was made on the head, and the bregma was exposed. A small burr hole was made in the skull to allow access to the PVN. The coordinates for the PVN, determined using the Paxinos and Watson atlas, were 1.5 mm posterior to the bregma, 0.4 mm lateral to the midline, and 7.8 mm ventral to the dura. A thin needle (0.2-mm outer diameter) connected to a 0.5-μl microsyringe (Hamilton) was lowered into the PVN. An inhibitor of NOS, N-monomethyl-l-arginin (l-NMMA), was injected into the PVN in three doses (50, 100, and 200 pmol) in random order. Subsequent injections were made at least 20 min after prior injections to allow MAP, HR, and RSNA to return to basal levels. In a separate group of rats (n = 5), the vehicle control, 100 nl artificial cerebrospinal fluid aCSF, was microinjected into the PVN, and RSNA, MAP, and HR were monitored.
Immunohistochemistry.
Rats were anesthetized with pentobarbital (65 mg/kg) and perfused transcardially with 150 ml heparinized saline followed by 250 ml of 4% paraformaldehyde in 0.1 M sodium phosphate buffer. The brain was removed and postfixed at 4°C for 4 h in 4% paraformaldehyde solution and then placed in 20% sucrose. The brain was blocked in the coronal plane, and sections of 30 μm thickness were cut with a cryostat. Sections were incubated with 10% normal donkey serum in PBS for 1 h at room temperature and then incubated with primary antibody against ACE2 (anti-goat, 1:200) and the nNOS receptor (mouse monoclonal antibody, 1:200, Santa Cruz Biotechnology) overnight at 4°C. After being washed with PBS, sections were incubated with Cy3- and Cy2-conjugated donkey anti-mouse secondary antibody (1:400, Jackson ImmunoResearch) for 2 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (Molecular Probes). After being washed with PBS and dried, sections were coverslipped with fluoromounting-G (SouthernBiotech). The distribution of ACE2 and nNOS immunofluorescence within the PVN were viewed using an Olympus fluorescence microscope equipped with a digital camera (Qimaging). Openlab software 4.0.3. (Improvision) was used to identify the total intensity of Cy3 (red) and Cy2 (green) and further the colocalization of staining (yellow). Three alternate sections (1.8 ± 0.1 mm posterior to the bregma) representing the PVN were analyzed in this way, and the mean data were then calculated. No staining was seen when PBS was used instead of the primary antibodies or when the mixture of antibodies and the appropriate control antigen was added in the above-described procedures.
Micropunch of the PVN and isolation of protein for Western blot analysis measurements.
After the animal had been euthanized, the brain was removed and quickly frozen on dry ice. Six serial coronal sections (100 μm) were cut using a cryostat, and, following the Palkovits technique, the PVN was bilaterally punched using a diethylpyrocarbonate-treated blunt 18-gauge needle attached to a syringe, such that there were 12 total punches/brain. The punching samples were placed in 100 ml protein extraction buffer (10 mM Tris, 1 mM EDTA, 1% SDS, 0.1% Triton X-100, and 1 mM PMSF), sonicated, and incubated for 30 min at 37°C to extract the protein.
Western blot analysis measurements of ACE2, Mas receptor, and nNOS protein.
The total protein concentration from the extracted protein described above was measured using a BCA Assay Kit (Pierce). Samples were adjusted to contain the same concentration of total protein, and equal volumes of 2× (4%) SDS sample buffer were then added. Samples were boiled for 3 min and then loaded onto a 7.5% SDS-PAGE gel (40 mg/20 ml per well). Gels were subjected to electrophoresis at 40 mA/gel for 60 min. Fractionated proteins on the gel were electrophoretically transferred to a polyvinylidene difluoride membrane (Millipore) at 300 mA for 90 min. The membrane was probed with primary antibodies to hACE2 (R&D Systems) and rat ACE2, nNOS, GAPDH (Santa Cruz Biotechnology), and the Mas receptor (Thermo Scientific) at 1:500 dilution, washed with Tris-buffered saline-Tween, and then probed with secondary antibodies (peroxidase-conjugated goat anti-goat or rabbit IgG, Pierce). An enhanced chemiluminescence substrate (Pierce) was applied to the membrane for 5 min followed by a 30-s exposure within an Epi Chemi II Darkroom (UVP BioImaging) for visualization using the Worklab digital imaging system (8-bit resolution). Kodak MI software was used to highlight the bands and quantify the signal. Expression of ACE2, the Mas receptor, or nNOS was calculated as the ratio of intensity of the ACE2, Mas receptor, or nNOS band, respectively, relative to the intensity of the GAPDH band. Each Western blot was performed with samples from all groups.
Cell culture and adenoviral transfection.
