

Abstract
Hypoxic-ischemic and traumatic brain injuries are leading causes of long-term mortality and disability in infants and children. Although several preclinical models using rodents of different ages have been developed, species differences in the timing of key brain maturation events can render comparisons of vulnerability and regenerative capacities difficult to interpret. Traditional models of developmental brain injury have utilized rodents at postnatal day 7–10 as being roughly equivalent to a term human infant, based historically on the measurement of post-mortem brain weights during the 1970s. Here we will examine fundamental brain development processes that occur in both rodents and humans, to delineate a comparable time course of postnatal brain development across species. We consider the timing of neurogenesis, synaptogenesis, gliogenesis, oligodendrocyte maturation and age-dependent behaviors that coincide with developmentally regulated molecular and biochemical changes. In general, while the time scale is considerably different, the sequence of key events in brain maturation is largely consistent between humans and rodents. Further, there are distinct parallels in regional vulnerability as well as functional consequences in response to brain injuries. With a focus on developmental hypoxicischemic encephalopathy and traumatic brain injury, this review offers guidelines for researchers when considering the most appropriate rodent age for the developmental stage or process of interest to approximate human brain development.
Keywords: Brain development, Human, Rodent, Traumatic brain injury, Immature, Hypoxia-ischemia
1. Introduction
Rodent models of ischemic and traumatic brain injury are frequently used in research laboratories, both to investigate the underlying mechanisms of injury vulnerability and evaluate potential therapeutic approaches. Perinatal hypoxic-ischemic encephalopathy (HI or HIE) accounts for 25% of developmental disabilities in children, occurring in 1% of all full-term births (Shevell et al., 2000). Perinatal asphyxia-induced brain injury is one of the most common causes of morbidity and mortality in term and preterm neonates, accounting for 23% of neonatal deaths globally (Lawn et al., 2005). Neonatal stroke, a cerebrovascular event which occurs between 28 weeks gestation and one postnatal month of age, may be either hemorrhagic or HI in origin and has been associated with consequences including cerebral palsy and behavioral abnormalities (Lee et al., 2005; Lynch, 2009). Traumatic brain injury (TBI) is a leading cause of long-term neurocognitive and psychosocial deficits in infants and young children worldwide (Mazzola and Adelson, 2002; Selassie et al., 2008), with an estimated 475,000 cases of TBI in 0–14 year old children each year in the US (Langlois et al., 2005). Rates are highest in children under 4 years of age, and TBI sustained during early childhood typically results in poorer outcomes and longer recovery times compared to children who sustain injury in later childhood or adolescence (Catroppa et al., 2008; Duval et al., 2008). Regardless of the injury type or mechanism, traumatic and ischemic injuries share many common pathological mechanisms (Kochanek, 1993), and it is increasingly evident that the developing brain responds differently to injury compared to the adult brain (Babikian et al., 2010; Blomgren et al., 2007; Claus et al., 2010; Giza et al., 2007; Hu et al., 2000; Potts et al., 2006; Qiu et al., 2007; Zhu et al., 2009, 2005). It is thus crucial that we gain a better understanding of the unique properties intrinsic to the developing brain and its response to insult (Giza et al., 2009). The number of paradigms to model the injured immature brain is growing, using different animals of varying ages (Balduini et al., 2000; Bittigau et al., 2003; Claus et al., 2010; Ikonomidou and Kaindl, 2011; Tai et al., 2009; Zhu et al., 2005). Yet questions of comparability across species continue to create controversy. Which ages in rodents best correspond to the premature, newborn at term, infant, child and adolescent human? Which aspects of brain development are most essential to equate to humans when using an animal model? Keeping in mind that no given model is likely to fully mimic the human disease or condition, we suggest that it is most important to accurately define and correlate general mechanisms of injury and neuroprotection, which are often dependent on the maturation stage of the nervous system (Hagberg et al., 2002a).
Here, we will review key events that accompany brain development in both rodents and humans to identify temporal ‘benchmarks’ where there is heightened vulnerability to injury during infancy, childhood and adolescence. Developmental changes in neuroanatomy, cell proliferation, synaptogenesis and myelination will be discussed, as well as differential immune responses seen at different ages. Lastly, the emergence of age-dependent behaviors in rodents and humans will be considered in relation to ongoing developmentally regulated molecular and anatomical changes. The impact of TBI or HIE at different developmental processes will be highlighted throughout, to emphasize the complex interplay between injury mechanisms superimposed upon maturation-related changes in brain structure and function.
2. Gross neuroanatomy
The first major event of central nervous system (CNS) development in all vertebrates is the formation of a specialized fold of ectodermal tissue called the neural tube, from which the spinal cord and brain subsequently differentiate. Neural tube formation occurs approximately mid-gestation in rodents, on gestational day (gd) 10.5–11 and 9–9.5 in rats and mice, respectively, with birth typically occurring on gd 20–21. In humans, this event occurs earlier during prenatal development, between gd 24 and 28 (3–4 weeks) out of a gestation period of 266–280 days (40 weeks) (DeSesso et al., 1999; Rice and Barone, 2000). The key stages of cortical development during fetal brain formation are remarkably conserved between mammalian species and have been extensively described elsewhere (Clancy et al., 2007; Finlay and Darlington, 1995; Molnár and Clowry, 2012; Monk et al., 2001). This has allowed for the implementation of an online database for translating early neurodevelopmental milestones between species. A collaboration between the University of Central Arkansas and Cornell University has resulted in a statistical-based algorithm which integrates data from 10 different mammalian species, across key events up to post-conception day 156 in humans (www.translatingtime.org) (Clancy et al., 2007). These collaborators concede the difficulty of extending such a model to encapsulate peri- and postnatal periods, when a maturing brain is more profoundly affected by activity and environmental influences.
In the postnatal brain, species' differences in brain development were historically assessed by the measurement of post-mortem tissue weights. Dobbing and Sands generated much of the groundwork in this area of comparative neuroanatomy, by characterizing the brain growth trajectories across seven mammalian species based on weight changes over time. In particular, they examined the timing of the brain growth spurt, defined as the total brain weight gain as a percentage of its adult weight, found to peak in humans around birth and postnatal day (pnd) 7 in rats (Dobbing and Sands, 1979). This likely founded the widespread ‘rule of thumb’ usage of 7-day-old rat pups to investigate perinatal scenarios (Vannucci, 1990; Yager and Ashwal, 2009). The rat cortex reaches approximately 90% of its adult weight by pnd 20, the typical age of weaning in rodents. In humans, brain weight reaches a similar plateau by 2–3 years of age (Dekaban et al., 1987; Dobbing and Sands, 1973, 1979). Thus, based on brain weights alone, pnd 20 in rats appears to correspond to a 2–3 year old human child (Table 1). Importantly, however, these early studies do not account for the considerable heterogeneity between different brain regions, which likely mature at different rates.
Table 1.
Summary of key developmental processes across comparable ages in humans and rodents.
| Human | Rodent | Developmental milestones | Reference (s) |
|---|---|---|---|
| 23–32 wk gestation (pre-term infant) | pnd 1–3 | Oligodendrocyte maturation state changes—predominance of mitotically active pre-OLsa. | Craig et al. (2003), Lodygensky et al. (2010), Dean et al. (2011a,b) |
| Immune system development. | Holsapple et al. (2003) | ||
| Establishment of the blood-brain barrier. | Engelhardt (2003), Daneman et al. (2010) | ||
| 36–40 wk gestation (term infant) | pnd 7–10 | Peak brain growth spurt. | Dobbing and Sands (1979), Bockhorst et al. (2008) |
| Peak in gliogenesis. | Catalani et al. (2002), Kriegstein and Alvarez-Buylla (2009) | ||
| Increasing axonal and dendritic density. | Cowan (1979), Bockhorst et al. (2008), Baloch et al. (2009) | ||
| Oligodendrocyte maturation state changes–switch to a pre-dominance of immature OLs. | Craig et al. (2003), Dean et al. (2011a,b) | ||
| Consolidation of the immune system. | Holsapple et al. (2003) | ||
| 2–3 year old | pnd 20–21 | Brain reaches 90–95% of adult weight. | Dobbing and Sands (1973, 1979), Dekaban et al. (1987), Giedd et al. (1999) |
| Peak in synaptic density at 50%> adult levels. | Huttenlocher (1979), Micheva and Beaulieu (1996) | ||
| Peak in myelination rate. | Keshavan et al. (2002) | ||
| Neurotransmitter and receptor changes. | Hedner et al. (1986), Romijn et al. (1991) | ||
| 4–11 year old | pnd 25–35 | Fractionation/specialization of prefrontal cortex neural networks (structural maturation). | Tsujimoto (2008) |
| Maximum volume of grey matter and cortical thickness. | Sowell et al. (1999), Bansal et al. (2008) | ||
| 12–18 year old | pnd 35–49 | Reduced synapse density, reaching a plateau at adult levels. | Huttenlocher (1979), Lidow et al. (1991) |
| Refinement of cognitive-dependent circuitry. Ongoing myelination; increasing white matter volume and fractional anisotrophy. | Giedd et al. (1999), Chahboune et al. (2007), Baloch et al. (2009), Brouwer et al. (2012) | ||
| 20 years + | pnd 60+ | Adult levels of neurotransmitters. | Romijn et al. (1991) |
| Adult levels of synaptic density. | Huttenlocher (1979) | ||
| Ongoing myelination and declining grey matter. | Lebel and Beaulieu (2011), Lebel et al. (2012) |
OL: oligodendrocyte.
Advancements in non-invasive imaging techniques during the 1990s stimulated a period of renewed interest in developmental brain growth in humans, with serial magnetic resonance imaging (MRI) allowing for the discrimination of gray and white matter and cortical thickness (Lenroot et al., 2007). By MRI, brain volume in typically developing children reaches 90–95% of its adult size by the age of 6, slightly later than earlier estimates from post-mortem studies, before peaking at 10.5 years in females and 14.5 years in males (Bansal et al., 2008; Giedd et al., 1999; Lenroot and Giedd, 2006) (Fig. 1). Of note, dramatic changes are evident in both cortical and subcortical structures during childhood and adolescence. Using conventional MRI sequences, the intensity of gray and white matter during the first 6 months of life in humans are reversed from the adult (i.e. gray matter appears lighter than white matter). Between 6 and 12 months, a regionally specific transition period is evident during which gray and white matter are poorly differentiated, consistent with a decrease in water content and the accumulation of myelin (Inder and Huppi, 2000; Paus et al., 2001). White matter subsequently increases linearly across most brain regions with increasing age, beginning towards the end of the second trimester and continuing well into the third decade of life (Giedd et al., 1999). Changes in gray matter volumes tend to be region-specific, and follow an inverted U-shaped trajectory during childhood. Gray matter in the frontal lobe undergoes protracted structural development, to reach its maximal volume at 11–12 years of age, followed by a slow decline during adolescence and early adulthood (Bansal et al., 2008; Sowell et al., 1999). Gray matter in the temporal lobe follows a similar non-linear development, reaching its maximum size at 16–17 years with a slight decline thereafter. Two independent MRI studies of cohorts aged 7–19 and 9–22 years have similarly demonstrated that most cortical regions follow a trajectory of early thickening during childhood and adolescence, followed by thinning with advancing age (Raznahan et al., 2011; Shaw et al., 2006), likely to reflect synaptic pruning. Gray matter volume continues to decline with increasing age, stabilizing to a plateau by age 50, whereas white matter volume peaks at approximately 37 years of age (Lebel et al., 2012).
Fig. 1.
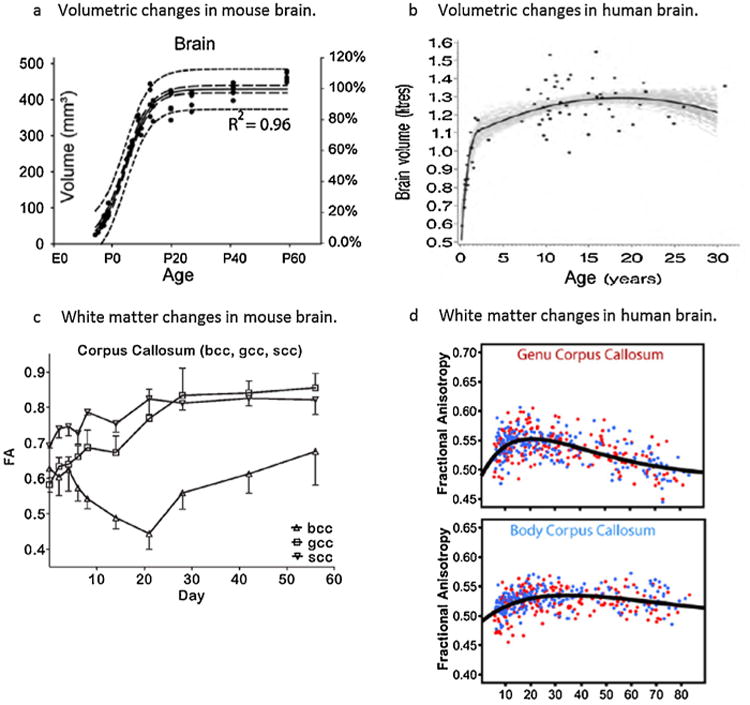
Temporal changes in postnatal brain development in rodents and humans, as assessed by magnetic resonance imaging. (a) Total mouse brain volumes on the left vertical axis (also expressed as % adult volume; right axis) measured between embryonic day 12 and pnd 60. Measurements were fitted to a sigmoidal model with a 95% confidence interval (long dash lines) and 95% prediction band (short dash lines). Reprinted from Chuang et al. (2011), with permission from Elsevier. (b) Total human brain volumes measured between 3 monthsand30yearsofage, in a total of 71 males (females follows a similar trajectory; data not shown). Black dots represent the observed data; the black line indicates the estimated median growth curve, and gray lines represent a set of 100 credible growth curves as determined by simulations. Reprinted from Groeschel et al. (2010), with permission from Elsevier. (c) Fractional anisotrophy (FA) in the corpus callosum of rats between pnd 0 and pnd 56 (n = 4–6/time point). bcc, gcc and scc represent the body, genu and splenium of the corpus callosum, respectively. From Bockhorst et al. (2008); reprinted with permission from Wiley Interscience. (d) FA in the corpus callosum of human patients aged 5–83 years (n = 403); males and females are indicated by blue and red dots, respectively. Note that FA typically peaks at ∼20-30 years of age (early adulthood), which is roughly comparable to ∼pnd 60 in rodents. From Lebel et al. (2012); reprinted with permission from Elsevier.
The past 5 years have seen a rapid increase in high quality imaging of developing rodents, particularly in mice. Some investigators have employed MRI to generate 3D reconstructions of the developing mouse brain, showing a plateau in the volume of most brain structures by pnd 20, consistent with findings from earlier post-mortem brain analysis (Chuang et al., 2011). Baloch and colleagues used diffusion tensor imaging (DTI) to generate an atlas of mouse brain neuroanatomical development in C57Bl/6J mice aged 2–40 days. They demonstrated a rapid drop in fractional anisotropy within the first postnatal week (Baloch et al., 2009), likely due to increasing dendrite density and complexity (including apical dendritic retraction) at this time (Cowan, 1979; Molnár and Rutherford, 2013). Another study in Wistar rats similarly demonstrated the greatest structural changes in gray matter within the first 5 postnatal days (Bockhorst et al., 2008).
