

Abstract
There has been significant recent progress in the computational design of protein interactions including the creation of novel heterodimers, homodimers, nanohedra, fibril caps and a protein crystal. Essential to these successes has been the use of innovative strategies for finding binding modes that are achievable, i.e. identifying binding partners and docked conformations that can be successfully stabilized via sequence optimization and backbone refinement. In many cases this has involved the use of structural motifs commonly found at naturally occurring interfaces including alpha helices inserted into hydrophobic grooves, beta-strand pairing, metal binding, established helix packing motifs, and the use of symmetry to form cooperative interactions. Future challenges include the creation of hydrogen bond networks and antibody-like interactions based on the redesign of protein surface loops.
Introduction
Engineering of protein-protein interactions can generate pair-wise complexes, multi-component complexes, and interaction networks with extensive structural and functional diversity. Practical applications for designed interactions are numerous and include the creation of competitive inhibitors, affinity reagents, biosensors, scaffolding molecules and nanomaterials [2-20].
Directed evolution methods and selection strategies such as phage display are effective for generating new target-binding interactions using antibody scaffolds [21] and non-antibody scaffolds [22], and binders identified with these approaches have shown promise in clinical trials [23]. Computational interface design is an alternative approach that is currently not as robust as experimental selection and screening, but offers precise control of binding location and binding mode and can be applied to design goals that are more difficult for selection technologies such as homomeric assemblies, arrays, fibril caps, and multi-specificity.
Methods in computational protein design have been used to redesign the affinity and specificity of naturally occurring interactions [24-27] as well as create new interactions from scratch (Figure 1, Table I). This review focuses on novel interfaces that have been designed in the last two years. In most cases, new interactions have been created by mutating the amino acids on the surfaces of naturally occurring proteins so that favorable interactions occur upon formation of the target complex. In these simulations there are three possible degrees of freedom, the relative rigid-body orientation of the proteins, the backbone conformation of residues at the interface, and the amino acid sequence of the interacting residues [28]. Sequence optimization is typically performed with rotamer-based side-chain sampling methods such as simulated annealing or dead end elimination [29,30]. In most studies, backbone sampling has not been performed or only small perturbations have been allowed. Accurate remodeling of the protein backbone is a challenging goal in its own right. As a more conservative approach for allowing alternative backbones at the interface, several methods computationally consider a large set of proteins as the template for interface design.
Figure 1. Types of protein complexes designed using computational methods.
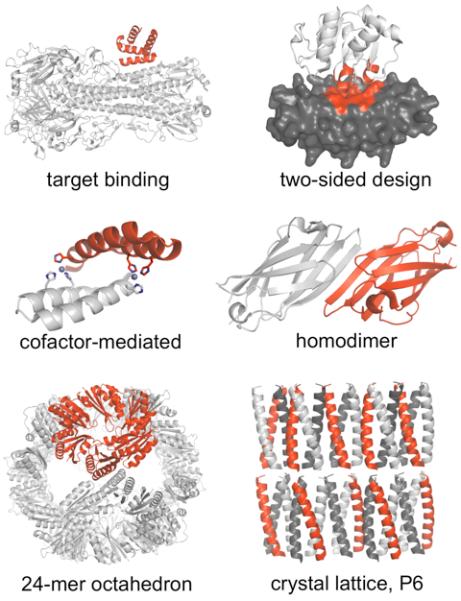
Target binding [32]: a helical scaffold (red) redesigned to bind the stem region of influenza hemagglutinin (white). Two-sided design [48]: redesigned scaffolds were ankyrin repeat protein (gray) and a coenzyme A binding protein (PH1109, white), interface contacts in red. Cofactor-mediated binding [39]: A helical hairpin designed for zinc-mediated homodimerization. Histidine residues (sticks) coordinate zinc (spheres). Homodimer [38]: the γ-adaptin appendage domain – a monomer with an exposed beta strand – redesigned to allow intermolecular beta-sheet formation. Nanohedra [17]: a native trimer (red) redesigned to form an octamer of trimers, a 24-mer octahedron. P6 crystal lattice [45]: a previously designed coiled-coil homotrimer modified to form a predetermined crystal lattice in a rare space group, P6.