NG108-15 (neuroblastoma X glioma) hybrid cells were grown in DMEM supplemented with 10% FBS, penicillin G, and streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. Cells were seeded in six-well plates and grown until 60–70% confluent before treatment with the indicated concentrations of AdACE2 or AdEGFP in a dose-dependent manner (2.5 × 106, 1.25 × 105, and 6.25 × 105 plaque-forming units/ml, 24 h) for 24 h. Cells were washed twice with ice-cold PBS, homogenized with sonication in lysis buffer (10 mM Tris, 1 mM EDTA, 1% SDS, 0.1% Triton X-100, and 1 mM PMSF). The lysate was incubated at 37°C for 30 min. The protein content of the lysate was estimated using the BCA method with BSA as the standard (Pierce). NG-108 cell protein lysates (30–40 μg) were processed to measure nNOS, ACE2, and Mas receptor protein by Western blot analysis. The distribution of ACE2 and nNOS in the cultured cells was observed by immunohistochemisty.
Data analysis.
Data are presented as means ± SE. Data were subjected to two-way ANOVA followed by a comparison for individual group differences using the Newman-Keuls test. Statistical significance was indicated by P values of <0.05.
RESULTS
General data.
Table 1 shows the salient characteristics of the sham and CHF rats used in the present study. Heart weight and body weight were significantly higher in CHF rats compared with sham rats (P < 0.05). This indicates that there was myocardial hypertrophy and retention of water in the rats with CHF. The CHF group displayed an average myocardial infarct size of >30% over the LV. Sham rats had no observable damage to the myocardium. LVEDP was significantly elevated in CHF rats compared with sham rats (P < 0.05).
Table 1.
Characteristics of sham and CHF rats tranfected with AdEGFP or AdACE2
| Sham + AdEGFP | CHF + AdEGFP | Sham + AdACE2 | CHF + AdACE2 | |
|---|---|---|---|---|
| No. of rats | 6 | 9 | 6 | 7 |
| Body weight, g | 392 ± 24 | 409 ± 15 | 386 ± 17 | 406 ± 25 |
| Heart weight, g | 1.2 ± 0.1 | 2.0 ± 0.2* | 1.2 ± 0.1 | 1.7 ± 0.1* |
| Infarct size, % of the epicardial LV | 0 | 37 ± 4* | 0 | 36 ± 4* |
| LV end-diastolic pressure, mmHg | 2.1 ± 1.0 | 27.8 ± 4.4* | 1.7 ± 0.8 | 25.8 ± 3.8* |
| Basal mean arterial pressure, mmHg | 91 ± 5 | 93 ± 7 | 93 ± 3 | 88 ± 3 |
| Basal heart rate, beats/min | 354 ± 11 | 348 ± 15 | 368 ± 15 | 363 ± 11 |
| Basal integrated renal sympathetic nerve activity, μV•s | 3.4 ± 0.6 | 4.8 ± 0.5* | 3.5 ± 0.5 | 4.3 ± 0.6 |
Values are means ± SE. Sham rats, sham-operated rats; CHF rats, rats with congestive heart failure; AdEGFP, adenovirus containing enhanced green fluorescent protein; AdACE2, adenovirus with angiotensin-converting enzyme 2; LV, left ventricular.
P < 0.05 vs. sham groups.
Before the microinjection of l-NMMA into the PVN, MAP and HR of the sham + AdEGFP group was not significantly different from that found in the CHF + AdEGFP group. The basal RSNA in CHF + AdEGFP rats was significantly higher than that in sham + AdEGFP rats (P < 0.05). After adenoviral transfection, there were no significant changes in the basal MAP, HR, and RSNA between the AdACE2 and AdEGFP-treated groups in both sham and CHF rats.
ACE2, Mas receptor, and nNOS expression in the PVN.
We investigated the distribution of ACE2 and nNOS in the PVN (sham and CHF rats, n = 4 rats/group). As shown in the representative examples shown in Fig. 1, A and B, at the −1.8-mm level of the bregma, ACE2 immunostaining was detected in the PVN area as well as in the outside of the PVN. Most nNOS immunostaining was detected in the PVN. ACE2 and nNOS immunoreactivity were partially colocalized in the PVN. ACE2 expression has been showed to be located in the neuronal cytoplasm and endothelial cells in the brain (7, 16). Immunofluorescence for ACE2 and nNOS was significantly decreased in the PVN from rats with CHF compared with sham rats. Consistent with these results, Western blot analysis showed that ACE2 (0.16 ± 0.04 vs. 0.30 ± 0.06, P < 0.05; Fig. 2B) and nNOS (0.5 ± 0.1 vs. 1.1 ± 0.1, P < 0.05; Fig. 3) protein levels were significantly lower in CHF rats compared with sham rats (n = 4 rats/group). Mas receptor protein levels were significantly decreased in CHF rats compared with sham rats (Fig. 2B).