One very notable difference between the human and rodent developing brain is gyrification, which is essentially absent in the rodent brain. A considerable amount of cortical folding begins at approximately 15 weeks gestation in humans, with the major sulci distinguishable by 28 weeks, and most gyri being fully formed by birth (Dubois et al., 2008). Sulcal and gyral patterns continue to increase in complexity postnatally, which is thought to be due to changes in cell density and maturation of subcortical fiber tracts (Levine and Barnes, 1999). This added complexity should be kept in mind during cross-species anatomical comparisons, although the functional significance of gyrification is still open to debate (White et al., 2010).
Brain injury that occurs early during development results in significant and persistent decreases in cortical and hippocampal volumes. The type and distribution of human brain lesions differ markedly between premature and term babies, likely attributed to the stage of brain maturation and subsequent regional vulnerability, as described elsewhere (Miller and Ferriero, 2009; Vexler and Yenari, 2009; Yager and Thornhill, 1997). Atrophy of both gray and white matter is also obvious in models of perinatal HIE. This atrophy is attributed to both loss of ischemic infarcted tissue and impaired development of the surrounding tissue over time (Li et al., 2011, 2010). The immature brain is considerably more resistant to hypoxia and a lack of adenosine-5′ -triphosphate than the adult brain, presumably because of the lower density of axons and dendrites over which a membrane gradient must be maintained. As such, obtaining a similarly sized injury between age groups requires different durations of hypoxia-ischemia (Zhu et al., 2005). Moreover, different mechanisms of injury are activated in the immature (pnd 5 and 9) brain versus the adult, the most obvious difference being that apoptotic mechanisms are several-fold more pronounced in immature animals (Zhu et al., 2005). It is becoming increasingly recognized that the developing brain shows marked susceptibility to both oxidative stress and neuronal apoptosis which may underlie this age-dependent injury vulnerability (Bayir et al., 2006; Blomgren and Hagberg, 2006; Blomgren et al., 2007; Blomgren et al., 2003; Ikonomidou and Kaindl, 2011; Potts et al., 2006).
Neuroimaging studies have consistently documented widespread brain atrophy after TBI during infancy and childhood (Wilde et al., 2005). There are reduced volumes in both gray and white matter, particularly within vulnerable regions such as the hippocampus, which are reflected by enlarged ventricular volumes (Berryhill et al., 1995; Wilde et al., 2005). Significant global thinning of cortical gray matter has also been reported amongst older brain-injured children (aged 9–16 years), compared to age-matched typically developing children, which is correlated with deficits in working memory (Merkley et al., 2008). Global brain atrophy is similarly evident in rodents, following experimental TBI at different stages of brain development. In the adult rat brain, substantial progressive atrophy of the cortex, hippocampus, thalamus and septum is evident over one year after fluid percussion injury, coinciding with pronounced ventriculomegaly ipsilateral to the site of injury (Smith et al., 1997). Similar regional vulnerability has also been demonstrated in younger animals. Traumatic injury induced by controlled cortical impact at pnd 21, an age thought to approximate the toddler-aged child (Yager and Thornhill, 1997), causes progressive neuronal loss resulting in reduced cortical and subcortical volume as mice mature to adulthood (Pullela et al., 2006). Contusive brain injury at pnd 11 and pnd 17, described as being equivalent to the human infant and toddler respectively, likewise results in substantial atrophy of the cortex and hippocampus, with concurrent enlargement of the lateral ventricles and greater tissue loss reported in younger animals (Huh and Raghupathi, 2007; Raghupathi and Huh, 2007).
3. Cell proliferation
The generative capacity of the immature brain varies by brain region and cell type. In general, cell proliferative processes between rodents and humans are remarkably parallel, although the time scales are substantially different (Bayer et al., 1993). The neonatal mammalian brain contains a temporary ‘subplate zone,’ a layer of glutamatergic and gamma-aminobutyric acid (GABA)-ergic neurons between the immature cerebral cortex and white matter regions, which acts as a source of new neurons during development (Kostović et al., 1989). Although the human and rodent subplate share common gene expression patterns, there are species differences in structural organization and complexity; the majority of cells form a single compact layer in mice, whereas they are dispersed throughout a larger zone in humans (Wang et al., 2010). In rats, neurogenesis (the generation of new neurons) in most cortical and subcortical regions begins at gd 9.5 and is completed by pnd 15 (Babikian et al., 2010; Rice and Barone, 2000). In humans, cortical neurogenesis occurs predominantly during gestation but may continue up to 2.5 years of age (Herschkowitz et al., 1997; Prins and Hovda, 2003) (Fig. 2). The hippocampus develops perinatally in both rodents and humans. While the majority of hippocampal proper pyramidal cells are generated prenatally, only around 15% of granule cells are present in the rat dentate gyrus at birth (Diamond, 1990), with neurogenesis in this region peaking between gd 14–17 (Rice and Barone, 2000). In comparison, hippocampal neurogenesis peaks around gd 60 in humans (Clancy et al., 2007), with 80% of dentate gyrus granule cells in primates being generated prior to birth (Rakic and Nowakowski, 1981).
Fig. 2.
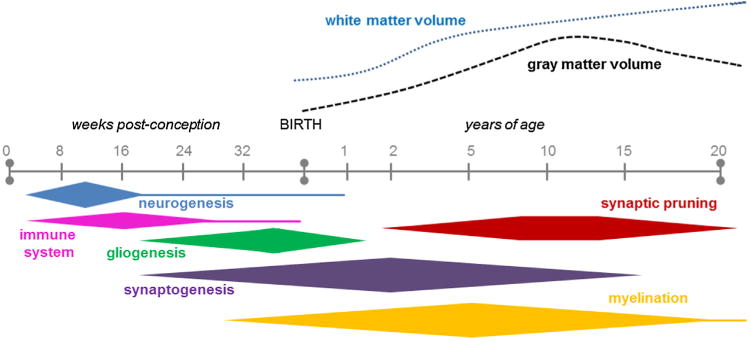
The time course of key neurodevelopmental processes in humans, during gestation and up to 20 years of age (not to scale), with the associated changes in white and gray matter volumes over time. Adapted from Lenroot and Giedd (2006). Note that there may be considerable variability in developmental timing between different cortical and subcortical regions.
By birth in humans, radial glia begin to differentiate into glial fibrillary protein (GFAP)-expressing astrocytes (Kriegstein and Alvarez-Buylla, 2009; Sanai et al., 2011). This transition from neurogenesis to astrogenesis is mediated by several soluble factors including members of the interleukin (IL)-6 cytokine family and bone morphogenic proteins (Miller and Gauthier, 2007). The subventricular zone (SVZ) of the lateral ventricles is another source of new astrocytes and oligodendrocytes during the early postnatal weeks, producing cells which migrate radially towards overlying structures including neocortex (Marshall et al., 2003). Along with the subgranular zone (SGZ) of the hippocampus dentate gyrus, the SVZ is traditionally thought to be responsible for the restricted ongoing neurogenesis in the adult mammalian brain (Eriksson et al., 1998). However, recent findings indicate that the migration of immature neurons along the SVZ peaks early during postnatal development in humans, and is largely depleted after18 months of age (Sanai et al., 2011).
In the rat, astrocytes undergo a rapid period of maturation in the first few postnatal weeks, which involves changes in morphology, connectivity and electrophysiological properties (Zhou et al., 2006). The greatest increase in GFAP-positive cell numbers in the hippocampus has been reported at pnd 11–16, with a further smaller increase thereafter, to reach adult numbers by one month of age (Catalani et al., 2002). Most recently, it has been observed that expression of the metabotropic glutamate receptor mGluR5 by astrocytes is also developmentally regulated, suggesting that signaling between neurons and glia in the immature and adult brains may be fundamentally different (Sun et al., 2013).
Similarly in humans, gliogenesis continues throughout fetal and postnatal periods (Roessmann and Gambetti, 1986). This peak in gliogenesis coincides with the rapid growth of blood vessels, elaboration of dendrites and the establishment of synapses, suggesting a likely coordination between glial, vascular and synaptic growth to ensure that appropriate glial–vasculature and glial–neuronal interactions are instituted (Bautch and James, 2009; Wise and Jones, 1976). Supporting this hypothesis is the observation that synapses form in earnest only after astrocytes appear in the developing brain (Barker and Ullian, 2010). Further, both structurally and by the release of numerous molecules, blood vessels provide a supportive environment in regions of neurogenesis. In the embryonic brain, this ‘vascular niche’ is thought to influence neurogenesis, neuronal migration and neurite extension (Bautch and James, 2009; Stubbs et al., 2009).
After experimental injury to the adult brain, increased neurogenesis is typically stimulated in the SVZ and SGZ, with a proportion of newborn neurons migrating towards the injury site (Parent et al., 2002; Richardson et al., 2007; Urrea et al., 2007) (also see (Kernie and Parent, 2010) for a concise review). The extent to which injury-induced neurogenesis contributes to recovery and neuronal replacement is a field of intense ongoing research. The integration of newly generated neuronsin the hippocampus can be temporally correlated with the recovery of cognitive function after fluid percussion injury in rats (Kleindienst et al., 2005; Sun et al., 2007), which is abolished when these cells are selectively ablated (Blaiss et al., 2011). Interestingly, there are conflicting findings as to whether neurogenesis is increased or decreased after injury to the immature brain, likely dependent on the age, injury mechanisms, location and severity. The SVZ and SGZ may show inherent vulnerability to injury associated with their neurogenic potential and/or their highly vascularized nature during early brain development (Baburamani et al., 2012; Ballabh, 2010). Using a rat cryoinjury model to mimic human brain contusions, a more robust increase in SVZ proliferation and neuroblast production was seen after injury at pnd 6 and 10 compared to pnd 21, suggesting that the age at which the injury occurs considerably affects the regenerative capacity (Covey et al., 2010). Similarly, neurogenesis is impaired in mice after TBI at pnd 21, which show reduced proliferation of SGZ cells and limited precursor cell survival at 6 weeks after injury (Potts et al., 2009). Comparing the impact of HI injury in the mouse brain at pnd 9 and 21, this insult disrupts the growth of the granule cell layer (GCL) in the hippocampus in pnd 9 brains, whereas in pnd 21 brains, where the GCL had reached its full size, the volume was unaffected by HI (Qiu et al., 2007). As expected, in the absence of injury, hippocampal BrdU incorporation and neurogenesis are several-fold higher in the younger (pnd 9) brains compared to at pnd 21. However, the regenerative response to HI is paradoxical–neurogenesis is decreased in the pnd 9 and increased in the pnd 21 injured brain (Qiu et al., 2007) (Fig. 3). In combination, this study suggests that the regenerative capacity of the rodent brain by three weeks of age is reduced to a level equivalent to or even lower than that in adulthood. This inability of the still-developing brain to compensate for neurons lost due to injury, particularly to the hippocampal dentate gyrus, is likely to contribute to the long-term deficits in hippocampal memory formation commonly seen in brain-injured rodents and patients. However, due to technical challenges associated with investigating neurogenesis in the living human brain, it remains to be seen whether similar changes in the neurogenic response occur after injury during human brain development.
Fig. 3.
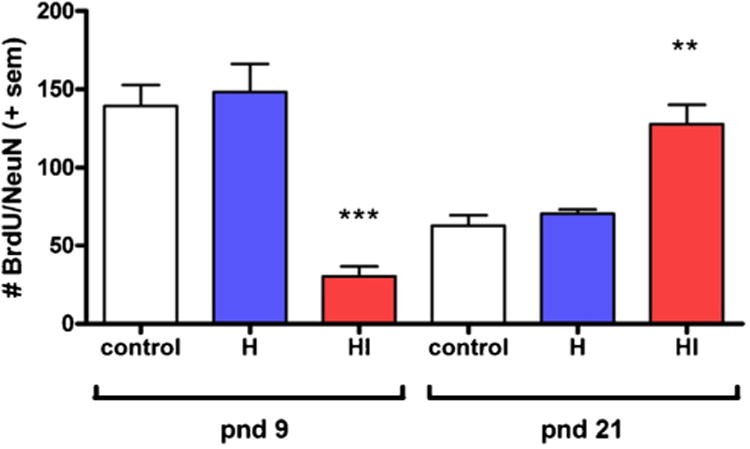
BrdU/NeuN double-labeling to identify newly generated neurons in the granule cell layer of the mouse hippocampus. In the absence of injury (control), neurogenesis is several-fold higher in the younger (pnd 9) brains compared to at pnd 21. However, the response to hypoxia-ischemia (HI) is paradoxical-neurogenesis is decreased after injury at pnd 9, but increased in the pnd 21 injured brain. H: hypoxia only. Asterisks indicate statistical significance compared to age-matched controls. Modified from Qiu et al. (2007).
4. Synaptogenesis and neurotransmission
Synaptogenesis refers to the biochemical and morphological changes which enable the formation of synapses between neurons. Across mammalian species, neurons present at birth undergo a period of overproduction of their arborization and synaptic contacts to increase synaptic density, followed by an elimination or pruning phase of refinement. This activity-dependent pruning of excess synapses is hypothesized to contribute to plasticity and be a mechanism by which the cortical circuitry is refined, to allow for more efficient processing of adult cognition. It is a fundamental developmental strategy, common to most regions of the mammalian CNS including humans, primates and rodents (Andersen, 2003).
Synaptic formation in the developing human brain was first investigated by Huttenlocher in 1979 who demonstrated that synapse density differs with age and brain region. While the proliferation of synapses begins around 20 weeks of gestation, density increases rapidly after birth, particularly within the early postnatal months, to reach a level approximately 50% higher than that seen in adults by 2 years of age (Herschkowitz et al., 1997; Huttenlocher, 1979) (Fig. 2). The timing of this synapse proliferation is region-dependent; for example, synaptic density peaks in the primary visual cortex as earlyas8–12 months of age, compared to 2–4 years of age in the prefrontal cortex (Huttenlocher et al., 1982; Lenroot and Giedd, 2006).