Table I. Summary of recent computer-based designed interactions.
| Goal | Strategy | System | PDB codes | Ref |
|---|---|---|---|---|
| Interface de novo design | ||||
| Target binding | Known hydrophobic groove, hotspots | Hemaglutinin | 3R2X | [32] |
| Target binding | Known hydrophobic groove | GoLoco peptide, Gαil | 2XNS | [33] |
| Target binding | Known hydrophobic groove | PAK1 | [12] | |
| Target binding, multi- specificity |
Dataset-driven scoring of coiled-coils | bZIP coiled-coil | [8] | |
| Fibril inhibition | Strand pairing and hydrophobic sidechains | Tau and PAP fibrils | [7] | |
| Two-sided design | Aromatic sidechain hydrogen bond | Ankyrin and PH1109 | 3Q9U, 3Q9N, 3QA9 | [48] |
| Homodimer | Strand pairing and hydrophobic sidechains | Native monomer | 3ZY7 | [38] |
| Homodimer | Metal binding and hydrophobic sidechains | Native monomer | 3V1A, 3V1B, 3V1C, 3V1D, 3V1E, 3V1F |
[39] |
| Heterotetramer | Nonnatural zinc chromophore | Heterotetramer coiled-coil | [43] | |
| Transmembrane targeting |
GxxGxxG motif in a beta helix | Beta-peptide | [46] | |
| Transmembrane | Porphyrin binding | Four-helix bundle | [42] | |
| Octahedron, 24-mer | Hydrophobic sidechains, hydroxyl hbonds | Native trimer | 3VCD, 4DDF | [17] |
| Tetrahedron, 12-mer | Hydrophobic sidechains | Native trimer | 4EGG, 4DCL | [17] |
| Nanotube coating | Helix Ala/Gly interaction with carbon ring | Coiled-coil hexamer | 3S0R | [16] |
| Array, P6 crystal | Superhelix stacking, GxxG helix | Previous trimer lattice | 3V86, 4DAC | [45] |
|
| ||||
| Scaffold design for new binding complexes | ||||
| Target binding | Grafting | HIV epitopes 2F5 and 4E10 | 3RPT, 3RU8 and 3RI0, 3RHU, 3RFN |
[3] |
| Target binding | Grafting | MMP-14 and TIMP-2 | not released | [52] |
| Target binding | PAK1 biosensor | Autoinhib. helical domain | [13] | |
| Nanohedron | Genetic fusion, rigid helix linker | Native dimer, trimer, helix | 3VDX, 4D9J | [35,55] |
| Arrays, 1D and 2D | Genetic fusion, rotational symmetry | (TEM, AFM data) | [56] | |
|
| ||||
| Interface redesign | ||||
| Specificity | Second-site suppressor mutations | GoLoco peptide and Gai, UbcH7 and E6AP |
[57] [14] |
|
| Specificity | Flexible backbone design to replace Phe | GTPase/GEF (1KI1) | 3QBV | [14] |
| Multi-specificity | Structure-based modeling and library screening |
Bcl-x(L) | [58] | |
| Affinity | Noncanonical amino acid at a native interface | Calpain, calpastatin | [59] | |
| Affinity | Sequence design with backbone perturbation | PDZ peptide and CAL | [2] | |
| Affinity | Hydrophobic interface design | Cytochrome cb562 | 3HNI, 3HNJ, 3HNL | [60] |
| Affinity | Two hydrophobic mutations | PCNA and polymerase | [9] | |
| Homologous target | Computationally-directed library | Ubc12 and E6AP | [61] | |
| Hexamer | Augmentation of coiled-coil tetramer | GCN4-p1 starting tetramer | 3R4A, 3R4H, 3R3K, 3R46, 3R48 |
[37] |
| Metal switching | Replace native interactions with metal coordination residues |
Ferritin cage | 4DYX, 4DYY, 4DYZ, 4DZ0 |
[62] |
Despite continued improvements in conformational searching and energy evaluation, interface design remains an imposing challenge due to limitations in these two areas [31]. In particular, the design of interfaces that make use of cooperative hydrogen bonding networks has been especially challenging. As a result, successful designs have relied on carefully chosen strategies in which a desired high-probability binding mode limits the conformational search and is somewhat robust to inaccuracies in energy calculations. Here we review the emerging strategies for designing a desired binding mode (Figure 2).
Figure 2. Types of interaction motifs used in de novo designed protein interactions.