Fig. 1.

A: immunofluorescent photomicrographs from sections of the paraventricular nucleus (PVN; −1.8-mm level of the bregma) region stained for angiotensin (ANG)-converting enzyme (ACE)2 and neuronal nitric oxide (NO) synthase (nNOS). Immunofluorescent signals of ACE2 (red) and nNOS (green) staining decreased within the PVN in rats with congestive heart failure (CHF) compared with sham-operated (sham) rats. Blue spots show nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI). Bar = 100 μm. B: quantification of the immunostaining intensity of ACE2 and nNOS. *P < 0.05 vs. sham rats.
Fig. 2.

A: representative Western blot analysis results showing human (h)ACE2 protein expression in the PVN of AdACE2-transfected rats but very low levels in AdEGFP-tranfected rats. B: representative Western blot analysis results showing ACE2 and Mas receptor protein expression in punched PVN samples. *P < 0.05 vs. sham + AdEGFP rats; #P < 0.05 vs. CHF + AdEGFP rats.
Fig. 3.

Representative Western blot analyis results showing nNOS protein expression in punched PVN samples. *P < 0.05 vs. sham + AdEGFP rats; #P < 0.05 vs. CHF + AdEGFP rats.
Three days after adenoviral microinjection, we evaluated hACE2 protein expression in the PVN by Western blot analysis. The results showed hACE2 protein expression in the PVN of AdACE2 rats but very low levels in AdEGFP rats (Fig. 2A). We then evaluated the efficacy of AdACE2 gene transfer in the PVN by comparing rat ACE2 protein levels of the PVN infected with AdACE2 or with AdEGFP in sham and CHF rats (n = 4 rats/group). There was a significant increase in the intensity of the protein bands of rat ACE2 (Fig. 2B) in the AdACE2-infected PVN compared with the AdEGFP-infected PVN in both sham (0.30 ± 0.06 vs. 0.50 ± 0. P < 0.05) and CHF (0.16 ± 0.04 vs. 0.41 ± 0.09, P < 0.05) groups. Overexpression of ACE2 also caused an increase in Mas receptor expression in CHF + AdACE2 rats (Fig. 2B). An example of the differences in levels of ACE2 and Mas receptor protein of the AdACE2-infected versus AdEGFP-infected PVN is shown in Fig. 2B.
To determine if the overexpression of ACE2 regulates nNOS expression in vivo, we measured nNOS protein levels in the PVN after adenoviral transfection. Sample gels showing nNOS and GADPH protein in the four experimental groups are shown in Fig. 3. The results showed that in the CHF + AdACE2 group, relative nNOS expression was significantly greater than in the CHF + AdEGFP (1.0 ± 0.1 vs. 0.5 ± 0.1, P < 0.05) group and not different from the sham + AdACE2 (1.0 ± 0.2 vs. 1.2 ± 0.2, P > 0.05) or sham + AdEGFP (1.0 ± 0.2 vs. 1.1 ± 0.1, P > 0.05) groups.
NO-mediated sympathetic responses.
To assess the effects of ACE2 overexpression on NO-mediated sympathoinhibition, RSNA, MAP, and HR were recorded after adenoviral transfection. Typical tracings of the RSNA, MAP, and HR responses to the administration of l-NMMA (200 pmol) into the PVN in the four groups of rats (sham + AdEGFP, sham + AdACE2, CHF + AdEGFP, and CHF + AdACE2, n = 5 rats/group) are shown in Fig. 4. Typically, microinjection of l-NMMA produces responses in RSNA, MAP, and HR over a 10-min period, as previously described (44). RSNA, MAP, and HR responses to microinjection of l-NMMA (200 pmol) into the PVN were significantly blunted (RSNA: 16 ± 3% vs. 32 ± 2%, MAP: 6 ± 1 vs. 15 ± 3 mmHg, and HR: 20 ± 7 vs. 9 ± 2 beats/min, P < 0.05) in CHF + AdEGFP rats compared with sham + AdEGFP rats. AdACE2 significantly ameliorated the blunted RSNA, MAP, and HR responses to l-NMMA in rats with CHF (RSNA: 28 ± 3% vs. 16 ± 3%, MAP: 9 ± 1 vs. 6 ± 1 mmHg, and HR: 20 ± 5 vs. 9 ± 2 beats/min, P < 0.05) compared with CHF + AdEGFP rats. There were no significant differences in the responses to l-NMMA between the sham + AdACE2 and sham + AdEGFP groups (Figs. 4 and 5). There were no significant changes in RSNA, MAP, and HR in the sham + AdACE2 group after viral transfer compared with the sham + AdEGFP group. AdEGFP demonstrated no significant effects on the responses of RSNA, MAP, and HR to l-NMMA in both sham and CHF groups (Fig. 5). The vehicle control (100 nl artificial cerebrospinal fluid) microinjected into the PVN had no effects on RSNA, MAP, and HR.