In rodents, the critical period of synaptogenesis occurs during the first three postnatal weeks of life, peaking during week 2. In the molecular layer of the rat dentate gyrus, synapse numbers prior to pnd 4 are less than 1% of adult levels, however a subsequent growth spurt results in complete synapse numbers by pnd 25 (Crain et al., 1973). This synaptogenesis coincides with robust astrogenesis, and is likely supported by the early astrocytic release of synapse-forming factors such as thrombospondins 1 and 2 (Christopherson et al., 2005). Synaptic density in the rat and mouse somatosensory cortex is low in the first postnatal week, followed by an abrupt increase beginning at pnd 10, to reach equivalence with adult numbers by pnd 30 (De Felipe et al., 1997; Micheva and Beaulieu, 1996; Rice and Barone, 2000). In situ hybridization in the rat brain has also demonstrated that N-Methyl-D-aspartic acid (NMDA) subunit expression increases dramatically from birth, peaking at approximately pnd 20 in rat hippocampus and cortex (Zhong et al., 1995). This increase in synaptic density is correlated with an increase in synaptic responses and dramatic changes in the functional properties of neurons, such as an increase in the peak amplitude of action potentials (Zhang, 2006). While the entire process appears to be completed in the rat brain by 3–4 weeks of age, synaptic elimination and remodeling in humans is more prolonged and continues well into adolescence (Huttenlocher, 1979; Petanjek et al., 2011; Uylings and van Eden, 1990). Between the ages of 2 and 7 years, neuronal density in layer III of the prefrontal cortex decreases from 55% to approximately 10% above adult levels (Huttenlocher, 1979). During later childhood (7–15 years of age), synaptic density in the frontal cortex decreases by approximately 40% (Lidow et al., 1991). In parallel, reactivity for the presynaptic marker synaptophysin increases in the human brain from age 5 to peak in late childhood, before decreasing to adult levels by mid-adolescence (∼16 years) (Glantz et al., 2007). These synaptic changes occur in the absence of any significant neuronal loss, and likely reflect the refinement and maturation of neural circuitries during childhood and early adolescence.
In addition to changes in synaptic density, age-dependent changes in many neurotransmitter systems have been reported (Herschkowitz et al., 1997). NMDA receptor density in the rat temporal cortex increases gradually from pnd 1 to peak at adult levels by pnd 28, while in the human frontal cortex, NMDA receptor binding follows a similar developmental trajectory to peak between 1 and 2 years of age (McDonald and Johnston, 1990). In the rat cortex, post-synaptic glutamate receptors increase rapidly from pnd 5 to pnd 20 (50% of adult levels), followed by a continual increase to plateau by pnd 40–50 (Sanderson and Murphy, 1982). As glutamate receptors have been implicated in the modulation of cell death after perinatal brain injury (Lea and Faden, 2001), it is likely that age-specific vulnerability is somewhat dependent on the developmental expression of these neurotransmitter networks. 5-hydroxytryptamine (5-HT, or serotonin) increases in the human brain during the first two years of life, before declining to adult levels by the age of five (Hedner et al., 1986). In mice, cortical 5-HT rises to double the adult level during the first postnatal week, coinciding with an up-regulation of receptor expression and function, which is hypothesized to be involved in the establishment of physiological functions such as sleep, appetite, learning and memory (Zhang, 2006). Romijn et al. (1991) sought to determine markers of ‘functional comparability’ between rats and humans, as determined by the levels of neurotransmitters and their synthesizing enzymes. For example, these investigators calculated that levels of the key GABA-synthesizing enzyme, glutamate decarboxylase at 40 weeks gestation in humans was 16.2% of the adult human level. A similar calculation to determine 16.2% of the adult rat levels corresponds to 7.4 days of age in the rat. In contrast, the relative levels of the choline acetyltransferase enzyme at birth compared with adulthood in humans was strikingly later, at pnd 20.4 in the rat cortex (Romijn et al., 1991). Collectively, these studies highlight some of the disparities between different developmental milestones, and serve as a reminder that equating ages based upon a single milestone or event may lead to misinterpretation.
Another fundamental difference between the immature and adult brain is a maturation shift in the actions of GABA signaling. Activation of the GABAa receptor in immature neurons triggers depolarization and excitation, compared to the classic inhibitory role of this system in the adult brain (Ben-Ari et al., 2012). Excitatory GABA in the immature brain is thought to play a crucial role in many developmental processes including neuronal differentiation and dendritic arborization (Ben-Ari et al., 2012; Tyzio et al., 2007). Underlying this functional change is the revelation that the concentration of intracellular chloride is intrinsically higher in the developing brain compared to at adulthood, attributed to developmentally regulated expression of specific chloride transporter molecules including NKCC1 and KCC2, which import and export chloride, respectively (Blaesse et al., 2009). Expression of NKCC1 is high during embryonic and early postnatal life, while KCC2 activity is minimal, resulting in the accumulation of chloride in immature neurons. KCC2 expression increases towards the end of the second postnatal week in the rat cortex (Ben-Ari et al., 2007; Tyzio et al., 2007), and at about 40 weeks post-conception in the human cortex (Dzhalaetal., 2005), in parallel with a decrease in spontaneous transients indicative of the ongoing maturation of neuronal connectivity (Vanhatalo et al., 2005).
Interference with neuronal function and excitability by brain injury in adults can lead to post-traumatic epilepsy, at a reported frequency between 4 and 53% (Frey, 2003). Developmental HI injury has likewise been implicated in epileptogenesis. There are several lines of evidence to support this position. HI induced in pnd 7 rats results in epilepticform activity and mossy fiber sprouting in the injured hippocampus (Williams et al., 2004). Moreover, a recent study revealed a dramatic decrease in postsynaptic densities associated with glutamatergic axons and oligodendrocyte precursor cell synapses after HI in pnd 6 mice, suggesting that these newly formed synapses are highly vulnerable to white matter injury in the developing brain (Shen et al., 2012). Neurotransmitter changes during early postnatal development, coupled with the high number of glutamatergic synapses during the peak of synaptogenesis, renders the immature brain inherently more prone to seizure activity compared to an older brain. Further, it has been proposed that GABA-mediated depolarization of neurons during brain injury to the immature brain may facilitate neuronal excitability underlying the age-dependent susceptibility to acute seizures after HI and TBI (Ben-Ari et al., 2012; Dzhala et al., 2012).
The etiology, mechanisms and occurrence of epilepsy after TBI in children are also poorly understood. One retrospective study calculated an 11% incidence of developing post-traumatic epilepsy within 10 years in a Swedish pediatric population (Emanuelson and Uvebrant, 2009). In the adult rodent brain, post-traumatic epileptic seizures have been associated with an impairment of hippocampal synaptic plasticity and disruption in the hippocampal circuitry (Hunt et al., 2010; Zhang et al., 2011). In the postnatal immature brain, it is likely that such disruption may have additional consequences by interfering during critical periods of synaptic formation and pruning (Fig. 4). The development of a pediatric post-traumatic epilepsy model, in which electroencephalographic epileptic activity and behavioral seizures are seen in >85% of rats after controlled cortical impact model at pnd 17, may elucidate some of the underlying mechanisms (Statler et al., 2009).
Fig. 4.
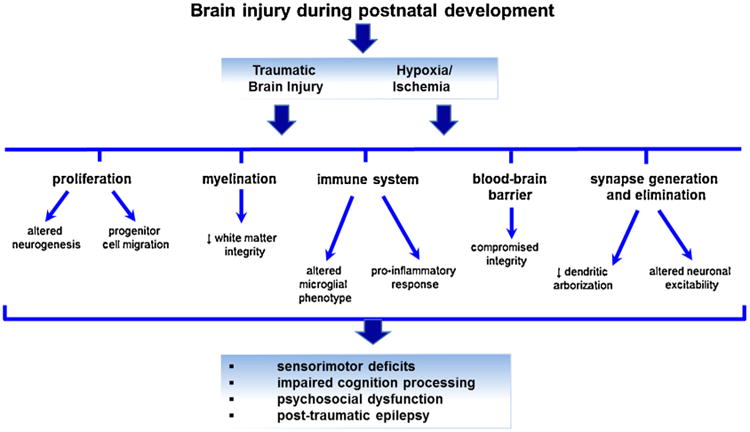
Traumatic or hypoxic-ischemic injury to the early postnatal brain can impact many key neurodevelopmental processes which are undergoing maturation changes during this time, resulting in functional consequences including sensorimotor, psychosocial and cognitive deficits.
5. Myelination
The formation of myelin sheaths, generated by interfascicular oligodendrocytes in the CNS, is essential for the propagation and speed of neurotransmission in the mammalian brain. It is now generally accepted that myelination is a prolonged process which continues well into childhood and adolescence in specific brain regions (Fig. 2). An increased understanding of oligodendrocyte maturation stages in the normally developing human and rodent brain over the past decade has also highlighted the vulnerability of white matter to brain injury. Technological advances including refinement of conventional MRI as well as diffusion tensor imaging have stimulated a recent resurgence of clinical research into white matter development, as well as a shift towards longitudinal within-subject design rather than traditional cross-sectional studies (Lebel and Beaulieu, 2011).
Oligodendrocyte development begins in utero in humans. Preoligodendrocytes (‘Pre-OLs,’ defined as non-myelinating, mitotically active oligodendrocyte precursors) are the dominant cell type between 18 and 28 weeks post-conception, with a shift towards ‘immature OLs’ (post-mitotic, myelinating) being most prevalent by 28–40 weeks (Craig et al., 2003; Dean et al., 2011a). Injury to the immature periventricular white matter, either in premature infants or in utero in infants who are later born at term, appears to be the most common cause of cerebral palsy (Volpe, 2003). White matter is more vulnerable than gray matter in the premature infant, and the period of greatest vulnerability is between 23 and 32 weeks gestation. Based on clinical studies, the most common causes of white matter lesions are HIE or infections, or a combination of the two (Hagberg et al., 2002b). White matter is also vulnerable to injury in term infants, older infants and children, but the topography of injury and pathogenesis differ from periventricular white-matter injury associated with prematurity (Cowan et al., 2003). Back and colleagues have examined oligodendrocyte maturation by immunofluorescence labeling in young rats and mice, estimating that white matter development and axonal outgrowth in the rodent CNS at pnd 1–3 corresponds to 23–32 weeks gestation in human infants, while pnd 7 is analogous to 32–36 weeks (Craig et al., 2003) (Table 1). Based on this reasoning, a pnd 7 rodent thus represents a preterm infant—as such, some researchers now consider rodents at pnd 10 to be more comparable to a term infant, at least in terms of white matter development (Hagberg et al., 2002a). Myelination is well underway in rodents by pnd 10–14 and peaks at approximately pnd 20, when mature OL markers including myelin basic protein are detectable (Wiggins, 1986).
Myelination, as with synaptogenesis, occurs in a conserved and region-dependent pattern, with synthesis beginning in the peripheral nervous system, brain stem and spinal cord (Inder and Huppi, 2000). In the brain, myelination generally proceeds from inferior to superior and posterior to anterior, commencing in the occipital lobe followed by the temporal and frontal lobes (Tasker, 2006; Volpe, 2000). These changes in myelination are readily identifiable in vivo by MR imaging: in particular, T1 and T2 weighted images that highlight fat (myelin) and gray matter, respectively. As the human brain matures, increasing myelination is accompanied by a decrease in brain water content, resulting in an inversion of contrast such that the T1-weighted signal becomes stronger (Lodygensky et al., 2010). This is particularly evident by 28 weeks gestation in humans, a time corresponding with the above-mentioned shift from pre-OLs to immature OLs, the latter being able to produce myelin.
Myelination in humans was conventionally believed to be essentially complete by 3–5 years of age (Dietrich et al., 1988; Nakagawa et al., 1998). Although most major tracts are significantly myelinated by early childhood, it is now well recognized that axons continue to myelinate into the second and third decades of life. This ongoing myelination likely contributes to the linear increase in total white matter volume, which increases by 12% between 4 and 22 years of age (Giedd et al., 1999). However, other factors including axonal diameter, axonal packing and axonal pruning are also likely to contribute to overall changes in white matter during development. A recent study which scanned the brains of twins at 9 and again at 12 years of age demonstrated increases in white matter volume, surface area, fiber bundle expansion and fractional anisotropy across the three-year period, with a significant influence of inheritability on such changes (Brouwer et al., 2012). Fractional anisotropy is considered to be a measure of microstructural organization and directionality, or an indirect indicator of myelination and axonal diameter. In general, fractional anisotropy increases with advancing age and increasing myelination in the cortex, while the mean diffusivity, a measure of water diffusion, tends to decrease.
These ongoing changes in white matter in the human brain are region-specific in both their timing and reported effects on cognitive development. Between 8 and 18 years of age, increased fractional anisotropy in the left frontal lobe was positively correlated with the development of working memory capacity, while an increase in the left temporal lobe was associated with improvements in reading ability (Nagy et al., 2004). Several recent studies employing diffusion-weighted imaging found that the improvement in spatial working memory performance through late childhood and early adolescence was associated with ongoing white matter microstructural changes within fronto-parietal brain regions, including the superior longitudinal fasciculus, the white matter underlying the dorsolateral prefrontal cortex (Østby et al., 2011; Vestergaard et al., 2011). The rate of myelination in the corpus callosum is highest during childhood and declines thereafter, but continues well past an individual's 20th year (Keshavan et al., 2002). Further, different regions of the corpus callosum appear to develop at different rates, with the genu and splenium showing peaks in fractional anisotropy earlier than the callosum body (Lebel et al., 2012) (Fig. 1). These changes are speculated to relate to faster and more effective communication between brain regions, and reflect the increasing cognitive capacity seen during adolescence (Lenroot and Giedd, 2006). In general, maturation of commissural and projection fibers (e.g. corpus callosum and corticospinal tract) occurs the earliest, while association fibers (e.g. the inferior and superior longitudinal fasciculi and cingulum) continue maturing at later ages (Lebel et al., 2012). Frontal-temporal connections demonstrate the most prolonged development, well past the twentieth year of life (Lebel and Beaulieu, 2011).
In both rats and mice, DTI has demonstrated the continuation of fiber maturation in the corpus callosum as well as the internal and external capsules until at least pnd 30–40, consistent with the known timing of myelination in rodents (Baloch et al., 2009; Bockhorst et al., 2008). Two independent studies in C57Bl/6 mice reported that the fractional anisotropy in the corpus callosum genu and splenium reached a plateau at pnd 40–45, the latest times examined in their respective studies (Baloch et al., 2009; Chahboune et al., 2007). In contrast, little change was seen in the somatosensory cortex across time between pnd 15 and 45, consistent with previous findings that this region matures earlier and remains stable from approximately pnd 10 onwards (Chahboune et al., 2007). One notable weakness of the rodent imaging studies performed to date is a lack of data from pnd 60 onwards, which would provide insight into the ongoing maturation of white matter structures throughout adulthood compared to what is known in humans.
There has been considerable attention afforded to mechanisms by which injury to the pre- and perinatal brain has differential consequences on white matter development, primarily depending on the developmental stage of oligodendrocytes. White matter in humans prior to 32 weeks gestation, thought to be equivalent to rats at pnd 1–3, is particularly sensitive to damage from HI and metabolic insults, and is likely to result from the specific vulnerability of immature OLs to oxidative stress, NMDA receptor over-stimulation, and the presence of free radicals (Back et al., 2001; Huang et al., 2009a; Johnston, 1995). Disruption of myelination after prenatal brain injury may reflect a depletion of the pre-OL pool required to generate mature myelinating oligodendrocytes (Dean et al., 2011b; Segovia et al., 2008).