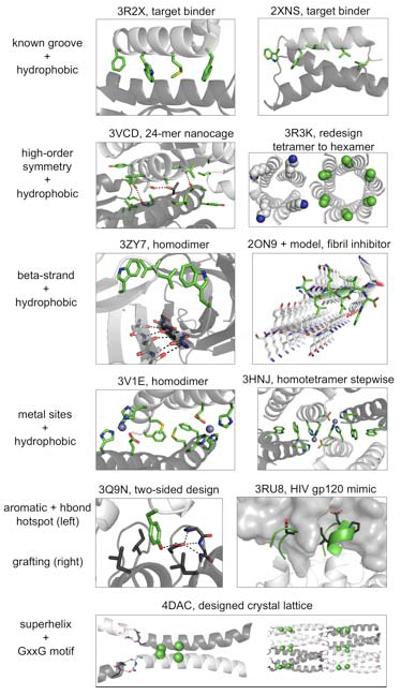
Known groove: influenza hemagglutinin binding (left), Gαi1 binding (right). High-order symmetry: a trimer redesigned to form a 24-mer octahedron (left), a coiled-coil tetramer mutated to form a coiled-coil hexamer (right). Beta-strand: a monomer redesigned to form a strand-mediated homodimer (left), amyloid fibril formation inhibited with a designed binding peptide (right). Metal coordination: a monomer redesigned to form a zinc-mediated homodimer (left), a stepwise process converted a crystal-contact tetramer to a solution-phase tetramer by placing histidines at crystal contacts. Subsequently, the zinc-mediated tetramer was improved by computational design (right). Aromatic and hbond hotspot: tyrosine was used to form a hydrophobic and hydrogen bonding hotspot interaction with a preordered aspartate side chain in a hydrophobic pocket. Grafting: discontinuous side-chain and backbone interaction motifs from a known antibody-antigen pair were grafted onto an unrelated scaffold. Superhelix and glycine crossing: a previously designed and crystallized coiled-coil trimer was redesigned to form superhelix stacking interactions and helical glycine crossing interactions, generating the intended honeycomb-like P6 crystal lattice.
Helix in a known binding groove
An alpha helix docked in a hydrophobic groove is a favorable scenario for interface design [12,32,33]. Overall shape complementarity of a helix for a concave groove guides the binding orientation and avoids the error-prone challenge of designing polar contacts from scratch.
As a prominent example, broad inhibition of influenza viral fusion [6] was accomplished starting with computational methods to bind the hydrophobic groove in the conserved stem region rather than the variable head domain of hemagglutinin [32]. The first design step positioned disembodied hydrophobic side chains at the target stem to serve as hotspots, and the second design step docked protein scaffolds to connect these disembodied hotspot side chains. By building outwards from native-like and de novo hydrophobic side-chain hotspots, two moderate-affinity binders for hemagglutinin were produced (Kd > 5 μM), and the crystal structure of an affinity-matured variant demonstrated high accuracy in the intended binding location and orientation [32].
Secondly, the C-terminal coil region of the GoLoco peptide binds a hydrophobic groove in the heterotrimeric G-protein alpha subunit (Gαi1), and this peptide coil was redesigned to form a helix and bind the same hydrophobic groove. Rather than start from side-chain hotspots, this study used simultaneous backbone design by fragment insertion and sequence design of the GoLoco peptide. Binding of the redesigned helix was ~2-fold weaker than the native coil, but the crystal structure demonstrated accurate backbone and side-chain positions [33].
High-order symmetry (O, T, C6, D2)
High-order symmetry helps specify a binding orientation because off-target binding modes are unlikely to be compatible with cooperative symmetric assembly. Also, high-order symmetry enables design of large complexes using a small number of designed contacts. The first approach to design a large nanocage used native dimers and trimers fused by a rigid helix [34,35]. Building on this result, instead of using a rigid linker, a designed interface was used as the rigid connection between native oligomers. In this study, the outer surfaces of native trimers were redesigned to self-associate and accurately form a 24-mer octahedron and a 12-mer tetrahedron. These large nanohedra resulted from one designed protein interface with mostly hydrophobic interactions [17].
Hydrophobic shape complementarity in a symmetric system was also used to design a hexameric channel. Classical coiled-coils feature core hydrophobic side-chain interdigitation and peripheral electrostatic complementarity, but surprisingly, a six-membered classical coiled-coil had not been previously observed. Inspired by an approach of hydrophobic expansion [36], a designed coiled-coil tetramer was mutated (peripheral lysines to alanines) to modulate the symmetric hydrophobic complementarity and produced a hexameric channel-forming assembly [37].
Strand pairing
Hydrophobic contacts will usually need help from polar contacts to specify a desired binding mode. Strand pairing is an effective strategy to generate new polar contacts – it provides cooperative formation of multiple hydrogen bonds to specify binding orientation, and these backbone-backbone hydrogen bonds avoid the entropy loss of side-chain hydrogen bonds. The following two studies combine strand pairing with hydrophobic side-chain interdigitation.
Amyloid fibrils are associated with human disease and can form via repeating strand-strand interactions. To cap and inhibit amyloid fibril formation, short peptides were designed to form beta-strand hydrogen bonds and interdigitating hydrophobic side-chain interactions at the fibril end. A designed D-peptide inhibited fibril formation by the tau peptide of Alzheimer’s disease, and a designed noncanonical L-peptide inhibited fibril formation by a PAP peptide fragment associated with HIV transmission. Strand pairing was a central design element, and the specific side-chain interactions were critical for binding in both cases [7].