Fig. 4.
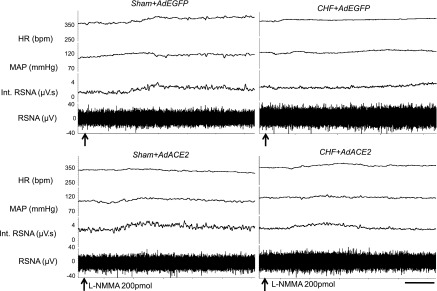
Segments of original recordings demonstrating the representative responses of heart rate [HR; in beats/min (bpm)], mean arterial pressure (MAP; in mmHg), integrated (Int) renal sympathetic nerve activity (RSNA; in μV·s), and RSNA (in μV) to microinjections of N-monomethyl-l-NMMA (l-NMMA; arrows) into the PVN. Bar = 2 min.
Fig. 5.

Mean RSNA, MAP, and HR responses to l-NMMA injected into the PVN. *P < 0.05 vs. sham + AdEGFP rats at the same dose of l-NMMA; #P < 0.05 vs. CHF + AdEGFP rats within the same dose of l-NMMA.
nNOS, ACE2, and Mas receptor expression in the NG-108 cell line.
To determine if overexpression of ACE2 regulates NOS expression in vitro, neuronal NG-108 cells were treated with AdACE2 in the absence or presence of the Mas receptor blocker A-779. Changes in nNOS, ACE2, and Mas receptor protein expression were analyzed by Western blot analysis after 24 h of viral transfection (Fig. 6). nNOS protein expression was upregulated significantly even at the lower concentrations of AdACE2 compared with the control (without AdACE2). The highest dose showed an ∼60% increase in nNOS protein expression compared with the control dose. To ascertain whether this increase of nNOS is mediated through the Mas receptor, NG-108 cells were incubated with A-779, a specific Mas receptor blocker. Treatment with A-779 significantly abrogated the increase in nNOS expression by AdACE2. The results shown in Fig. 6 also demonstrate that the Mas receptor was effectively blocked in the experiments and that AdACE2 in the presence of A-779 effectively increased ACE2 expression.
Fig. 6.

Protein expression of nNOS (A) and ACE2 and Mas receptors (B) measured by Western blot analysis in AdACE2-treated NG-108 cells with or without A-779. *P < 0.05 vs. control (without AdACE2).
Using immunofluorescent staining, ACE2 and nNOS interactions and their subcellular localization were studied in NG-108 cells (Fig. 7). The intensity of ACE2 staining and nNOS staining was also increased with AdACE2 treatment. AdACE2-treated cells showed more ACE2 and nNOS, both colocalizing in the cytoplasm, compared with the control (AdEGFP-treated cells).
Fig. 7.

Immunofluorescent photomicrographs of AdACE2/AdEGFP-treated NG-108 cells stained for ACE2 and nNOS. Immunofluorescent signals of ACE2 (red) and nNOS (green) staining increased with AdACE2 treatment. Blue spots show nuclei stained with DAPI. Bar = 50 μm.
The ACE2-ANG-(1–7)-Mas mechanism of NO-mediated sympathetic responses.
To further test the ACE2-ANG-(1–7)-Mas mechanism stimulating nNOS activity in vivo, the effects of the Mas receptor blocker A-779 on NO-mediated RSNA, MAP, and HR responses in a group of normal rats (n = 4) were examined. The result showed that A-779 reduced l-NMMA responses in normal rats (Fig. 8). This suggests that the effect of ACE2 on NO activity is through Mas receptors. This is also consistent with other results showing that NO is known to be released after the stimulation of Mas receptors in the brain and the periphery (3, 25).
Fig. 8.

Mean RSNA (A), MAP [blood pressure (BP); B], and HR (C) responses to l-NMMA injected into the PVN after the Mas receptor blocker A-779. *P < 0.05 vs. normal rats without A-779.
DISCUSSION
In the present study, we showed that there was a concomitant decrease in ACE2 and nNOS within the PVN of rats with CHF. Moreover, AdACE2 transfection significantly increased nNOS protein levels in the PVN in CHF rats. AdACE2 transfection also significantly improved attenuated RSNA, MAP, and HR responses to administrations of the NOS inhibitor l-NMMA within the PVN in rats with CHF compared with CHF rats transfected with control AdEGFP. Furthermore, nNOS protein expression increased after increasing amounts of AdACE2 transfection in neuronal NG-108 cells in a dose-dependent manner. Taken together, these data support our hypothesis that the upregulation of ACE2 by gene transfer significantly upregulates nNOS production. The effect of ACE2 on the NO-mediated mechanism in the PVN may play an important contributing role in the altered balance and tone of sympathetic outflow in the CHF condition.