Less understood is how TBI to the perinatal and postnatal human brain impacts the development of myelination and its integrity. TBI can cause white matter damage either directly via focal hemorrhagic or non-hemorrhagic lesions, or by diffuse injury resulting in axonal shearing (Tasker, 2006). Diffuse axonal injury is a common finding after TBI, however its contribution to neurobehavioral impairments is unclear, as is the relationship between myelin damage as compared to axonal damage. A study of adult patients with mild, moderate or severe TBI suggests that all severities can result in a degree of axonal damage, with irreversible myelin damage being more apparent with more severe injuries, and predictive of greater cognitive impairments (Kraus et al., 2007). Impaired cognitive processing speeds in brain-injured adults are strongly dependent upon the structural integrity of white matter tracts associated with parietal and temporal cortices in particular (Turken et al., 2008) (Fig. 4). In children, white matter integrity remains abnormal for at least 12 months after a moderate-to-severe TBI, with reduced fractional anisotropy as compared to children with orthopedic injuries only in the corpus callosum genu, posterior limb of the internal capsule, superior longitudinal fasciculus and superior fronto-occipital fasciculus (Wozniak et al., 2007; Yuan et al., 2007). Levin and colleagues further demonstrated that this reduction in fractional anisotropy, evident by DTI in the corpus callosum of children at least one year post-injury, reflects a decrease in axonal fiber density and myelination (Wilde et al., 2006). Further, a higher fractional anisotropy is related to increased cognitive processing speeds and better functional outcomes, indicating ongoing disruption of brain connectivity (Wilde et al., 2006). This finding is in agreement with earlier studies of children and adolescents at least 6 years after injury in which functional measures were found to be dependent on the extent of corpus callosum atrophy (Verger et al., 2001). Further, neuropsychological outcomes have been correlated with atrophy specifically in posterior corpus callosal regions after pediatric head injury (Ewing-Cobbs et al., 2008). Such changes have been modeled in young rats, in which an earlier developmental age at injury (pnd 1) results in greater corpus callosum loss and poorer neurobehavioral outcomes compared to injury at a slightly later age (pnd 5) (Threlkeld et al., 2007). White matter atrophy after adult brain injury may be attributed to acute tissue loss or delayed secondary de-afferentation; in children, however, this scenario is super-imposed upon a still-developing system, such that atrophy may also reflect a failure in growth or loss of growth potential (Tasker, 2006).
6. Innate and adaptive immunity
The immune system can have a profound effect on brain development, function and behavior, including modulation of brain activities such as temperature, sleep patterns and feeding behaviors (Steinman, 2004). On a global level, it has been suggested that the worldwide distribution of cognitive ability is determined in part by variation in the intensity of infectious diseases (Eppig et al., 2010). Of particular relevance to the developing brain, it has recently been demonstrated that microglia play a crucial role in normal postnatal synaptic pruning via complement signaling (Schafer et al., 2012). Further, members of the tumor necrosis factor receptor super family have been implicated in the regulation of neuronal progenitor proliferation, differentiation and patterning (Saliba and Henrot, 2001; Twohig et al., 2011). These findings highlight the complex interplay between the CNS and immune response, which is likely to influence both normal brain development as well as the brain's response to injury (Bilbo and Schwarz, 2012).
Development of the immune system differs in rodents and humans, as does the systems' response to injury at different ages. This is exemplified in studies involving thymectomies and interleukins. In humans, the immune system is predominantly established by birth, such that thymectomies within the first few postnatal days are tolerated (Holsapple et al., 2003). In contrast, few B and T cells are present in rodents at birth (Marshall-Clarke et al., 2000), prior to an increase at 2–3 weeks of age corresponding with the onset of antigen responsiveness (Spear et al., 1973). Thus, an early postnatal thymectomy (<36 h) in mice causes a failure in T cell development and leads to wasting syndrome. By pnd 21, B cell numbers in rodents are comparable to adult levels, however T cells are still reduced (Ladics et al., 2000). In humans, neonates show reduced levels of IL-12 and interferon (IFN)γ, whereas circulating levels of IL-1β, IL-6, IL-23 and IL-10 are increased compared to in adults, suggesting that the infant innate immune system is armed for protection against extracellular bacterial pathogens as opposed to intracellular pathogens and viruses (Prendergast et al., 2012). A comprehensive inter-species comparison of pre- and perinatal immune system development can be found elsewhere (Holsapple et al., 2003).
Microglia, the resident immuno-competent cells of the CNS, begin to colonize the mouse brain around embryonic day 9.5, or slightly later in the rat brain (Bilbo and Schwarz, 2012). Derived from progenitor cells in the yolk sac, the early rat microglial population proliferates rapidly and begins to express characteristic myeloid markers including CD11b, F4/80 and the fractalkine receptor, CX3CR1, by approximately gd 15–16 (Ginhoux et al., 2010). A 20-fold increase in microglial cell numbers occurs after birth, between pnd 0–11 (Alliot et al., 1999). Cells typically become more ramified as they fully differentiate, to exhibit a mature resting phenotype in the rat brain by pnd 30. Thus microglia continue to undergo morphological alterations across development in a region-specific fashion, suggestive of ongoing maturational changes (Schwarz and Bilbo, 2012). In humans, the first macrophage-like cells appear around gestational week 4–6, with pronounced accumulation of short-processed amoeboid cells observed by 13–24 weeks (Hutchins et al., 1990; Male and Rezaie, 2001). As in rodents, fetal microglia typically localize to highly vascularized regions in the brain, and begin to take on a more mature phenotype at a time corresponding with neuronal development, suggesting that maturation of neurons may play a role in maintaining microglia in a surveillance phenotype (Harry and Kraft, 2012).
Several chemokines are up-regulated in the rat cortex and hippocampus at birth compared to in the adult brain, including CCL2 (monocyte chemoattractant protein-1, MCP-1), CCL3, CCL6, CCL7, CCL12 and CXCL6. These chemokines are hypothesized to play a role in attracting immature microglia into the brain during early development (Bilbo and Schwarz, 2012; Schwarz et al., 2012). Further, basal levels of many cytokines are significantly higher at birth compared to in the brains of adult rats, such as IL-1β which is almost six fold higher in the newborn hippocampus (Schwarz et al., 2012). The nature of an immune response to a specific injury can therefore be entirely different in the immature versus the adult rodent brain. For example, when adult rat brains are subjected to ionizing radiation, IL-6 levels increase, microglia adopt an activated (ED1+) phenotype and remain ED1+ for an extended period of time (Monje et al., 2003). When immature (pnd 9) rat brains are irradiated, IL-6 levels decrease (Kalm et al., 2009a), microglia are already ED1+ in control brains (a non-toxic phenotype) and this is not altered by the injury (Kalm et al., 2009b). Unlike the chronic inflammation observed in the adult brain (Monje et al., 2003), inflammation in the pnd 9 rat brain is transient (Kalm et al., 2009a).
The innate immune response to injury shows age-related differences, with the immature human and rodent brain responding differently relative to adults both in terms of cytokine production and inflammatory cell infiltration. For example, neutrophil infiltration is more prominent and protracted in the mouse brain subjected to TBI at pnd 21 compared to the adult brain (Claus et al., 2010). This observation is consistent with others who have also demonstrated age-related differences in how the brain responds to inflammatory stimuli. Injection of IL-1β into the parenchyma of pnd 21 rats results in pronounced neutrophil recruitment, elevated chemokine production, reduced expression of the tight junction protein claudin-1, and increased blood-brain barrier disruption, which is not seen in similarly-treated adults (Anthony et al., 1997, 1998; Campbell et al., 2007; Lawson and Perry, 1995). Such evidence has led to the hypothesis that there is a ‘window of susceptibility’ during which the developing brain is more susceptible to inflammatory stimuli than the adult brain (Fig. 4).
While rodent models provide avenues for studying the innate immunity in the periphery as well as in the CNS, studies of pediatric brain-injured patients are more limited in scope. Systemically, the immune response of human neonates differs from the adult, with a bias towards Th2 responses as well as higher productionofIL-1 and IL-10 from peripheral blood monocytes after stimulation with pathogens (Levy, 2007). After TBI, several cytokines have been detected in the cerebrospinal fluid and serum at elevated levels in pediatric patients, including IL-1α, IL-6, IL-12, TNFα, MIP-1α and IL-8 (Amick et al., 2001; Berger et al., 2009; Buttram et al., 2007; Whalen et al., 2000). Elevated IL-1β has also been detected after injury in young brain-injured patients, however at much lower levels than those previously published in adults, suggesting a differential response (Giza et al., 2007). Whether such distinctions are beneficial or detrimental to the young injured brain remains to be determined. Post-injury up-regulation of IL-6 and nerve growth factor, for example, has been associated with better neurological outcomes after childhood TBI, which may reflect an endogenous attempt at neuroprotection (Chiaretti et al., 2008).
7. Blood-brain barrier development
Historically, barrier mechanisms in the neonatal brain were commonly considered to be immature and leaky. It is now known that the blood-brain barrier is established and functional during embryogenesis in both rodents and humans, and is tightly regulated by pericyte-endothelial cell interactions (Daneman et al., 2010; Saunders et al., 2012). The blood-brain barrier and choroid plexus of the developing mammalian brain contain some features which are not seen later during adulthood, such as a specialized system of tubulo-endoplasmic reticulum which transports proteins directly across the epithelial cells and likely contributes to the higher concentrations of plasma-derived proteins found in the cerebrospinal fluid of neonates (Saunders et al., 2012). Tight junctions in cerebral vessels and the choroid plexus are present very early in embryonic development, reportedly as soon as endothelial cells begin to differentiate, and are sufficiently formed to exclude proteins from the brain by birth (Engelhardt, 2003; Kniesel et al., 1996). However, capillaries within the rat cortex do not exhibit an adult-like appearance until pnd 14–21 (Cornford and Cornford, 1986), showing ongoing structural and molecular organization with increasing coverage of vessels with astrocytic end-feet across the first three postnatal weeks (Xu and Ling, 1994). In parallel, the rate of iron transport into the brain appears to be developmentally regulated, with iron transportation peaking in the rat within the first few weeks of life (Moos and Morgan, 2002). Despite this, free iron is detectable in the plasma of normal human neonates, suggesting that the capacity for iron transport is saturated (Berger et al., 1995). This may underlie the increased magnitude of iron accumulation in the immature injured brain compared to adults, which can have a potentially greater deleterious effect relative to in the adult brain due to reduced antioxidant defenses in the developing brain (Potts et al., 2006). Rodent studies of both HIE and early TBI have demonstrated blood-brain barrier disruption as a consequence (Baburamani et al., 2012; Pop and Badaut, 2011). The barrier was reportedly more vulnerable to a HI insult in rats at pnd 7 compared to pnd 21 (Muramatsu et al., 1997), suggesting an age-dependent susceptibility. In line with this finding, permeability of the blood-brain barrier to proteins is transiently increased after systemic inflammation in the early postnatal rat (pnd 0 and 8), but not at a later stage of development (pnd 20) (Stolp et al., 2005). In contrast, recent evidence of differential gene expression by endothelial cells after neonatal ischemic stroke compared to in adults, coinciding with greater preservation of tight junction proteins and reduced albumin leakage after injury in the pnd 7 rat brain (Fernández-López et al., 2012), highlights our incomplete understanding of age-dependent responses of the blood-brain barrier to injury.
8. Age-dependent behavioral phenotypes
The species comparisons presented here thus far have been based on anatomical, molecular and biochemical changes across brain development. Equally important are age-dependent behaviors which can be assessed when modeling human brain injury in rodents. Although it is unrealistic to explicitly link biological processes of CNS development with the behavioral capacities they control, certain behaviors can in fact be temporally correlated with the maturation of specific neuronal regions or processes. Several lines of evidence support this position in both humans and rodents. In humans, an increase in cortical inhibition in the brain stem at 2– 3 months of age corresponds with a decrease in spontaneous crying and the disappearance of primary neonatal reflexes such as the palmer grasp reflex (Herschkowitz et al., 1997). The first establishment of hippocampal-dependent recognition memory in infants at 8 weeks of age occurs around the same time as an increase in mossy cells in the hippocampal dentate gyrus (Seress and Mrzljak, 1992). Further, an increase in synaptic density and dendritic arborization at approximately 7–10 months of age coincides with a rapid improvement in working memory, or the ability to retrieve recently acquired memories and use this knowledge appropriately (Herschkowitz et al., 1997). It is possible to map the age at which certain behaviors occur in the human with that of the rodent. Adult-like locomotor function is achieved in humans by 3–4 years, compared to pnd 15–16 in rats (Wood et al., 2003). The visual system of humans is functional by birth but undergoes considerable postnatal development, achieving 20/20 visual acuity by 4–6 months of age, and ongoing maturation of contrast sensitivity for several years after birth. In contrast, rodents are born with their eyes closed, and their eyelids do not open until pnd 10-15. Despite this distinction, phototactic responses can be observed in rats as early as pnd 5, and visual evoked potentials by pnd 11, suggesting that their visual system might be rapidly developing at a roughly equivalent rate to species born with their eyes open (Wood et al., 2003).
During childhood and adolescence, while it is generally understood that changes in structural organization and cognitive maturation occurs concurrently, the links between ongoing brain development and emerging functions are less defined. Working memory mediated by the prefrontal cortex increases dramatically between the ages of 4 and 7 years, and performance on tasks requiring frontally mediated inhibitory control show substantial improvement between 5 and 11 years of age (Tsujimoto, 2008). These advancements in cognition likely result from the functional maturation of the brain at this time, including the aforementioned synaptic pruning and ongoing myelination. Further, this period of cognitive development coincides with the prefrontal cortex dissociating or ‘fractionating’ into focused, functionally specialized networks associated with specific processes (Tsujimoto, 2008). The extent to which the rodent brain parallels this timing of specialization remains unclear.
In rodents, the timingof ‘childhood’ has been best characterized by an increase in socialization following weaning at approximately pnd 21. Behaviors guided by social cognition change dramatically across childhood and adolescence and are paralleled by functional reorganization of relevant brain regions including the medial prefrontal cortex, anterior cingulate cortex, amygdala and anterior insula (Blakemore, 2008). Spontaneous social play behavior including mutual investigation and wrestling is common between young rodents, and is likely to be an important precursor to normal social functioning during adulthood (Wills et al., 1983). Several investigators have designated pnd 15–25 as the ‘socialization period’ during which such amicable and playful behaviors are particularly prevalent (Terranova and Laviola, 2005). Sexual maturity in rats and mice is reached between pnd 35–56, depending on the sex and strain (Lambert, 2009), which triggers a shift towards more aggressive and mate-seeking orientated adult-type behaviors. In humans, children begin to imitate and pretend play at approximately 2.5 years of age, and begin interacting socially by about 4 years, with an increase across subsequent childhood and adolescence periods (Wood et al., 2003) (Table 2).
Table 2.