Secondly, a monomeric protein with a surface-exposed beta strand was redesigned to form a symmetric beta-strand-mediated homodimer. To supplement the strand pair, side chains were designed to form mostly polar or mostly hydrophobic interactions. The successful design (Kd = 1 μM) had hydrophobic side-chain interactions. A crystal structure revealed highly accurate positioning of the strand-pair atoms and highly accurate binding orientation, with mostly accurate positioning of the hydrophobic side chains. Thus, the strand-strand polar contacts were primarily responsible for the high accuracy of the overall binding orientation, and the interdigitating hydrophobic side chains were critical for binding [38].
Metal binding
Metal binding is another strategy for generating new polar contacts – metal coordination geometry is well-defined and coordination bonds are stronger than hydrogen bonds. Metal binding sites and a small hydrophobic protein interface were designed together to achieve high affinity and a predetermined binding orientation. In the absence of metal, an unanticipated binding orientation was observed, highlighting the importance of polar contacts to govern the binding mode. Addition of zinc improved affinity by >100-fold and recovered the intended binding orientation [39]. As evidenced in this study and other zinc-binding studies [20,40], a three-His one-Asp/Glu motif is favored over a four-His zinc coordination motif.
As another example of metal cofactor-mediated protein assembly, multi-heme arrays in a helix bundle were designed with the goal of artificial electron transport through a membrane. A previous method for designing water-soluble multi-porphyrin binding helical bundles [41] was extended to design porphyrin proteins in a membrane (PRIME) [42]. In this study, two FeIII diphenyl porphyrins were coordinated by a bis-histidine motif with second-shell threonine hydrogen bonds from helices arranged in a tetrameric bundle with D2 symmetry. This approach combined classic coiled-coil side-chain motifs, metal coordination, and symmetry to govern complex formation. Similar methods have also been used to achieve selective binding of an artificial porphyrin chromophore [43].
Super-helix and helix glycine motifs
Two types of helical interaction motifs were used to design a predetermined protein crystal lattice in the P6 space group. Crystal lattices in the rarely observed P6 space group feature planar layering. Contacts between layers were formed by a stacking helix-to-helix end-to-end interaction – this pseudocontiguous “super helix” crystal contact required N-terminal acetyl and C-terminal amide groups and was derived from the previously observed P321 crystal lattice of the original three-helix bundle design [44]. Contacts across a layer were formed by GxxxG helical glycine interactions for helix association with tight packing and a predictable crossing angle [45]. This is the first example of a de novo designed protein crystal.
Hexameric helical assemblies coating carbon nanotube surfaces were also designed using a superhelical motif. The second major feature was the use of repeating alanine or glycine residues to interact with the repeating depressions in 6-membered carbon ring centers of the nanotube surface. The peptide was labeled with gold particles to generate TEM images to confirm a patterned array of protein coating the nanotube. Nanotube binding is not amenable to crystallization, but a crystal structure of a tetrameric form of the designed protein showed repeating hydrophobic and polar side-chain interactions between adjacent helices [16].
Thirdly, non-natural beta peptides were designed to form beta helices that bind an alpha helical transmembrane domain. The designed beta helices featured outward-facing apolar residues with lysine and tryptophan residues near the lipid headgroup region. A GxxGxxG motif in the designed beta-helix was used to target a GxxxG motif in the target transmembrane helix [46].
Electrostatic patterning of coiled coils
Electrostatic patterning in coiled-coils was used in some of the studies already discussed [37,45], but this feature is most prominent in a designed interaction that inhibits a bZIP transcription factor called BZLF1 [8]. The BZLF1 transcription factor homodimerizes (A:A) to bind DNA, so the designed competitive interaction (A:B) inhibits transcription. Given the structural similarity of the inhibitor and target, inhibitor designs are likely to homodimerize (B:B) and lose potency, so self-association of these inhibitory peptides was explicitly disfavored. The method combined experimental data from known bZIP coiled-coils, physics-based calculations, and machine learning to generate a primary sequence scoring function for rapid design with multi-specificity [47]. The best sequences for BZLF1 binding featured charge-charge repulsions that disfavored B:B homodimerization and charge-charge attractions that favored A:B complex formation [8].
Hydrogen bond from an aromatic sidechain
Although it is a challenging design goal, side-chain hydrogen bonds can be used as hotspots. In two-sided design of a de novo binding pair, tyrosine and tryptophan residues were intended to simultaneously form aromatic/hydrophobic hotspots and a hydrogen bond [48]. One designed pair bound tightly (Kd = 120 nM). Structure determination of an affinity-matured variant revealed that the actual binding orientation was flipped 180° compared to the model, and that the tryptophan hotspot did not form. However, the tyrosine hotspot at the center of the interface did form and was the pivot point between the actual and intended binding orientation, suggesting that the intended tyrosine interaction motif was a strong hotspot.