Previous studies (24, 36, 43) have demonstrated that CHF is associated with increased neuronal activity in the PVN. Moreover, we (23, 41) have demonstrated that nNOS gene expression and protein as well as the number of nNOS-positive cells in the PVN are significantly reduced in rats with CHF. The inhibitory system (NO) in the PVN is diminished in rats with CHF. The initial causes for the decrease in nNOS (inhibitory system) remain to be examined. Stimulation of the PVN with ANG II has been shown to increase RSNA. An alternate pathway for ANG II is via the ACE2 pathway to produce ANG-(1–7), which, via the Mas receptor, could diminish the effects of ANG II type 1 receptor activation. There is evidence in endothelial cells showing that activation of Mas receptors increases endothelial NOS (27). In a human endothelial cell line, overexpression of ACE2 prevented ANG II suppression of insulin-stimulated NO generation (45). However, it is not clear how this pathway is altered in rats with CHF. The immunoreactivity of ACE2 indicates the presence of ACE2 in the PVN and colocalization with nNOS in neurons. Our data show that there is a concomitant decrease in both nNOS and ACE2 within the PVN of rats with CHF. Since there was a concomitant decrease in nNOS and ACE2, there may be some causal link between these two enzymes. Furthermore, upregulating levels of ACE2 via viral transfection restored levels of nNOS in the PVN. The functional responses of RSNA to l-NMMA suggest that increasing ACE2 also improves the responses to endogenous NO responses. Taken together, these data suggest that ACE2 upregulation restores the decreased levels of nNOS within the PVN and improves the endogenous NO-mediated inhibition in rats with CHF.
ACE2 is a carboxypeptidase that cleaves ANG II to ANG-(1–7). It appears to negatively regulate the renin-angiotensin system. In the heart, ACE2 deficiency results in a severe impairment of cardiac contractility and the upregulation of hypoxia-induced genes (13). ACE2 has also been found to be upregulated in failing hearts (12). In the kidney, ACE2 protein levels are significantly decreased in hypertensive rats, suggesting a negative regulatory role of ACE2 in blood pressure control (4). Relatively few studies have addressed ACE2 expression in the brain. In the CHF condition, the central renin-angiotensin system is activated (42, 49). Our data show that there were decreased levels of ACE2 and Mas receptors in the PVN of rats with CHF, supporting the idea that an imbalance of ACE2 and ACE is involved in the activation of the central renin-angiotensin system during CHF.
ACE2 has been suggested to participate in the central regulation of blood pressure and altered sympathetic nerve activity in hypertension. Overexpression of ACE2 in the brain reduces hypertension by improving the arterial baroreflex and autonomic function in hypertensive animals (37, 38). Injection of the ACE2 inhibitor MLN-4760 into the NTS reduces the baroreceptor reflex sensitivity for HR control in rats (6). Our data show that the basal RSNA in CHF rats is significantly higher than that in sham rats. After adenoviral transfection, the improved responses of RSNA to l-NMMA microinjections indicate that ACE2 is involved in NO-mediated sympathoinhibition in CHF. The lack of changes in the basal levels of RSNA in CHF rats may be due to the fact that adenoviral transfection of the PVN may not be complete, resulting in an elevated basal tone. However, the origin of ACE2 in the PVN and the process by which nNOS is generated in neurons are currently unknown.
Overexpression of ACE2 exerts protective effects in local tissues, including in the brain (13). It was first shown that lentivirus-mediated ACE2 overexpression in the RVLM causes a long-term decrease in blood pressure in the spontaneously hypertensive rat (38). Subsequently, Feng et al. (10) also showed that ACE2 overexpression in the subfornical organ prevented ANG II-mediated pressor and drinking responses. These are the first pieces of evidence for the beneficial/therapeutic effects of ACE2 overexpression in the central nervous system. The present in vivo study corroborates and also provides evidence for the protective role of ACE2 on neural overexcitation by improving nNOS-mediated inhibitory mechanisms within the PVN in the CHF condition.
So how does the upregulation of ACE2 within the PVN restore levels of nNOS in the PVN of rats with CHF? To examine the direct effect of ACE2 on nNOS, we determined that in an isolated cell culture system, incubation of AdACE2 with NG-108 cells caused a dose-dependent increase in the expression of nNOS protein. Furthermore, coincubation with A-779 prevented upregulation of the AdACE2-induced increase in nNOS protein. These data are consistent with the idea that increased ACE2 expression in rats with CHF may have caused the increase in nNOS protein expression within the PVN of rats with CHF.