Summary of key behavioral phenotypes which emerge across comparable ages in humans and rodents.
| Human | Rodent | Behavioral phenotype | References |
|---|---|---|---|
| 2–3 year old | pnd 20–21 | Increased activity levels and sociability. Weaning. | Wills et al. (1983), Wood et al. (2003), Terranova and Laviola (2005), Blakemore (2008) |
| Increasing working memory. | Herschkowitz et al. (1997) | ||
| 4–11 year old | pnd 25–35 | Increased sociability. | Terranova and Laviola (2005) |
| Further development of working memory and inhibitory control. | Tsujimoto (2008) | ||
| 12–18 year old | pnd 35–49 | Adolescent-type behaviors including sociability, risk-taking, impulsivity. | Spear (2000), Sturman and Muoghaddam (2011) |
| Onset of sexual maturity. | Lambert (2009) | ||
| Increased cognitive control capacities. | Laviola et al. (2003), Raznahan et al. (2011) | ||
| >20 years | pnd >60 | Emergence of adult-type behaviors including reduced risk-taking, reduced impulsivity and increased parental tendencies. | Laviola et al. (2003) |
Behavioral changes which characterize the period of adolescence include an increase in social interactions, novelty-seeking, risk-taking tendencies, emotional instability and impulsivity (Sturman and Muoghaddam, 2011). Here, we define ‘adolescence’ as the transitional phase during which an individual develops from a child into an independent adult, coinciding with the hormonal changes associated with the onset of puberty. In the clinical setting, adolescence has been described as 9–18 years, 10–20 years, and even up to 25 years of age (Spear, 2000). In rodents, the terms ‘adolescence’ and ‘juvenile’ have been used to cover the whole time span from weaning at pnd 21 to adulthood at pnd 60 (Babikian et al., 2010; Spear, 2000). Narrower time windows, between pnd 30–45 or pnd 35–49, are more commonly employed as standard descriptions of adolescence or ‘periadolescence,’ being a period when mice exhibit increased locomotor and explorative activity (Laviola et al., 2003; Spear and Brake, 1983). Adolescence in both rodents and humans is also a period of ongoing refinement and maturation of neural circuitry, which continues through to adulthood. The dorsolateral prefrontal cortex is required for many cognitive control mechanisms, and circuitry changes in this region during adolescence are thought to underlie the characteristic tendencies for heightened risk-taking and impulsivity (Laviola et al., 2003; Raznahan et al., 2011). Further, changes in sleep behaviors including a shift in timing and reduction of slow wave sleep (particularly delta electroencephalography activity) in adolescence have been associated with synaptic pruning which is ongoing throughout this period (Colrain and Baker, 2011; Feinberg and Campbell, 2010).
As with many aforementioned indices of age-dependent vulnerability to ischemic and traumatic brain injuries, the age at the time of injury determines the extent of impact on functional outcomes. Behavioral consequences after brain injury have been extensively reported in children and adolescents, as in adults. Developmental HI can yield long-term functional and behavioral impairments, including developmental delays, motor deficits and reduced cognitive capacity which may persist across an individual's lifetime (Johnston and Hoon, 2006; van Handel et al., 2007). Ranging from hyperactivity and attention deficiencies to problems with cognition, executive function, learning and memory, TBI at a younger age is also associated with a poorer neurobehavioral outcomes, reflecting a heightened vulnerability of the immature brain (Anderson and Moore, 1995; Anderson et al., 2009; Catroppa et al., 2008; Levin et al., 2004; Wells et al., 2009).
Rodent models of HI and TBI have further highlighted the emergence of age-dependent behaviors. In HI models utilizing rats at pnd 3 or 7, sensorimotor function and performance in the Morris water maze are impaired by adulthood (Huang et al., 2009b; Ou-Yang et al., 2012), as well as cognitive ability by a reversal-learning task even after a mild HI insult (van der Kooij et al., 2010). In a separate study which compared varying lengths of HI induced at pnd 10, similarly poor performance in the Morris water maze was attributed to persistent deficits in both cued and spatial learning (McAuliffe et al., 2006). Comparable findings have been demonstrated in rodents after experimental TBI. Immature rodents are proficient at the Morris water maze by pnd 21–23, a task commonly used to assess hippocampally mediated spatial memory and place navigation (Bachevalier and Beauregard, 1993) Contusive brain injury at pnd 11 and pnd 17 in rats results in marked spatial learning deficits when tested one month later, with greater memory retention deficits reported in animals injured at the younger age (Huh and Raghupathi, 2007). In contrast, brain-injured pnd 17 rats are remarkably resistant to impairments in the Morris water maze compared to rats injured at pnd 28 or adulthood (Prins and Hovda, 1998), although this interpretation may be confounded by the onset of testing differing between the age groups (10 days post-injury for rats injured at pnd 17, compared to 24 h post-injury for pnd 28 rats). Mice subjected to TBI at pnd 21 show hyperactivity coupled with deficits in memory, cognition and social interactions upon reaching adulthood (Pullela et al., 2006; Semple et al., 2012). Injury induced in pnd 17 rats similarly manifests emerging behavioral problems including anxiety and motor deficits at adulthood (Ajao et al., 2012). Together, these findings are consistent with the hypothesis that the full extent of behavioral deficits may not become evident until maturity (Barker et al., 2010; Eslinger et al., 1992).
9. Future research
While there is a strong foundation for the use of age-specific rodent models to study injury to the developing human brain, there remain areas of research that require further elucidation. With the exception of neuroanatomical changes measured by MRI, there is a notable paucity of detailed mouse studies in many aspects of brain maturation and developmental behaviors, particularly in terms of neuronal proliferation and synaptogenesis. Findings in rats are commonly presumed to apply equally across all rodent species and strains; however this may not always be the case (Wang et al., 2011). Caution should be noted when extrapolating studies performed in the immature rat into the mouse, and across different strains, as such comparisons may not always be valid. Regardless of the strain and experimental design, the selection of appropriately aged animals will depend on the outcome or outcomes being measured, with an increasing number of variables requiring a more generalized age comparisons across several parameters.
There remain many gaps in our understanding of brain development in rodents and humans, despite the fact that these are the species that have been most thoroughly evaluated to date. Although less studied, there are advantages to working with larger animal models in which brain structures including white matter content more accurately mimics the human condition. To that end, sheep, cats, rabbits and swine have been utilized as models of perinatal and postnatal brain injury (Duhaime, 2006; Finnie, 2012). Biochemical, anatomical and behavioral characterizations in larger animal models to date are very limited in scope. However, there are notable advances using these models in terms of MR imaging (Conrad et al., 2012; Dean et al., 2013; Drobyshevsky et al., 2012; Winter et al., 2011), defining the magnitude and time course of injury responses (Duhaime et al., 2003, 2000) and white matter vulnerability (Back et al., 2012; Dean et al., 2013). Further, the emergence of species-appropriate behavioral paradigms aim to bridge the gap between large animal and human studies (Friess et al., 2007; Sullivan et al., 2012).
Finally, there are several complementary areas of exploration that will benefit our ability to model the developing human brain. Systemic elements including age-specific vascular function, blood pressure and intracranial pressure changes are likely to influence the immature brain's response to injury (Prins et al., 1996). Physiological and biomechanical properties (e.g. brain water content, skull elasticity), choice of anesthesia, and appropriate methods for the evaluation of functional recovery (taking into account age-appropriate behaviors) may all affect outcome measures in comparative studies (Prins and Hovda, 2003). Lastly, considerable differences in glucose utilization and cerebral blood flow have been detected in the immature brain compared to adult, indicative of altered responses of this system after stress or injury during different developmental stages, as described elsewhere (Babikian et al., 2010; Nehling, 1988; Prins, 2008).
10. Concluding remarks
The pronounced differences between rodents and humans should be considered when comparing the maturational age of the CNS during normal and disrupted development. However, there is also considerable cross-species alignment in terms of key developmental milestones, behavioral phenotypes and regional vulnerability to brain injury. It is now generally accepted that the maturation state of the brain, and in particular, specific processes of synaptogenesis and myelination, rather than chronological age, is the critical determinant of outcome after brain injury. Thus, comparisons can bemade which take into consideration the timing of indices of neurobiological development, to gauge the impact of specific insults at different developmental stages, and best model the process of interest to the investigator (Table 1). Developmentally related differences are of importance not only to our understanding of the healthy brain during maturation, but also to predict differential responses to injury and potential therapeutics. For example, successful targeting of pathological processes in the adult brain may not necessarily be predictive of similar success in the immature brain, in which there may be a unique response to injury and potentially broader developmental consequences.
In summary, we have presented key benchmarks of brain developmental processes, which occur in both rodent and humans during postnatal development, albeit along different timelines as described. Importantly, two distinct processes of cortical thinning by synaptic pruning and white matter increases by myelination continue throughout late childhood and adolescence, likely reflecting ongoing maturation of neural circuitry which underlies behavioral changes at these times. We conclude that there is sufficient evidence for selection of an age-appropriate rodent model that is predicated on biochemical and neuroanatomical changes during early postnatal development, as well as the emergence of age-specific behaviors. Ongoing research across the gamut of cerebral development in both humans and rodents in parallel will provide a greater understanding of how all these factors interact, and manifest as age-dependent behavioral changes. Ultimately, the aim is to exploit this knowledge to develop age-specific therapeutics, in order to treat injuries to the immature brain without simultaneously interfering with ongoing developmental processes.
Acknowledgments
Support was provided by NIH/NINDS RO1 NS050159 and NS077767, a Sir Keith Murdoch Fellowship from the American Australian Association, and the Neurobehavioral Core for Rehabilitation Research at the University of California San Francisco.
Abbreviations
- CNS
Central nervous system
- GAB
Agamma-Aminobutyric acid
- GCL
granule cell layer
- Gd
gestation day
- HI
hypoxia-ischemia/hypoxic-ischemic
- HIE
hypoxic-ischemic encephalopathy
- IL
interleukin
- MRI
magnetic resonance imaging
- NMDA
N-methyl-D-aspartate
- OL
oligodendrocyte
- pnd
postnatal day
- pre-OL
preoligodendrocyte
- SGZ
subgranular zone
- SVZ
subventricular zone
- TBI
traumatic brain injury
- 5-HT
5-hydroxytryptamine
Footnotes
The authors report no conflicts of interest.
Contributor Information
Bridgette D. Semple, Email: bridgette.semple@ucsf.edu.
Klas Blomgren, Email: klas.blomgren@ki.se.
Kayleen Gimlin, Email: kayleen.gimlin@ucsf.edu.
Donna M. Ferriero, Email: ferrierod@neuropeds.ucsf.edu.
Linda J. Noble-Haeusslein, Email: linda.noble@ucsf.edu.
References
- Ajao D, Pop V, Kamper J, Adami A, Rudobeck E, et al. Traumatic brain injury in young rats leads to progressive behavioral deficits coincident with altered tissue properties in adulthood. Journal of Neurotrauma. 2012;29:2060–2074. doi: 10.1089/neu.2011.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Research Development: Brain Research. 1999;117:145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Amick JE, Yandora KA, Bell M, Wisniewski SR, Adelson PD, et al. The Th1 versus Th2 cytokine profile in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatric Critical Care Medicine. 2001;2:260–264. doi: 10.1097/00130478-200107000-00013. [DOI] [PubMed] [Google Scholar]
- Andersen SL. Trajectories of brain development: point of vulnerability or window of opportunity? Neuroscience & Biobehavioral Reviews. 2003;27:3–18. doi: 10.1016/s0149-7634(03)00005-8. [DOI] [PubMed] [Google Scholar]
- Anderson V, Moore C. Age at injury as a predictor of outcome following pediatric head injury: a longitudinal perspective. Child Neuropsychology. 1995;1:187–202. [Google Scholar]
- Anderson V, Spencer-Smith M, Leventer R, Coleman L, Anderson P, et al. Childhood brain insult: can age at insult help us predict outcome? Brain. 2009;132:45–56. doi: 10.1093/brain/awn293. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Bolton SJ, Fearn S, Perry VH. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain. 1997;120:435–444. doi: 10.1093/brain/120.3.435. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Dempster R, Fearn Sea. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Current Biology. 1998;8:923–926. doi: 10.1016/s0960-9822(07)00373-9. [DOI] [PubMed] [Google Scholar]
- Babikian T, Prins ML, Cai Y, Barkhoudarian G, Hartonian I, et al. Molecular and physiological responses to juvenile traumatic brain injury: focus on growth and metabolism. Developmental Neuroscience. 2010;32:431–441. doi: 10.1159/000320667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baburamani AA, Ek CJ, Walker DW, Castillo-Melendez M. Vulnerability of the developing brain to hypoxic-ischemic damage: contribution of the cerebral vasculature to injury and repair? Frontiers in Physiology. 2012;3 doi: 10.3389/fphys.2012.00424. http://dx.doi.org/10.3389/fphys.2012.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachevalier J, Beauregard M. Maturation of medial temporal lobe memory functions in rodents, monkeys and humans. Hippocampus. 1993;3:191–201. [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. Journal of Neuroscience. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Riddle A, Dean J, Hohimer AR. The instrumentad fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics. 2012;9:359–370. doi: 10.1007/s13311-012-0108-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balduini W, De Angelis V, Cimino M. Long-lasting behavioral alterations following a hypoxic/ischemic brain injury in neonatal rats. Brain Research. 2000;859:318–325. doi: 10.1016/s0006-8993(00)01997-1. [DOI] [PubMed] [Google Scholar]
- Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatrics Research. 2010;67:1–8. doi: 10.1203/PDR.0b013e3181c1b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloch S, Verma R, Huang H, Khurd P, Clark S, et al. Quantification of brain maturation and growth patterns in C57Bl/6 mice via computational neuroanatomy of diffusion tensor images. Cerebral Cortex. 2009;19:675–687. doi: 10.1093/cercor/bhn112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Gerber AJ, Peterson BS. Brain morphometry using anatomical magnetic resonance imaging. Journal of American Academy of the Child & Adolescent Psychiatry. 2008;47:619–621. doi: 10.1097/CHI.0b013e31816c54ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker AJ, Ullian EM. Astrocytes and synaptic plasticity. Neuroscientist. 2010;16:40–50. doi: 10.1177/1073858409339215. [DOI] [PubMed] [Google Scholar]
- Barker LA, Andrade J, Morton N, Romanowski CA, Bowles DP. Investigating the ‘latent’ deficit hypothesis: age at time of head injury, implicit and executive functions and behavioral insight. Neuropsychologia. 2010;48:2550–2563. doi: 10.1016/j.neuropsychologia.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Bautch VL, James JM. Neurovascular Development: the beginning of a beautiful friendship. Cell Adhesion and Migration. 2009;3:199–204. doi: 10.4161/cam.3.2.8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timelines of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Bayir H, Kochanek PM, Kagan VE. Oxidative stress in immature brain after traumatic brain injury. Developmental Neuroscience. 2006;28:420–431. doi: 10.1159/000094168. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: A pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiological Review. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. The Neuroscientist. 2012;18:467–486. doi: 10.1177/1073858412438697. [DOI] [PubMed] [Google Scholar]
- Berger HM, Mumby S, Gutteridge JM. Ferrous ions detected in iron-overloaded cord blood plasma from preterm and term babies: implications for oxidative stress. Free Radical Research. 1995;22:555–559. doi: 10.3109/10715769509150327. [DOI] [PubMed] [Google Scholar]
- Berger RP, Ta'asan S, Rand A, Lokshin A, Kockanek PM. Multiplex assessment of serum biomarker concentrations in well-appearing children with inflicted traumatic brain injury. Pediatric Research. 2009;65:97–102. doi: 10.1203/PDR.0b013e31818c7e27. [DOI] [PubMed] [Google Scholar]
- Berryhill P, Lilly MA, Levin HS, Hillman GR, Mendelsohn D, et al. Frontal lobe changes after severe diffuse closed head injury in children: a volumetric study of magnetic resonance imaging. Neurosurgery. 1995;37:392–399. doi: 10.1227/00006123-199509000-00004. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Frontiers in Neuroendocrinology. 2012;33:267–286. doi: 10.1016/j.yfrne.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Felderhoff-Mueser U, GHansen HH, Ikonomidou C. Neuropathological and biochemical features of traumatic injury in the developing brain. Neurotoxicology Research. 2003;5:475–490. doi: 10.1007/BF03033158. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cationchloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Blaiss CA, Yu TS, Zhang G, Chen J, Dimchev G, et al. Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. Journal of Neuroscience. 2011;31:4906–4916. doi: 10.1523/JNEUROSCI.5265-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore SJ. The social brain in adolescence. Nature Review Neuroscience. 2008;9:267–277. doi: 10.1038/nrn2353. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radical Biology & Medicine. 2006;40:388–397. doi: 10.1016/j.freeradbiomed.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Leist M, Groc L. Pathological apoptosis in the developing brain. Apoptosis. 2007;12:993–1010. doi: 10.1007/s10495-007-0754-4. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Hallin U, Hagberg H. Mitochondria and ischemic reperfusion damage in the adult and in the developing brain. Biochemical & Biophysical Research Communications. 2003;304:551–559. doi: 10.1016/s0006-291x(03)00628-4. [DOI] [PubMed] [Google Scholar]
- Bockhorst KH, Narayana PA, Liu R, Ahobila-Vijjula P, Ramu J, et al. Early postnatal development of rat brain: in vivo diffusion tensor imaging. Journal of Neuroscience Research. 2008;86:1520–1528. doi: 10.1002/jnr.21607. [DOI] [PubMed] [Google Scholar]
- Brouwer RM, Mandl RC, Schnack HG, van Soelen IL, van Baal GC, et al. White matter development in early puberty: a longitudinal volumetric and diffusion tensor imaging twin study. PloS One. 2012;7:e32316. doi: 10.1371/journal.pone.0032316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttram SD, Wisniewski SR, Jackson EK, Adelson PD, Feldman K, et al. Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. Journal of Neurotrauma. 2007;24:1707–1717. doi: 10.1089/neu.2007.0349. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Carare-Nnadi RO, Losey PH, Anthony DC. Loss of the atypical inflammatory response in juvenile and aged rats. Neuropathology & Applied Neurobiology. 2007;33:108–120. doi: 10.1111/j.1365-2990.2006.00773.x. [DOI] [PubMed] [Google Scholar]
- Catalani A, Sabbatini M, Consoli C, Cinque C, Tomassoni D, et al. Glial fibrillary acidic protein immunoreactive astrocytes in developing rat hippocampus. Mechanics of Ageing & Development. 2002;123:481–490. doi: 10.1016/s0047-6374(01)00356-6. [DOI] [PubMed] [Google Scholar]
- Catroppa C, Anderson V, Morse S, Haritou F, Rosenfeld J. Outcome and predictors of functional recovery 5 years following pediatric traumatic brain injury (TBI) Journal of Pediatric Psychology. 2008;33:707–718. doi: 10.1093/jpepsy/jsn006. [DOI] [PubMed] [Google Scholar]
- Chahboune H, Ment LR, Stewart WB, Ma X, Rothman DL, Hyder F. Neurodevelopment of C57B/L6 mouse brain assessed byin vivo diffusion tensor imaging. NMR in Biomedicine. 2007;20:375–382. doi: 10.1002/nbm.1130. [DOI] [PubMed] [Google Scholar]
- Chiaretti A, Antonelli A, Mastrangelo A, Pezzotti P, Tortorolo L, et al. Interleukin-6 and nerve growth factor upregulation correlates with improved outcome inchildren with severe traumatic brain injury. Journal of Neurotrauma. 2008;25:225–234. doi: 10.1089/neu.2007.0405. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Chuang N, Mori S, Yamamoto A, Jiang H, Ye X, et al. An MRI-based atlas and database of the developing mouse brain. Neuroimage. 2011;54:80–89. doi: 10.1016/j.neuroimage.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy B, Kersh B, Hyde J, Darlington RB, Anand KJS, Finlay BL. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics. 2007;5:79–94. doi: 10.1385/ni:5:1:79. [DOI] [PubMed] [Google Scholar]
- Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Manvelyan H, et al. Age is a determinant of the inflammatory response and loss of cortical volume after traumatic brain injury. Developmental Neuroscience. 2010;32:454–465. doi: 10.1159/000316805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colrain IM, Baker FC. Changes in sleep as a function of adolescent development. Neuropsychology Review. 2011;21:5–21. doi: 10.1007/s11065-010-9155-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad MS, Dilger RN, Johnson RW. Brain growth of the domestic pig (Sus scrofa) from 2 to 24 weeks of age: a longitudinal MRI study. Developmental Neuroscience. 2012;34:291–298. doi: 10.1159/000339311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornford EM, Cornford ME. Nutrient transport and the blood-brain barrier in developing animals. Federation Proceedings. 1986;45:2065–2072. [PubMed] [Google Scholar]
- Covey MV, Jiang Y, Alli VV, Yang Z, Levison SW. Defining the critical period for neocortical neurogenesis after pediatric brain injury. Developmental Neuroscience. 2010;32:488–498. doi: 10.1159/000321607. [DOI] [PubMed] [Google Scholar]
- Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, et al. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361:736–742. doi: 10.1016/S0140-6736(03)12658-X. [DOI] [PubMed] [Google Scholar]
- Cowan WM. The development of the brain. Scientific American. 1979;241:113–133. [PubMed] [Google Scholar]
- Craig A, Ling Luo N, Beardsley DJ, Wingate-Pearse N, Walker DW, et al. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Experimental Neurology. 2003;181:231–240. doi: 10.1016/s0014-4886(03)00032-3. [DOI] [PubMed] [Google Scholar]
- Crain B, Cotman C, Taylor D, Lynch G. A quantitative electron microscopic study of synaptogenesis in the dentate gyrus of the rat. Brain Research. 1973;63 doi: 10.1016/0006-8993(73)90088-7. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felipe J, Marco P, Fairen A, Jones EG. Inhibitory synaptogenesis in mouse somatosensory cortex. Cerebral Cortex. 1997;7:619–634. doi: 10.1093/cercor/7.7.619. [DOI] [PubMed] [Google Scholar]
- Dean JM, McClendon E, Hansen K, Azimi-Zonooz A, Chen K, et al. Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Science Translational Medicine. 2013;16(168):ra7. doi: 10.1126/scitranslmed.3004669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JM, Moravec MD, Grafe M, Abend N, Ren J, et al. Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Developmental Neuroscience. 2011a doi: 10.1159/000327242. http://dx.doi.org/10.1159/000327242. [DOI] [PMC free article] [PubMed]
- Dean JM, Riddle A, Maire J, Hansen KD, Preston M, et al. An organotypic slice culture model ofchronic white matter injury with maturation arrest of oligodendrocyte progenitors. Molecular Neurodegeneration. 2011b;6:46. doi: 10.1186/1750-1326-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekaban AS. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Annals of Neurology. 1987;4:345–356. doi: 10.1002/ana.410040410. [DOI] [PubMed] [Google Scholar]
- DeSesso JM, Scialli AR, Holson JF. Apparent lability of neural tube closure in laboratory animals and humans. American Journal of Medical Genetics A. 1999;87:143–162. doi: 10.1002/(sici)1096-8628(19991119)87:2<143::aid-ajmg6>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Diamond A. Rate of maturation of the hippocampus and the developmental progression of children's performance on the delayed non-matching to sample and visual paired comparison tasks. Annals of New York Academy of Science. 1990;608:394–426. doi: 10.1111/j.1749-6632.1990.tb48904.x. [DOI] [PubMed] [Google Scholar]
- Dietrich RB, Bradley WG, Zaragoza EJt, Otto RJ, Taira RK, et al. MR evaluation of early myelination patterns in normal and developmentally delayed infants. AJR American Journal of Roentgenology. 1988;150:889–896. doi: 10.2214/ajr.150.4.889. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Quantitative growth and development of human brain. Archives of Diseases in Childhood. 1973;48:757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of brain growth spurt. Early Human Development. 1979;311:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Drobyshevsky A, Derrick M, Luo K, Zhang LQ, Wu YN, et al. Near-term fetal hypoxia-ischemia in rabbits: MRI can predict muscle tone abnormalities and deep brain injury. Stroke. 2012;43:2757–2763. doi: 10.1161/STROKEAHA.112.653857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois J, Benders M, Borradori-Tolsa C, Cachia A, Lazeyras F, et al. Primary cortical folding in the human newborn: an early marker of later functional development. Brain. 2008;131:2028–2041. doi: 10.1093/brain/awn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime AC. Large animal models of traumatic injury to the immature brain. Developmental Neuroscience. 2006;28:380–387. doi: 10.1159/000094164. [DOI] [PubMed] [Google Scholar]
- Duhaime AC, Hunter JV, Grate LL, Kim A, Golden J, et al. Magnetic resonance imaging studies of age-dependent responses to scaled focal brain injury in the piglet. Journal of Neurosurgery. 2003;99:542–548. doi: 10.3171/jns.2003.99.3.0542. [DOI] [PubMed] [Google Scholar]
- Duhaime AC, Margulies SS, Durham SR, O'Rourke MM, Golden JA, et al. Maturation-dependent response of the piglet brain to scaled cortical impact. Journal of Neurosurgery. 2000;93:455–462. doi: 10.3171/jns.2000.93.3.0455. [DOI] [PubMed] [Google Scholar]
- Duval J, Braun CMJ, Montour-Proulx I, Daigneault S, Rouleau I, Bgin J. Brain lesions and IQ: recovery versus decline depends onage of onset. Journal of Child Neurology. 2008;23:663–668. doi: 10.1177/0883073808314161. [DOI] [PubMed] [Google Scholar]
- Dzhala V, Valeeva G, Glykys J, Khazipov R, Staley K. Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. Journal of Neuroscience. 2012;32:4017–4031. doi: 10.1523/JNEUROSCI.5139-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, et al. NKCC1 transporter facilitates seizures in the developing brain. Nature Medicine. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Emanuelson I, Uvebrant P. Occurrence of epilepsy during the first 10 years after traumatic brain injury acquired in childhood up to the age of 18 years in the south western Swedish population-based series. Brain Injury. 2009;23:612–616. doi: 10.1080/02699050902973913. [DOI] [PubMed] [Google Scholar]
- Engelhardt B. Development of the blood-brain barrier. Cell & Tissue Research. 2003;314:119–129. doi: 10.1007/s00441-003-0751-z. [DOI] [PubMed] [Google Scholar]
- Eppig C, Fincher CL, Thornhill R. Parasite prevalence and the worldwide distribution of cognitive ability. Proceeings of Biological Science. 2010;277:3801–3808. doi: 10.1098/rspb.2010.0973. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, et al. Neurogenesis in the adult human hippocampus. Nature Medicine. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Eslinger PJ, Grattan LM, Damasio H, Damasio AR. Developmental consequences of childhood frontal lobe damage. Archives of Neurology. 1992;49:764–769. doi: 10.1001/archneur.1992.00530310112021. [DOI] [PubMed] [Google Scholar]
- Ewing-Cobbs L, Prasad MR, Swank P, Kramer L, Cox CSJ, et al. Arrested development and disrupted callosal microstructure following pediatric traumatic brain injury: relation to neurobehavioral outcomes. Neuroimage. 2008;42:1305–1315. doi: 10.1016/j.neuroimage.2008.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg I, Campbell IG. Sleep EEG changes during adolescence: an index of a fundamental brain reorganization. Brain & Cognition. 2010;72:56–65. doi: 10.1016/j.bandc.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Fernández-López D, Faustino J, Daneman R, Zhou L, Lee SY, et al. Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. Journal of Neuroscience. 2012;32:9588–9600. doi: 10.1523/JNEUROSCI.5977-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BL, Darlington RB. Linked regularities in the development and evolution of mammalian brains. Science. 1995;268:1578–1584. doi: 10.1126/science.7777856. [DOI] [PubMed] [Google Scholar]
- Finnie JW. Comparative approach to understanding traumatic injury in the immature, postnatal brain of domestic animals. Australian Veterinary Journal. 2012;90:301–307. doi: 10.1111/j.1751-0813.2012.00955.x. [DOI] [PubMed] [Google Scholar]
- Frey LC. Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia. 2003;44:11–17. doi: 10.1046/j.1528-1157.44.s10.4.x. [DOI] [PubMed] [Google Scholar]
- Friess SH, Ichord RN, Owens K, Ralston J, Rizol R, et al. Neurobehavioral functional deficits following closed head injury in the neonatal pig. Experimental Neurology. 2007;204:234–243. doi: 10.1016/j.expneurol.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, et al. Brain development during childhood and adolescence: a longitudinal MRI study. Nature Neuroscience. 1999;2:861–863. doi: 10.1038/13158. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giza CC, Kolb B, Harris NG, Asarnow RF, Prins ML. Hitting a moving target: Basic mechanisms of recovery from acquired developmental brain injury. Developmental Neurorehabilitation. 2009;12:255–268. doi: 10.3109/17518420903087558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giza CC, Mink RB, Madikians A. Pediatric traumatic brain injury: not just little adults. Current Opinion in Critical Care. 2007;131:143–152. doi: 10.1097/MCC.0b013e32808255dc. [DOI] [PubMed] [Google Scholar]