Grafting: scaffold redesign to accommodate known interface contacts
The hotspot side chain design strategy [32,48-50] is similar in spirit to a side-chain-only grafting strategy previously used to generate a new binding pair [51]. A more recent strategy grafted backbone and side-chain epitopes from HIV for possible applications in vaccine development. Grafting of 6- to 11-residue linear epitopes required aggressive computational backbone sampling and resulted in successful binders [3]. Additionally, a discontinuous epitope – two segments from HIV gp120 – were grafted onto an unrelated scaffold, and computationally-guided directed evolution generated a high-affinity binder that demonstrated close mimicry in one epitope and partial mimicry in the second epitope [4]. Interestingly, grafting studies [3,4,52] take atomic-level binding motifs directly from native interactions but are still susceptible to atomic-level inaccuracies, highlighting the challenge of accurately designing new interactions from scratch.
Conclusions
There are many routes to engineering new protein complexes, including redesign of native interfaces, directed evolution, genetic fusion, and empirical approaches (Table I). This short review emphasizes computer-based de novo interface design with a desired orientation. In the past two years, the field of protein interface design has advanced significantly. However, despite many new successful designs, a low success rate [1] burdens the experimental stage and calls for high-probability binding modes that can overcome current limitations in the conformational search and energy evaluation. So far, these strategies and binding modes include: helices in hydrophobic grooves, side chain interdigitation, high-order symmetry, strand pairing, metal binding, glycine-mediated helix crossing, coiled-coil electrostatic patterning, hydrogen-bonding aromatic sidechains, grafting of native interface fragments, and fusion of native proteins. Interfaces containing loops are common in nature [53] and directed evolution studies [54], but they have not yet been designed computationally due to many challenges: loop conformations are challenging to predict, loop flexibility results in entropic penalty upon binding, loops require backbone and side-chain polar atoms to have their hydrogen bonding potential satisfied, and this design goal would require simultaneous design of the scaffold backbone and interface contacts. Additionally, a hydrogen bond network has not yet been a major component of a de novo designed interaction, in part due to side-chain flexibility and the penalty for desolvating side-chain polar atoms. Designing these and other challenging types of interfaces will require continued improvements in conformational searching, energy evaluation, and experimental throughput, as well as continued innovation of strategies to control the binding mode.
Inset.
Currently, the major challenge in protein interface design is generating polar contacts [1]. While successful de novo interface designs to-date are predominantly hydrophobic, polar contacts are often required for monomer solubility and binding specificity. Successful strategies for designing hydrophobic and polar contacts have been:
Hydrophobic binding strategies:
Helices in binding grooves
High-order symmetry/avidity
Side chain interdigitation
Helix crossing at glycine residues
Polar binding strategies:
Strand pairing
Helix stacking
Metal binding
Electrostatic patterning (coiled-coils)
Tyrosine hotspots
Grafted backbone and side-chain motifs
Highlights.
Many computationally designed protein-protein interactions were reported over the past two years.
Recent design goals include heterodimers, homodimers, homooligomers, fibril caps, and arrays.
Diverse polar and nonpolar interaction motifs can control the binding orientation.
Crystal structures show accurately designed interactions compared to atomic level predictions.
Loops and hydrogen bond networks are outstanding interface design challenges.
Acknowledgments and Funding
We thank Dr. Joseph Harrison for his helpful comments on the review. We also thank the funding sources: National Institutes of Health grant GM073960 (B.K.), National Institutes of Health grant T32GM008570 for the UNC Program in Molecular and Cellular Biophysics, National Science Foundation graduate research fellowship (2009070950 to B.D.), UNC Royster Society Pogue fellowship (B.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
** Of outstanding interest
* Of special interest
- 1.Stranges PB, Kuhlman B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013;22:74–82. doi: 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts KE, Cushing PR, Boisguerin P, Madden DR, Donald BR. Computational Design of a PDZ Domain Peptide Inhibitor that Rescues CFTR Activity. Plos Computational Biology. 2012;8 doi: 10.1371/journal.pcbi.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3*.Azoitei ML, Ban YE, Julien JP, Bryson S, Schroeter A, Kalyuzhniy O, Porter JR, Adachi Y, Baker D, Pai EF, et al. Computational design of high-affinity epitope scaffolds by backbone grafting of a linear epitope. J Mol Biol. 2012;415:175–192. doi: 10.1016/j.jmb.2011.10.003. A grafting-based strategy was developed for 6 to 11-residue linear epitopes rather than only side chains. This required more aggressive scaffold redesign and led to successful transplantation of HIV epitopes onto protein scaffolds. Crystal structures in the absence of antibody demonstrate accurately grafted structures.