An intracellular signaling pathway has been proposed in which overexpression of ACE2 activates the insulin/Akt-NOS pathway (45). Gene transfer of ACE2 regulated ANG II-mediated impairment of insulin signaling involves the Akt-NOS phosphorylation pathway. ANG II regulates insulin signaling via a pathway involving impairment of phosphotidylinositol 3-kinase-dependent activation of Akt-NOS phosphorylation and NO generation. It has been indicated that in the CHF state, the plasma ANG II level is increased (18, 47). Although ANG II does not cross the blood-brain barrier, circulating levels of ANG II may affect the central nervous system, including the PVN, through some of the CVOs. The neurons in CVOs have projections to the PVN. We think that through this pathway, the circulating ANG II could effect local ANG II levels in the central nervous system (20). The inhibitory effects of ANG II on nNOS expression can be reversed by ACE2 gene transfer. This may explain why gene transfer of ACE2 increases nNOS in CHF rats more obviously than in sham rats.
On the other hand, ANG II induces the expression of NADPH oxidase subunits p22phox and p47phox, contributing to the enhancement of oxidative stress and reduction of NO bioavailability (39). Gene transfer of ACE2 strikingly suppressed the expression of NADPH oxidase subunits and increased NO bioavailability (45). Recently, Gwathmey et al. (15) showed a marked sensitivity of the intracellular NO response to ANG-(1–7), which implies that the renal nuclei expression of Mas receptors is functionally linked to NO formation. However, a direct effect of ACE2 activation on NOS expression has not been studied until our in vitro data in NG-108 cell cultures in the present study.
A limitation of the present study is that no data are provided to demonstrate the effects of sustained increases of ACE2 expression in the PVN on the progression of neurohumoral dysfunction that occurs during CHF development. Feng et al. (10) indicated in their study that ACE2 activity in the whole hypothalamus at different time points showed a significant increase as early as 3 days after AdACE2 infection and achieved a maximum 7 days after infection. ACE2 activity had returned to baseline at 14 and 28 days after AdACE2/AdEGFP infection. The time course and pattern of expression obtained with AdACE2/AdEGFP were similar to previous data using similar constructs (32, 33). According to these studies, we think that the vector used in this study does not produce a reliable long-term (>10 days) increase of ACE2 expression and that the effects of ACE2 expression shown in the present study cannot be extrapolated to indicate an impact on CHF progression beyond the timeframes studied here.
Figure 9 shows a working hypothesis of how ACE2-mediated changes in the PVN regulate sympathoexcitation through a NO-mediated mechanism. CHF causes a decrease in nNOS/NO-mediated mechanisms (14, 40) by a decrease in ACE2-ANG-(1–7). Gene transfer of AdACE2 restores Mas receptors in the PVN, which, in turn, increase nNOS, which leads to an improvement in NO-mediated sympathoinhibition in rats with CHF.
Fig. 9.

Schematic diagram illustrating ACE2-mediated changes in the PVN regulated by sympathoexcitation through NO: CHF causes a decrease in nNOS/NO-mediated mechanisms [by a decrease in ACE2-ANG-(1–7); filled arrows show the effect of CHF]. AdACE2 restores NO-mediated sympathoinhibition in rats with CHF (open arrows show the effect of AdACE2). AT1R, ANG II type 1 receptor; MasR, Mas receptor; RVLM, rostral ventrolateral medulla; IML, intermediolateral nucleus.
In conclusion, the results of the present study demonstrate a concomitant decrease in ACE2 and nNOS within the PVN of rats with CHF. Furthermore, the results indicate that overexpression of ACE2 normalizes the lower expression of nNOS in the PVN and prevents the sympathoexcitatory state commonly observed in rats with CHF. Overexpression of ACE2 also attenuated RSNA responses to l-NMMA microinjected into the PVN in rats with CHF. Taken together, these data indicate that the effects of ACE2 on the NO-mediated mechanism within the PVN may play a crucial and pivotally important role in the altered sympathoexcitatory/inhibitory balance, leading to enhance sympathetic tone and activation in the CHF condition.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
The authors thank Dr. Lazartigues for access to AdACE2 from the core facilities at the University of Iowa.