- Glantz L, Gilmore JH, Hamer RM, Lieberman JA, Jarskog LF. Synaptophysin and postsynaptic density protein 95inthe human prefrontal cortex from mid-gestation into early childhood. Neuroscience. 2007;149:582–591. doi: 10.1016/j.neuroscience.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Ichord R, Palmer C, Yager JY, Vannucci SJ. Animal models of developmental brain injury: relevance to human disease. A summary of the panel discussion from the Third Hershey Conference on Developmental Cerebral Blood Flow and Metabolism. Developmental Neuroscience. 2002a;24:364–366. doi: 10.1159/000069040. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Mental Retardation and Developmental Disabilities Research Reviews. 2002b;8:30–38. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Kraft AD. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicity. 2012;33 doi: 10.1016/j.neuro.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedner J, Lundell KH, Breese GR, Mueller RA, Hedner T. Developmental variations in CSF monoamine metabolites during childhood. Biology of the Neonate. 1986;49:190–197. doi: 10.1159/000242530. [DOI] [PubMed] [Google Scholar]
- Herschkowitz N, Kagan J, Zilles K. Neurobiological bases of behavioral development in the first year. Neuroped. 1997;28:296–306. doi: 10.1055/s-2007-973720. [DOI] [PubMed] [Google Scholar]
- Holsapple MP, West LJ, Landreth KS. Species comparison of anatomical and functional immune system development. Birth Defects Research. 2003;68:321–334. doi: 10.1002/bdrb.10035. [DOI] [PubMed] [Google Scholar]
- Hu BR, Liu CL, Ouyang Y, Blomgren K, Siesjö BK. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. JCBFM. 2000;20:1294–1300. doi: 10.1097/00004647-200009000-00003. [DOI] [PubMed] [Google Scholar]
- Huang Z, Liu J, Cheung PY, Chen C. Long-term cognitive impairment and myelination deficiency in a rat model of perinatal hypoxic-ischemic brain injury. Brain Research. 2009a;8:100–109. doi: 10.1016/j.brainres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Huang Z, Liu J, Cheung PY, Chen C. Long-term cognitive impairment and myelination deficiency in a rat model of perinatal hypoxic-ischemic brain injury. Brain Research. 2009b;1301:100–109. doi: 10.1016/j.brainres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Huh JW, Raghupathi R. Chronic cognitive deficits and long-term histopathological alterations following contusive brain injury in the immature rat. Journal of Neurotrauma. 2007;24:1460–1474. doi: 10.1089/neu.2006.3787. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN. Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. Journal of Neurophysiology. 2010;103:1490–1500. doi: 10.1152/jn.00957.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins KD, Dickson DW, Rashbaum WK, Lyman WD. Localization of morphologically distinct microglial populations in the developing human fetal brain: implications for ontogeny. Brain Research Development: Brain Research. 1990;55:95–102. doi: 10.1016/0165-3806(90)90109-c. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. Synaptic density in human frontal cortex—developmental changes and effects of aging. Brain Research. 1979;163:195–205. doi: 10.1016/0006-8993(79)90349-4. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, de Courten C, Garey LJ, Van der Loos H. Synaptogenesis in human visual cortex—evidence for synapse elimination during normal development. Neuroscience Letters. 1982;33:247–252. doi: 10.1016/0304-3940(82)90379-2. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Kaindl AM. Neuronal death and oxidative stress in the developing brain. Antioxidants & Redox Signal. 2011;14:1535–1550. doi: 10.1089/ars.2010.3581. [DOI] [PubMed] [Google Scholar]
- Inder TE, Huppi PS. In vivo studies of brain development by magnetic resonance techniques. Mental Retardation and Developmental Disabilities Research Reviews. 2000;6:59–67. doi: 10.1002/(SICI)1098-2779(2000)6:1<59::AID-MRDD8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Johnston MV. Neurotransmitters and vulnerability of the developing brain. Brain Development. 1995;17:301–306. doi: 10.1016/0387-7604(95)00079-q. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Hoon AHJ. Cerebral palsy. Neuromolecular Medicine. 2006;8:435–450. doi: 10.1385/NMM:8:4:435. [DOI] [PubMed] [Google Scholar]
- Kalm M, Fukuda A, Fukuda H, Ohrfelt A, Lannering B, et al. Transient inflammation in neurogenic regions after irradiation of the developing brain. Radiation Res. 2009a;171:66–76. doi: 10.1667/RR1269.1. [DOI] [PubMed] [Google Scholar]
- Kalm M, Lannering B, Björk-Eriksson T, Blomgren K. Irradiation-induced loss of microglia in the young brain. Journal of Neuroimmunology. 2009b;206:70–75. doi: 10.1016/j.jneuroim.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Parent JM. Forebrain neurogenesis after focal ischemic and traumatic brain injury. Neurobiology of Disease. 2010;37:267–274. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavan MS, Diwadkar VA, DeBellis M, Dick E, Kotwal R, et al. Development of the corpus callosum in childhood, adolescence and early adulthood. Life Science. 2002;70:1909–1922. doi: 10.1016/s0024-3205(02)01492-3. [DOI] [PubMed] [Google Scholar]
- Kleindienst A, McGinn MJ, Harvey HB, Colello RJ, Hamm RJ, Bullock MR. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. Journal of Neurotrauma. 2005;22:645–655. doi: 10.1089/neu.2005.22.645. [DOI] [PubMed] [Google Scholar]
- Kniesel U, Risau W, Wolburg H. Development of blood-brain barrier tight junctions in the rat cortex. Brain Research Development: Brain Research. 1996;96:229–240. doi: 10.1016/0165-3806(96)00117-4. [DOI] [PubMed] [Google Scholar]
- Kochanek P. Ischemic and traumatic brain injury: pathobiology and cellular mechanisms. Critical Care Medicine. 1993;21:S333–S335. doi: 10.1097/00003246-199309001-00016. [DOI] [PubMed] [Google Scholar]
- Kostović I, Lukinović N, Judas M, Bogdanović N, Mrzljak L, et al. Structural basis of the developmental plasticity in the human cerebral cortex: the role of the transient subplate zone. Metabolic Brain Disease. 1989;4:17–23. doi: 10.1007/BF00999489. [DOI] [PubMed] [Google Scholar]
- Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain. 2007;130:2508–2519. doi: 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annual Review of Neuroscience. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladics GS, Smith C, Bunn TL, Dietert RR, Anderson PK, et al. Characterization of an approach to developmental immunotoxicology assessment in the rat using SRBC as the antigen. Toxicology Methods. 2000;10:283–311. [Google Scholar]
- Lambert MS. Breeding Strategies for Maintaining Colonies of Laboratory Mice. TJ Laboratory; Bar Harbor: 2009. Breeding strategies for maintaining colonies of laboratory mice. [Google Scholar]
- Langlois JA, Rutland-Brown W, Thomas KE. The incidence of traumatic brain injury among children in the United States: differences by race. Journal of Head Trauma Rehabilitation. 2005;20:229–238. doi: 10.1097/00001199-200505000-00006. [DOI] [PubMed] [Google Scholar]
- Laviola G, Macri S, Morley-Fletcher S, Adriani W. Risk-taking behavior in adolescent mice: psychobiological determinants and early epigenetic influence. Neuroscience & Biobehavioral Reviews. 2003;27:19–31. doi: 10.1016/s0149-7634(03)00006-x. [DOI] [PubMed] [Google Scholar]
- Lawn JE, Cousens S, Zupan J, Team LNSS. 4 million neonatal deaths: When? Where? Why? Lancet. 2005;365:891–900. doi: 10.1016/S0140-6736(05)71048-5. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH. The unique characteristics of inflammatory responses in mouse brain are acquired during postnatal development. European Journal of Neuroscience. 1995;7:1584–1595. doi: 10.1111/j.1460-9568.1995.tb01154.x. [DOI] [PubMed] [Google Scholar]
- Lea PMT, Faden AI. Traumatic brain injury: developmental differences in glutamate receptor response and the impact on treatment. Mental Retardation and Developmental Disabilities Research Reviews. 2001;7:235–248. doi: 10.1002/mrdd.1033. [DOI] [PubMed] [Google Scholar]
- Lebel C, Beaulieu C. Longitudinal development of human brain wiring continues from childhood into adulthood. Journal of Neuroscience. 2011;31:10937–10947. doi: 10.1523/JNEUROSCI.5302-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Gee M, Camicioli R, Wieler M, Martin W, Beaulieu C. Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage. 2012;60:340–352. doi: 10.1016/j.neuroimage.2011.11.094. [DOI] [PubMed] [Google Scholar]
- Lee J, Croen LA, Lindan C, Nash KB, Yoshida CK, et al. Annals of Neurology. 2005;58:2. [Google Scholar]
- Lenroot RK, Giedd JN. Brain development in children and adolescence: Insights from anatomical magnetic resonance imaging. Neuroscience & Biobehavioral Reviews. 2006;30:718–729. doi: 10.1016/j.neubiorev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Lenroot RK, Gogtay N, Greenstein DK, Wells EM, Wallace GL, et al. Sexual dimorphism of brain developmental trajectories during childhood and adolescence. Neuroimage. 2007;36:1065–1073. doi: 10.1016/j.neuroimage.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HS, Zhang L, Dennis M, Ewing-Cobbs L, Schachar R, et al. Psychosocial outcome of TBI in children with unilateral frontal lesions. Journal of International Neuropsychological Society. 2004;10:305–316. doi: 10.1017/S1355617704102129. [DOI] [PubMed] [Google Scholar]
- Levine D, Barnes PD. Cortical maturation in normal and abnormal fetuses as assessed with prenatal MR imaging. Radiology. 1999;210:751–758. doi: 10.1148/radiology.210.3.r99mr47751. [DOI] [PubMed] [Google Scholar]
- Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nature Review in Immunology. 2007;7:379–390. doi: 10.1038/nri2075. [DOI] [PubMed] [Google Scholar]
- Li H, Li Q, Du X, Sun Y, Wang X, et al. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. JCBFM. 2011;31:2106–2115. doi: 10.1038/jcbfm.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Li H, Roughton K, Wang X, Kroemer G, et al. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death & Diseases 1. 2010 doi: 10.1038/cddis.2010.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidow MS, Goldman-Rakic PS, Rakic P. Synchronized overproduction of neurotransmitter receptors in diverse regions of the primate cerebral cortex. Proceedings of the National Academy of Science. 1991;88:10218–10221. doi: 10.1073/pnas.88.22.10218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodygensky GA, Vasung L, Sizonenko SV, Hü ppi PS. Neuroimaging of cortical development and brain connectivity in human newborns and animal models. Journal of Anatomy. 2010;217:418–428. doi: 10.1111/j.1469-7580.2010.01280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JK. Epidemiology and classification of perinatal stroke. Seminars in Fetal & Neonatal Medicine. 2009;14:245–249. doi: 10.1016/j.siny.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Male D, Rezaie P. Colonisation of the human central nervous system by microglia: the roles of chemokines and vascular adhesion molecules. Progress in Brain Research. 2001;132:81–93. doi: 10.1016/S0079-6123(01)32067-8. [DOI] [PubMed] [Google Scholar]
- Marshall-Clarke S, Reen D, Tasker L, Hassan J. Neonatal immunity: how well has it grown up? Immunology Today. 2000;21:35–41. doi: 10.1016/s0167-5699(99)01548-0. [DOI] [PubMed] [Google Scholar]
- Marshall CAG, Suzuki SO, Goldman JE. Gliogenic and neurogenic progenitors of the subventricular zone: Who are they, where did they come from, and where are they going? GLIA. 2003;43:52–61. doi: 10.1002/glia.10213. [DOI] [PubMed] [Google Scholar]
- Mazzola CA, Adelson PD. Critical care management in head trauma in children. Critical Care Medicine. 2002;30:S393–S401. doi: 10.1097/00003246-200211001-00003. [DOI] [PubMed] [Google Scholar]
- McAuliffe JJ, Miles L, Vorhees CV. Adult neurological function following neonatal hypoxia–ischemia in a mouse model of the term neonate: Water maze performance is dependent onseparable cognitive and motor components. Brain Research. 2006;1118:208–221. doi: 10.1016/j.brainres.2006.08.030. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Research Review. 1990;15:41–70. doi: 10.1016/0165-0173(90)90011-c. [DOI] [PubMed] [Google Scholar]
- Merkley TL, Bigler ED, Wilde EA, McCauley SR, Hunter JV, Levin HS. Diffuse changes in cortical thickness in pediatric moderate-to-severe traumatic brain injury. Journal of Neurotrauma. 2008;25:1343–1345. doi: 10.1089/neu.2008.0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Beaulieu C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. Journal of Comparative Neurology. 1996;373:340–354. doi: 10.1002/(SICI)1096-9861(19960923)373:3<340::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Miller SP, Ferriero DM. From selective vulnerability to connectivity: insights from newborn brain imaging. Trends in Neuroscience. 2009;32:496–505. doi: 10.1016/j.tins.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Clowry G. Cerebral cortical development in rodents and primates. Progress in Brain Research. 2012;195:45–70. doi: 10.1016/B978-0-444-53860-4.00003-9. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Rutherford M. Brain maturation after preterm birth. Science Translational Medicine. 2013;5:168ps2. doi: 10.1126/scitranslmed.3005379. http://dx.doi.org/10.1126/scitranslmed.3005379. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Monk CS, Webb SJ, Nelson CA. Prenatal neurobiological development: molecular mechanisms and anatomical change. Developmental Neuropsychology. 2001;19:211–236. doi: 10.1207/S15326942DN1902_5. [DOI] [PubMed] [Google Scholar]
- Moos T, Morgan EH. A morphological study of the developmentally regulated transport of iron into the brain. Developmental Neuroscience. 2002;24:99–105. doi: 10.1159/000065702. [DOI] [PubMed] [Google Scholar]
- Muramatsu K, Fukuda A, Togari H, Wada Y, Nishino H. Vulnerability to cerebral hypoxic-ischemic insult in neonatal but not in adult rats is in parallel with disruption of the blood-brain barrier. Stroke. 1997;28:2281–2289. doi: 10.1161/01.str.28.11.2281. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Westerberg H, Klingberg T. Maturation of white matter is associated with the development of cognitive functions during childhood. Journal of Cognitive Neuroscience. 2004;16:1227–1233. doi: 10.1162/0898929041920441. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Iwasaki S, Kichikawa K, Fukusumi A, Taoka T, et al. Normal myelination of anatomic nerve fiber bundles: MR analysis. AJNR American Journal of Neuroradiology. 1998;19:1129–1136. [PMC free article] [PubMed] [Google Scholar]
- Nehling A. Cerebral energy metabolism, glucose transport and blood flow: changes with maturation and adaptation to hypoglycaemia. Diabetes & Metabolism. 1988;23:18–29. [PubMed] [Google Scholar]
- Østby Y, Tamnes CK, Fjell AM, Walhovd KB. Morphometry and connectivity of the fronto-parietal verbal working memory network in development. Neuropsychologia. 2011;49:3854–3862. doi: 10.1016/j.neuropsychologia.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Ou-Yang FL, Zhou XZ, Fang SZ, Cai YQ, Li H. Long-term behavioral and ultrastructural alterations following hypoxic-ischemic brain damage in neonatal rats. Chinese Journal of Contemporary Pediatrics. 2012;14:380–384. (in Chinese) [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Annals of Neurology. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Paus T, Collins DL, Evans AC, Leonard G, Pike B, Zijdenbos A. Maturation of white matter in the human brain: a review of magnetic resonance studies. Brain Research Bulletin. 2001;54:255–266. doi: 10.1016/s0361-9230(00)00434-2. [DOI] [PubMed] [Google Scholar]