- 4.Azoitei ML, Correia BE, Ban YEA, Carrico C, Kalyuzhniy O, Chen L, Schroeter A, Huang PS, McLellan JS, Kwong PD, et al. Computation-Guided Backbone Grafting of a Discontinuous Motif onto a Protein Scaffold. Science. 2011;334:373–376. doi: 10.1126/science.1209368. [DOI] [PubMed] [Google Scholar]
- 5.Keeffe JR, Gnanapragasam PN, Gillespie SK, Yong J, Bjorkman PJ, Mayo SL. Designed oligomers of cyanovirin-N show enhanced HIV neutralization. Proc Natl Acad Sci U S A. 2012;108:14079–14084. doi: 10.1073/pnas.1108777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7*.Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Munch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–U117. doi: 10.1038/nature10154. D-peptide and noncanonical L-peptides were designed to cap the ends of amyloid-like fibrils. Hydrophobic side-chain interdigitation and strand pairing interactions inhibited fibril formation.
- 8.Chen TS, Reinke AW, Keating AE. Design of peptide inhibitors that bind the bZIP domain of epstein-barr virus protein BZLF1. Journal of Molecular Biology. 2011;408:304–320. doi: 10.1016/j.jmb.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fridman Y, Gur E, Fleishman SJ, Aharoni A. Computational protein design suggests that human PCNA-partner interactions are not optimized for affinity. Proteins. 2012 doi: 10.1002/prot.24190. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds KA, Hanes MS, Thomson JM, Antczak AJ, Berger JM, Bonomo RA, Kirsch JF, Handel TM. Computational redesign of the SHV-1 beta-lactamase/beta-lactamase inhibitor protein interface. J Mol Biol. 2008;382:1265–1275. doi: 10.1016/j.jmb.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Der BS, Edwards DR, Kuhlman B. Catalysis by a de novo zinc-mediated protein interface: implications for natural enzyme evolution and rational enzyme engineering. Biochemistry. 2012;51:3933–3940. doi: 10.1021/bi201881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jha RK, Leaver-Fay A, Yin SY, Wu YB, Butterfoss GL, Szyperski T, Dokholyan NV, Kuhlman B. Computational Design of a PAK1 Binding Protein. Journal of Molecular Biology. 2010;400:257–270. doi: 10.1016/j.jmb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jha RK, Wu YI, Zawistowski JS, MacNevin C, Hahn KM, Kuhlman B. Redesign of the PAK1 autoinhibitory domain for enhanced stability and affinity in biosensor applications. J Mol Biol. 2011;413:513–522. doi: 10.1016/j.jmb.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapp GT, Liu S, Stein A, Wong DT, Remenyi A, Yeh BJ, Fraser JS, Taunton J, Lim WA, Kortemme T. Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. Proc Natl Acad Sci U S A. 2012;109:5277–5282. doi: 10.1073/pnas.1114487109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haidar JN, Pierce B, Yu Y, Tong WW, Li M, Weng ZP. Structure-based design of a T-cell receptor leads to nearly 100-fold improvement in binding affinity for pepMHC. Proteins-Structure Function and Bioinformatics. 2009;74:948–960. doi: 10.1002/prot.22203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16**.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. A peptide helical hexamer was designed to coat a carbon nanotube surface. Repeating helical glycine or alanine residues packed into the dimples of 6-membered rings of the nanotube, and intermolecular contacts between peptides were designed.
- 17**.King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, Gonen T, Yeates TO, Baker D. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. doi: 10.1126/science.1219364. Native trimers were redesigned to form a 24-mer octahedron and a 12-mer tetrahedron. Use of symmetry required only one designed interface to construct of these large nanohedra.
- 18.Lai YT, King NP, Yeates TO. Principles for designing ordered protein assemblies. Trends Cell Biol. 2012 doi: 10.1016/j.tcb.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Brodin JD, Ambroggio XI, Tang CY, Parent KN, Baker TS, Tezcan FA. Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nature Chemistry. 2012;4:375–382. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laganowsky A, Zhao ML, Soriaga AB, Sawaya MR, Cascio D, Yeates TO. An approach to crystallizing proteins by metal-mediated synthetic symmetrization. Protein Science. 2011;20:1876–1890. doi: 10.1002/pro.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sidhu SS. Antibodies for all: The case for genome-wide affinity reagents. FEBS Lett. 2012;586:2778–2779. doi: 10.1016/j.febslet.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 22.Gilbreth RN, Koide S. Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr Opin Struct Biol. 2012;22:413–420. doi: 10.1016/j.sbi.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wurch T, Pierre A, Depil S. Novel protein scaffolds as emerging therapeutic proteins: from discovery to clinical proof-of-concept. Trends in Biotechnology. 2012;30:575–582. doi: 10.1016/j.tibtech.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Kortemme T, Baker D. Computational design of protein-protein interactions. Curr Opin Chem Biol. 2004;8:91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Mandell DJ, Kortemme T. Computer-aided design of functional protein interactions. Nat Chem Biol. 2009;5:797–807. doi: 10.1038/nchembio.251. [DOI] [PubMed] [Google Scholar]
- 26.Mandell DJ, Kortemme T. Backbone flexibility in computational protein design. Curr Opin Biotechnol. 2009;20:420–428. doi: 10.1016/j.copbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 27.Karanicolas J, Kuhlman B. Computational design of affinity and specificity at protein-protein interfaces. Curr Opin Struct Biol. 2009;19:458–463. doi: 10.1016/j.sbi.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C, Bradley P, Baker D. Protein-protein docking with backbone flexibility. J Mol Biol. 2007;373:503–519. doi: 10.1016/j.jmb.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 29.Dahiyat BI, Mayo SL. Protein design automation. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desjarlais JR, Clarke ND. Computer search algorithms in protein modification and design. Curr Opin Struct Biol. 1998:8–471. doi: 10.1016/s0959-440x(98)80125-5. [DOI] [PubMed] [Google Scholar]
- 31.Fleishman SJ, Whitehead TA, Strauch EM, Corn JE, Qin S, Zhou HX, Mitchell JC, Demerdash ON, Takeda-Shitaka M, Terashi G, et al. Community-wide assessment of protein-interface modeling suggests improvements to design methodology. J Mol Biol. 2011;414:289–302. doi: 10.1016/j.jmb.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. A helical scaffold was redesigned to bind the conserved stem region of influenza hemagglutinin. Disembodied and docked hydrophobic side-chains were the starting point of design.