REFERENCES
- 1.Averill DB, Diz DI. Angiotensin peptides and baroreflex control of sympathetic outflow: pathways and mechanisms of the medulla oblongata. Brain Res Bull 2000: 119–128, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Benter IF, Ferrario CM, Morris M, Diz DI. Antihypertensive actions of angiotensin-(1–7) in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 269: H313–H319, 1995 [DOI] [PubMed] [Google Scholar]
- 3.Calka J, Block CH. Angiotensin-(1–7) and nitric oxide synthase in the hypothalamo-neurohypophysial system. Brain Res Bull 30: 677–685, 1993 [DOI] [PubMed] [Google Scholar]
- 4.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveria-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417: 822–828, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Diz DI, Arnold AC, Nautiyal M, Isa K, Shaltout HA, Tallant EA. Angiotensin peptides and central autonomic regulation. Curr Opin Pharmacol 11: 131–137, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diz DI, Garcia-Espinosa MA, Gegick S, Tommasi EN, Ferrario CM, Ann Tallant E, Chappell MC, Gallagher KP. Injections of angiotensin-converting enzyme 2 inhibitor MLN4760 into nucleus tractus solitarii reduce baroreceptor reflex sensitivity for heart rate control in rats. Exp Physiol 93: 694–700, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 292: R373–R381, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drexler H, Lu W. Endothelial dysfunction of hindquarter resistance vessels in experimental heart failure. Am J Physiol Heart Circ Physiol 262: H1640–H1645, 1992 [DOI] [PubMed] [Google Scholar]
- 9.Elased KM, Cunha TS, Gurley SB, Coffman TM, Morris M. New mass spectrometric assay for angiotensin-converting enzyme 2 activity. Hypertension 47: 1010–1017, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Feng Y, Yue X, Xia H, Bindom SM, Hickman PJ, Filipeanu CM, Wu G, Lazartigues E. Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res 102: 729–736, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferguson AV, Washburn DLS. Angiotensin II: a peptidergic neurotransmitter in central autonomic pathways. Prog Neurobiol 54: 169–192, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Goulter AB, Goddard MJ, Allen JC, Clark KL. ACE2 gene expression is up-regulated in the human failing heart. BMC Med 19: 19, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grobe JL, Der Sarkissian S, Stewart JM, Meszaros JG, Raizada MK, Katovich MJ. ACE2 overexpression inhibits hypoxia-induced collagen production by cardiac fibroblasts. Clin Sci (Lond) 113: 357–364, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Guggilam A, Patel KP, Haque M, Ebenezer PJ, Kapusta DR, Francis J. Cytokine blockade attenuates sympathoexcitation in heart failure: cross-talk between nNOS, AT-1R and cytokines in the hypothalamic paraventricular nucleus. Eur J Heart Fail 10: 625–634, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gwathmey TM, westwood BM, Pirro NT, Tang L, Rose JC, Diz DI, Chappell MC. Nuclear angiotensin-(1–7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Renal Physiol 299: F983–F989, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol 108: 923–932, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleiber AC, Zheng H, Schultz HD, Peuler JD, Patel KP. Exercise training normalizes enhanced glutamate-mediated sympathetic activation from the PVN in heart failure. Am J Physiol Regul Integr Comp Physiol 294: R1863–R1872, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu JL, Irvine S, Reid IA, Patel KP, Zucker IH. Chronic exercise reduces sympathetic nerve activity in rabbits with pacing-induced heart failure–a role for angiotensin II. Circulation 102: 1854–1862, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Mancia G. Sympathetic activation in congestive heart failure. Europ Heart J 11, Suppl A: 3–11, 1990 [DOI] [PubMed] [Google Scholar]
- 20.Osborn JW, Fink GD, Sved AF, Toney GM, Raizada MK. Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9: 228–235, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Packer M. Neurohormonal interactions and adaptations in congestive heart failure. Circulation 77: 721–730, 1988 [DOI] [PubMed] [Google Scholar]
- 22.Patel KP. Neural regulation in experimental heart failure. Baillieres Clin Neurol 6: 283–296, 1997 [PubMed] [Google Scholar]
- 23.Patel KP, Zhang K, Zucker IH, Krukoff TL. Decreased gene expression of neuronal nitric oxide synthase in hypothalamus and brainstem of rats in heart failure. Brain Res 734: 109–115, 1996 [PubMed] [Google Scholar]
- 24.Patel KP, Zhang PL, Krukoff TL. Alterations in brain hexokinase activity associated with heart failure in rats. Am J Physiol Regul Integr Comp Physiol 265: R923–R928, 1993 [DOI] [PubMed] [Google Scholar]