- 33*.Sammond DW, Bosch DE, Butterfoss GL, Purbeck C, Machius M, Siderovski DP, Kuhlman B. Computational design of the sequence and structure of a protein-binding peptide. Journal of the American Chemical Society. 2011;133:4190–4192. doi: 10.1021/ja110296z. A native coil that binds a hydrophobic groove was redesigned to form a helix and bind the same hydrophobic groove. Simultaneous design of the helical backbone structure and sequence was performed.
- 34.Padilla JE, Colovos C, Yeates TO. Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci U S A. 2001;98:2217–2221. doi: 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai YT, Cascio D, Yeates TO. Structure of a 16-nm Cage Designed by Using Protein Oligomers. Science. 2012;336:1129–1129. doi: 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Zheng Q, Deng YQ, Cheng CS, Kallenbach NR, Lu M. A seven-helix coiled coil. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15457–15462. doi: 10.1073/pnas.0604871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaccai NR, Chi B, Thomson AR, Boyle AL, Bartlett GJ, Bruning M, Linden N, Sessions RB, Booth PJ, Brady RL, et al. A de novo peptide hexamer with a mutable channel. Nature Chemical Biology. 2011;7:935–941. doi: 10.1038/nchembio.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38**.Stranges PB, Machius M, Miley MJ, Tripathy A, Kuhlman B. Computational design of a symmetric homodimer using beta-strand assembly. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:20562, 20567. doi: 10.1073/pnas.1115124108. A native monomer with an exposed beta-strand was redesigned to form a strand-mediated homodimer. A hydrophobic variant formed the intended dimer, a polar variant did not lead to binding.
- 39*.Der BS, Machius M, Miley MJ, Mills JL, Szyperski T, Kuhlman B. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J Am Chem Soc. 2012;134:375–385. doi: 10.1021/ja208015j. A native helical hairpin monomer was designed to form a zinc-mediated homodimer. Two different design goals were combined: zinc binding and protein-protein interaction. Zinc binding improved affinity >100-fold and controlled the binding orientation.
- 40.Salgado EN, Faraone-Mennella J, Tezcan FA. Controlling protein-protein interactions through metal coordination: assembly of a 16-helix bundle protein. J Am Chem Soc. 2007;129:13374–13375. doi: 10.1021/ja075261o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McAllister KA, Zou HL, Cochran FV, Bender GM, Senes A, Fry HC, Nanda V, Keenan PA, Lear JD, Saven JG, et al. Using alpha-helical coiled-coils to design nanostructured metalloporphyrin arrays. Journal of the American Chemical Society. 2008;130:11921–11927. doi: 10.1021/ja800697g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korendovych IV, Senes A, Kim YH, Lear JD, Fry HC, Therien MJ, Blasie JK, Walker FA, DeGrado WF. De Novo Design and Molecular Assembly of a Transmembrane Diporphyrin-Binding Protein Complex. Journal of the American Chemical Society. 2010;132:15516–15518. doi: 10.1021/ja107487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fry HC, Lehmann A, Saven JG, DeGrado WF, Therien MJ. Computational Design and Elaboration of a de Novo Heterotetrameric alpha-Helical Protein That Selectively Binds an Emissive Abiological (Porphinato)zinc Chromophore. Journal of the American Chemical Society. 2010;132:3997–4005. doi: 10.1021/ja907407m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogihara NL, Weiss MS, Degrado WF, Eisenberg D. The crystal structure of the designed trimeric coiled coil coil-VaLd: implications for engineering crystals and supramolecular assemblies. Protein Sci. 1997;6:80–88. doi: 10.1002/pro.5560060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45**.Lanci CJ, MacDermaid CM, Kang SG, Acharya R, North B, Yang X, Qiu XJ, DeGrado WF, Saven JG. Computational design of a protein crystal. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7304–7309. doi: 10.1073/pnas.1112595109. A previously designed coiled-coil homotrimer was redesigned to form a predefined honeycomb-like crystal lattice in the P6 space group. A super-helix stacking interaction in the “vertical” dimension required N-terminal acetyl and C-terminal amide modifications. Glycine helix-crossing motifs in the “horizontal” dimension were also used.