- 25.Porsti I, Bara AT, BUsse R, Hecker M. Release of nitric oxide by angiotensin-(1–7) from porcine coronary endothelium: implications for a novel angiotensin receptor. Br J Pharmacol 111: 652–654, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakima A, Averill DB, Gallagher PE, Kasper SO, Tommasi EN, Ferrario CM, Diz DI. Impaired heart rate baroreflex in older rats: role of endogenous angiotensin-(1–7) at the nucleus tractus solitarii. Hypertension 46: 333–340, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Sampaio WO, dos Santos Souza RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1–7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 49: 185–192, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Santos RA, Campagnole-Santos MJ, Andrade SP. Angiotensin-(1–7): an update. Regul Pept 91: 45–62, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Santos RA, Ferreira AJ, Simoes E. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1–7)-Mas axis. Exp Physiol 93: 519–527, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Shiavone MT, Santos RA, Brosnihan KB, Khosla MC, Ferrario CM. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1–7) heptapeptide. Proc Natl Acad Sci USA 85: 4095–4098, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silva AQ, Santos RA, Fontes MA. Blockade of endogenous angiotensin-(1–7) in the hypothalamic paraventricular nucleus reduces renal sympathetic tone. Hypertension 46: 341–348, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Sinnayah P, Lazartigues E, Sakai K, Sharma RV, Sigmund CD, Davisson RL. Genetic ablation of angiotensinogen in the subfornical organ of the brain prevents the central angiotensinergic pressor response. Circ Res 99: 1125–1131, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Sinnayah P, Lindley TE, Staber PD, Cassell MD, Davisson BL, Davisson RL. Selective gene transfer to key cardiovascular regions of the brain: comparison of two viral vector systems. Hypertension 39: 603–608, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Smith CJ, Sun D, Hoegler C, Roth BS, Zhang X, Zhao G, Xu XB, Kobari Y, Pritchard K, Jr, Sessa WC, Hintze TH. Reduced gene expression of vascular endothelial NO synthase and cyclooxygenase-1 in heart failure. Circ Res 78: 58–64, 1996 [DOI] [PubMed] [Google Scholar]
- 35.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275: 33238–33243, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Vahid-Ansari F, Leenen FH. Pattern of neuronal activation in rats with CHF after myocardial infarction. Am J Physiol Heart Circ Physiol 275: H2140–H2146, 1998 [DOI] [PubMed] [Google Scholar]
- 37.Xia H, Feng Y, Obr TD, Hickman PJ, Lazartigues E. Angiotensin II type 1 receptor-mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension 53: 210–216, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension 49: 926–931, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Yoshida K, Kobayashi N, Ohno T, Fukushima H, Matsuoka H. Cardioprotective effect of angiotensin II type 1 receptor antagonist associated with bradykinin-endothelial nitric oxide synthase and oxidative stress in Dahl salt-sensitive hypertensive rats. J Hypertens 25: 1633–1642, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Zhang K, Li YF, Patel KP. Blunted nitric oxide-mediated inhibition of renal nerve discharge within PVN of rats with heart failure. Am J Physiol Heart Circ Physiol 281: H995–H1004, 2001 [DOI] [PubMed] [Google Scholar]
- 41.Zhang K, Zucker IH, Patel KP. Altered number of diaphorase (NOS) positive neurons in the hypothalamus of rats with heart failure. Brain Res 786: 219–225, 1998 [DOI] [PubMed] [Google Scholar]
- 42.Zhang WG, Huang BS, Leenen FHH. Brain renin-angiotensin system and sympathetic hyperactivity in rats after myocardial infarction. Am J Physiol Heart Circ Physiol 276: H1608–H1615, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Zhang ZH, Francis J, Weiss RM, Felder RB. The renin-angiotensin-aldosterone system excites hypothalamic paraventricular nucleus neurons in heart failure. Am J Physiol Heart Circ Physiol 283: H423–H433, 2002 [DOI] [PubMed] [Google Scholar]
- 44.Zheng H, Mayhan WG, Bidasee KR, Patel KP. Blunted nitric oxide-mediated inhibition of sympathetic nerve activity within the paraventricular nucleus in diabetic rats. Am J Physiol Regul Integr Comp Physiol 290: R992–R1002, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Zhong JC, Yu XY, Lin QX, Li XH, Xiao DZ, Lin SG. Enhanced angiotensin converting enzyme 2 regulates the insulin/Akt signalling pathway by blockade of macrophage migration inhibitory factor expression. Br J Pharmacol 153: 66–74, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou LM, Shi Z, Gao J, Han Y, Yuan N, Gao XY, Zhu GQ. Angiotensin-(1–7) and angiotension II in the rostral ventrolateral medulla modulate the cardiac sympathetic afferent reflex and sympathetic activity in rats. Pflügers Arch 459: 681–688, 2010 [DOI] [PubMed] [Google Scholar]
- 47.Zucker IH, Hackley JF, Cornish KG, Hiser BA, Anderson NR, Kieval R, Irwin ED, Serdar DJ, Peuler JD, Rossing MA. Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension 50: 904–910, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Zucker IH, Wang W, Brändle M, Schultz HD, Patel KP. Neural regulation of sympathetic nerve activity in heart failure. Prog Cardiovasc Dis 37: 397–414, 1995 [DOI] [PubMed] [Google Scholar]
- 49.Zucker IH, Wang W, Pliquett RU, Liu JL, Patel KP. The regulation of sympathetic outflow in heart failure. The roles of angiotensin II, nitric oxide, and exercise training. Ann NY Acad Sci 940: 431–443, 2001 [PubMed] [Google Scholar]