- 46.Shandler SJ, Korendovych IV, Moore DT, Smith-Dupont KB, Streu CN, Litvinov RI, Billings PC, Gai F, Bennett JS, DeGrado WF. Computational Design of a beta-Peptide That Targets Transmembrane Helices. Journal of the American Chemical Society. 2011;133:12378–12381. doi: 10.1021/ja204215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–U852. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48*.Karanicolas J, Corn JE, Chen I, Joachimiak LA, Dym O, Peck SH, Albeck S, Unger T, Hu W, Liu G, et al. A de novo protein binding pair by computational design and directed evolution. Mol Cell. 2011;42:250–260. doi: 10.1016/j.molcel.2011.03.010. Two-sided design of a protein binding pair used aromatic side chains forming hydrogen bonds (tyrosine and tryptophan) as hotspots. After affinity maturation and crystallization, the actual structure was flipped relative to the intended orientation – the intended tyrosine hotspot interaction formed and was the pivot point between the intended and actual orientation.
- 49.Lewis SM, Kuhlman BA. Anchored Design of Protein-Protein Interfaces. Plos One. 2011:6. doi: 10.1371/journal.pone.0020872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleishman SJ, Corn JE, Strauch EM, Whitehead TA, Karanicolas J, Baker D. Hotspot-centric de novo design of protein binders. Journal of Molecular Biology. 2011;413:1047–1062. doi: 10.1016/j.jmb.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S, Liu SY, Zhu XL, Liang HH, Cao AN, Chang ZJ, Lai LH. Nonnatural protein-protein interaction-pair design by key residues grafting. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5330–5335. doi: 10.1073/pnas.0606198104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tlatli R, Nozach H, Collet G, Beau F, Vera L, Stura E, Dive V, Cuniasse P. Grafting of functional motifs onto protein scaffolds identified by PDB screening – An efficient route to design optimizable protein binders. FEBS J. 2012 doi: 10.1111/febs.12056. [DOI] [PubMed] [Google Scholar]
- 53.Guharoy M, Chakrabarti P. Secondary structure based analysis and classification of biological interfaces: identification of binding motifs in protein-protein interactions. Bioinformatics. 2007;23:1909–1918. doi: 10.1093/bioinformatics/btm274. [DOI] [PubMed] [Google Scholar]
- 54.Hackel BJ, Kapila A, Wittrup KD. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol. 2008;381:1238–1252. doi: 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lai YT, Cascio D, Yeates T. Crystal structure of a 16 nm, half-megadalton protein cage designed by fusing symmetric oligomeric domains. Protein Science. 2012;21:115–116. doi: 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- 56.Sinclair JC, Davies KM, Venien-Bryan C, Noble MEM. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nature Nanotechnology. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- 57.Sammond DW, Eletr ZM, Purbeck C, Kuhlman B. Computational design of second-site suppressor mutations at protein-protein interfaces. Proteins-Structure Function and Bioinformatics. 2010;78:1055–1065. doi: 10.1002/prot.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen TS, Palacios H, Keating AE. Structure-Based Redesign of the Binding Specificity of Anti-Apoptotic Bcl-x(L) J Mol Biol. 2013;425:171–185. doi: 10.1016/j.jmb.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Renfrew PD, Choi EJ, Bonneau R, Kuhlman B. Incorporation of Noncanonical Amino Acids into Rosetta and Use in Computational Protein-Peptide Interface Design. Plos One. 2012:7. doi: 10.1371/journal.pone.0032637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salgado EN, Ambroggio XI, Brodin JD, Lewis RA, Kuhlman B, Tezcan FA. Metal templated design of protein interfaces. Proc Natl Acad Sci U S A. 2010;107:1827–1832. doi: 10.1073/pnas.0906852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guntas G, Purbeck C, Kuhlman B. Engineering a protein-protein interface using a computationally designed library. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19296–19301. doi: 10.1073/pnas.1006528107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huard DJ, Kane KM, Tezcan FA. Re-engineering protein interfaces yields copper-inducible ferritin cage assembly. Nat Chem Biol. 2013;9:169–176. doi: 10.1038/nchembio.1163. [DOI] [PubMed] [Google Scholar]