- Petanjek Z, Judaš M, Šimic G, Rasin MR, Uylings HB, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proceedings of Natuonal Academy of Sciences of United States of America. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop V, Badaut J. A neurovascular perspective for long-term changes after brain trauma. Translaional Stroke Research. 2011;2:533–545. doi: 10.1007/s12975-011-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts M, Koh SE, Whetstone W, Walker B, Yoneyama T, et al. Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. Neuroreport. 2006;3:143–153. doi: 10.1016/j.nurx.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Rola R, Claus CP, Ferriero DM, Fike JR, Noble-Haeusslein LJ. Glutathione peroxidase overexpression does not rescue impaired neurogenesis in the injured immature brain. Journal of Neuroscience Research. 2009;87:1848–1857. doi: 10.1002/jnr.21996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast AJ, Klenerman P, Goulder PJR. The impact of differential antiviral immunity in children and adults. Nature Review in Immunology. 2012;12:636–648. doi: 10.1038/nri3277. [DOI] [PubMed] [Google Scholar]
- Prins ML. Cerebral metabolic adaption and keton metabolism after brain injury. JCBFM. 2008;28:1–16. doi: 10.1038/sj.jcbfm.9600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Hovda DA. Traumatic brain injury in the developing rat: effects of maturation on Morris water maze acquisition. Journal of Neurotrauma. 1998;15:799–811. doi: 10.1089/neu.1998.15.799. [DOI] [PubMed] [Google Scholar]
- Prins ML, Hovda DA. Developing experimental models to address traumatic brain injury in children. Journal of Neurotrauma. 2003;20:123–137. doi: 10.1089/08977150360547053. [DOI] [PubMed] [Google Scholar]
- Prins ML, Lee SM, Cheng CL, Becker DP, Hovda DA. Fluid percussion brain injury in the developing and adult rat: a comparative study of mortality, morphology, intracranial pressure and mean arterial blood pressure. Brain Research Development: Brain Research. 1996;95:272–282. doi: 10.1016/0165-3806(96)00098-3. [DOI] [PubMed] [Google Scholar]
- Pullela R, Raber J, Pfankuch T, Ferriero DM, Claus CP, et al. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Developmental Neuroscience. 2006;28:396–409. doi: 10.1159/000094166. [DOI] [PubMed] [Google Scholar]
- Qiu L, Zhu C, Wang X, Xu F, Eriksson PS, et al. Less neurogenesis and inflammationin the immature than inthe juvenile brain after cerebral hypoxia-ischemia. JCBFM. 2007;27:785–794. doi: 10.1038/sj.jcbfm.9600385. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Huh JW. Diffuse brain injury in the immature rat: evidence for an age-at-injury effect on cognitive function and histopathologic damage. Journal of Neurotrauma. 2007;24:1596–1608. doi: 10.1089/neu.2007.3790. [DOI] [PubMed] [Google Scholar]
- Rakic P, Nowakowski RS. The time of origin of neurons in the hippocampal region of the rhesus monkey. Journal of Comparative Neurology. 1981;196:99–128. doi: 10.1002/cne.901960109. [DOI] [PubMed] [Google Scholar]
- Raznahan A, Lerch JP, Lee N, Greenstein D, Wallace GL, et al. Patterns of coordinated anatomical change in human cortical development: A longitudinal neuroimaging study of maturational coupling. Neuron. 2011;72:873–884. doi: 10.1016/j.neuron.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environment Health Perspective. 2000;108:511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RM, Sun D, Bullock MR. Neurogenesis after traumatic brain injury. Neurosurgery Clinic of North America. 2007;18:169–181. doi: 10.1016/j.nec.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Roessmann U, Gambetti P. Astrocytes in the developing human brain. An immunohistochemical study. Acta Neuropathol. 1986;70:308–313. doi: 10.1007/BF00686089. [DOI] [PubMed] [Google Scholar]
- Romijn HJ, Hofman MA, Gramsbergen A. At what age is the developing cerebral cortex of the rat comparable to that of the full-term newborn human baby? Early Human Development. 1991;26:61–67. doi: 10.1016/0378-3782(91)90044-4. [DOI] [PubMed] [Google Scholar]
- Saliba E, Henrot A. Inflammatory mediators and neonatal brain damage. Biology of the Neonate. 2001;79:224–227. doi: 10.1159/000047096. [DOI] [PubMed] [Google Scholar]
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:382–386. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson C, Murphy S. Glutamate binding in the rat cerebral cortex during ontogeny. Deveopmental Brain Research. 1982;2:329–339. doi: 10.1016/0165-3806(81)90042-0. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Liddelow SA, Dziegielewska KM. Barrier mechanisms in the developing brain. Frontiers in Pharmacology. 2012;3 doi: 10.3389/fphar.2012.00046. http://dx.doi.org/10.3389/fphar.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD. Sex, glia, and development: interactions in health and disease. Hormonal Behavior. 2012;62:243–253. doi: 10.1016/j.yhbeh.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. Journal of Neurochemistry. 2012;120:948–963. doi: 10.1111/j.1471-4159.2011.07630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia KN, McClure M, Moravec MD, Luo NL, Wan Y, et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Annals of Neurology. 2008;63:520–530. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selassie AW, Zaloshnja E, Langlois JA, Miller T, Jones P, Steiner C. Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. Journal of Head Trauma Rehabilitation. 2008;23:123–131. doi: 10.1097/01.HTR.0000314531.30401.39. [DOI] [PubMed] [Google Scholar]
- Semple BD, Canchola SA, Noble-Haeusslein LJ. Deficits in social behavior emerge during development after pediatric traumatic brain injury in mice. Journal of Neurotrauma. 2012;29:2672–2683. doi: 10.1089/neu.2012.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seress L, Mrzljak L. Postnatal development of mossy cells in the human dentate gyrus: a light microscopic Golgi study. Hippocampus. 1992;2:127–141. doi: 10.1002/hipo.450020205. [DOI] [PubMed] [Google Scholar]
- Shaw P, Greenstein D, Lerch J, Clasen L, Lenroot R, et al. Intellectual ability and cortical development in children and adolescents. Nature. 2006;440:676–679. doi: 10.1038/nature04513. [DOI] [PubMed] [Google Scholar]
- Shen Y, Liu XB, Pleasure DE, Deng W. Axon–glia synapses are highly vulnerable to white matter injury in the developing brain. Journal of Neuroscience Research. 2012;90:105–121. doi: 10.1002/jnr.22722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevell MI, Majnemer A, Rosenbaum P, Abrahamowicz M. Etiologic yield of subspecialists' evaluation of young children with global developmental delay. Journal of Pediatrics. 2000;136:593–598. doi: 10.1067/mpd.2000.104817. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. Journal of Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Thompson PM, Holmes CJ, Jernigan TL, Toga AW. In vivo evidence for post-adolescent brain maturation in frontal and striatal regions. Nature Neuroscience. 1999;2:859–861. doi: 10.1038/13154. [DOI] [PubMed] [Google Scholar]
- Spear L. Modeling adolescent development and alcohol use in animals. Alcohol Research & Health. 2000;24:115–123. [PMC free article] [PubMed] [Google Scholar]
- Spear L, Brake SC. Periadolescence: age-dependent behavior and psychopharmacological responsitivity in rats. Developmental Psychobiology. 1983;16:83–109. doi: 10.1002/dev.420160203. [DOI] [PubMed] [Google Scholar]
- Spear PG, Wang AL, Rutishauser U, Edelman GM. Characterization of splenic lymphoid cells in fetal and newborn mice. Journal of Experimental Medicine. 1973;138:557–573. doi: 10.1084/jem.138.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE. A potential model of pediatric posttraumatic epilepsy. Epilepsy Research. 2009;86:221–223. doi: 10.1016/j.eplepsyres.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. Elaborate interactions between the immune and nervous systems. Nature Immunology. 2004;5:575–581. doi: 10.1038/ni1078. [DOI] [PubMed] [Google Scholar]
- Stolp HB, Dziegielewska KM, Ek CJ, Habgood MD, Lane MA, et al. Breakdown of the blood-brain barrier to proteins in white matter of the developing brain following systemic inflammation. Cell and Tissue Research. 2005;320:369–378. doi: 10.1007/s00441-005-1088-6. [DOI] [PubMed] [Google Scholar]
- Stubbs D, DeProto J, Nie K, Englund C, Mahmud I, et al. Neurovascular congruence during cerebral cortical development. Cerebral Cortex. 2009;19:132–141. doi: 10.1093/cercor/bhp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman DA, Muoghaddam B. The neurobiology of adolescence: Changes in brain architecture, functional dynamics, and behavioral tendencies. Neuroscience & Biobehavioral Reviews. 2011;35:1704–1712. doi: 10.1016/j.neubiorev.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan S, Friess SH, Ralston J, Smith C, Propert KJ, et al. Behavioral deficits and axonal injury persistence following rotational head injury are directional dependent. Journal of Neurotrauma. 2012 doi: 10.1089/neu.2012.2594. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, McGinn MJ, Zhou Z, Harvey HB, Bullock MR, Colello RJ. Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Experimental Neurology. 2007;204:264–272. doi: 10.1016/j.expneurol.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai WC, Burke KA, Dominguez JF, Gundamraj L, Turman JEJ. Growth deficits in a postnatal day 3 rat model of hypoxic-ischemic brain injury. Behavioral Brain Research. 2009;202:40–49. doi: 10.1016/j.bbr.2009.03.043. [DOI] [PubMed] [Google Scholar]
- Tasker RC. Changes in white matter late after severe traumatic brain injury in childhood. Developmental Neuroscience. 2006;28:302–308. doi: 10.1159/000094156. [DOI] [PubMed] [Google Scholar]
- Terranova ML, Laviola G. Scoring of social interactions and play in mice during adolescence. Current Protocols in Toxicology. 2005;(26):13.1.1–13.10.11. doi: 10.1002/0471140856.tx1310s26. [DOI] [PubMed] [Google Scholar]
- Threlkeld SW, Rosen GD, Fitch RH. Age at developmental cortical injury differentially alters corpus callosum volume in the rat. BMC Neuroscience. 2007;8:94. doi: 10.1186/1471-2202-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto S. The prefrontal cortex: Functional neural development during early childhood. Neuroscientist. 2008;14:345–358. doi: 10.1177/1073858408316002. [DOI] [PubMed] [Google Scholar]
- Turken A, Whitfield-Gabrieli S, Bammer R, Baldo JV, Dronkers NF, Gabrieli JD. Cognitive processing speed and the structure of white matter pathways: convergent evidence from normal variation and lesion studies. Neuroimage. 2008;42:1032–1044. doi: 10.1016/j.neuroimage.2008.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twohig JP, Cuff SM, Yong AA, Wang EC. The role of tumor necrosis factor receptor superfamily members in mammalian brain development, function and homeostasis. Reviews in Neuroscience. 2011;22:509–533. doi: 10.1515/RNS.2011.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, Khazipov R. Timing of the developmental switch in GABAA mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia. 2007;48:96–105. doi: 10.1111/j.1528-1167.2007.01295.x. [DOI] [PubMed] [Google Scholar]
- Urrea C, Castellanos DA, Sagen J, Tsoulfas P, Bramlett HM, Dietrich WD. Widespread cellular proliferation and focal neurogenesis after traumatic brain injury in the rat. Restorative Neurology & Neuroscience. 2007;25:65–76. [PubMed] [Google Scholar]
- Uylings HB, van Eden CG. Qualitative and quantitative comparison of the prefrontal cortex in rat and in primates, including humans. Progress in Brain Reserach. 1990;85:31–62. doi: 10.1016/s0079-6123(08)62675-8. [DOI] [PubMed] [Google Scholar]
- van der Kooij MA, Ohl F, Arndt SS, Kavelaars A, van Bel F, Heijnen CJ. Mild neonatal hypoxia-ischemia induces long-term motor- and cognitive impairments in mice. Brain Behavior & Immunology. 2010;24:850–856. doi: 10.1016/j.bbi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- van Handel M, Swaab H, de Vries LS, Jongmans MJ. Long-term cognitive and behavioral consequences of neonatal encephalopathy following perinatal asphyxia: a review. European Journal of Pediatrics. 2007;166:645–654. doi: 10.1007/s00431-007-0437-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhatalo S, Palva JM, Andersson S, Rivera C, Voipio J, Kaila K. Slow endogenous activity transients and developmental expression of K+-Cl- cotransporter 2 in the immature human cortex. European Journal of Neuroscience. 2005;22:2799–2804. doi: 10.1111/j.1460-9568.2005.04459.x. [DOI] [PubMed] [Google Scholar]
- Vannucci RC. Experimental biology of cerebral hypoxia-ischemia: Relation to perinatal brain damage. Pediatric Research. 1990;27:317–326. doi: 10.1203/00006450-199004000-00001. [DOI] [PubMed] [Google Scholar]
- Verger K, Junque C, Levin HS, Jurado MA, Perez-Gomez M, et al. Correlation of atrophy measures on MRI with neuropsychological sequelae in children and adolescents with traumatic brain injury. Brain Injury. 2001;15:211–221. doi: 10.1080/02699050010004059. [DOI] [PubMed] [Google Scholar]
- Vestergaard M, Madsen KS, Baaré WF, Skimminge A, Ejersbo LR, et al. White matter microstructure in superior longitudinal fasciculus associated with spatial working memory performance in children. Journal of Cognitive Neuroscience. 2011;23:2135–2146. doi: 10.1162/jocn.2010.21592. [DOI] [PubMed] [Google Scholar]
- Vexler ZS, Yenari MA. Does inflammation after stroke affect the developing brain differently than adult brain? Developmental Neuroscience. 2009;31:378–393. doi: 10.1159/000232556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Overview: normal and abnormal human brain development. Mental Retardation and Developmental Disabilities Research Reviews. 2000;6:1–5. doi: 10.1002/(SICI)1098-2779(2000)6:1<1::AID-MRDD1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Cerebral white matter injury of the premature infant-more common than you think. Pediatrics. 2003;112:176–180. doi: 10.1542/peds.112.1.176. [DOI] [PubMed] [Google Scholar]
- Wang WZ, Hoerder-Suabedissen A, Oeschger FM, Bayatti N, Ip BK, et al. Subplate in the developing cortex of mouse and human. Journal of Anatomy. 2010;217:368–380. doi: 10.1111/j.1469-7580.2010.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WZ, Oeschger FM, Montiel JF, García-Moreno F, Hoerder-Suabedissen A, et al. Comparative aspects of subplate zone studied with gene expression in sauropsids and mammals. Cerebral Cortex. 2011;21:2187–2203. doi: 10.1093/cercor/bhq278. [DOI] [PubMed] [Google Scholar]
- Wells R, Minnes P, Phillips M. Predicting social and functional outcomes for individuals sustaining pediatric traumatic brain injury. Developmental Rehabilitation. 2009;12:12–23. doi: 10.1080/17518420902773109. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Kockanek PM, Wisniewski SR, Bell MJ, et al. Interleukin-8 is increased in cerebrospinal fluid of children with severe head injury. Critical Care Medicine. 2000;28:929–934. doi: 10.1097/00003246-200004000-00003. [DOI] [PubMed] [Google Scholar]
- White T, Su S, Schmidt M, Kao CYGS. The development of gyrification in childhood and adolescence. Brain Cognition. 2010;72:36–45. doi: 10.1016/j.bandc.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins RC. Myelination: a critical stage of development. Neurotoxicology. 1986;7:103–120. [PubMed] [Google Scholar]
- Wilde EA, Chu Z, Bigler ED, Hunter JV, Fearing MA, et al. Diffusion tensor imaging in the corpus callosum in children after moderate to severe traumatic brain injury. Journal of Neurotrauma. 2006;23:1412–1426. doi: 10.1089/neu.2006.23.1412. [DOI] [PubMed] [Google Scholar]
- Wilde EA, Hunter JV, Newsome MR, Scheibel RS, Bigler ED, et al. Frontal and temporal morphometric findings on MRI in children after moderate to severe traumatic brain injury. Journal of Neurotrauma. 2005;22:333–344. doi: 10.1089/neu.2005.22.333. [DOI] [PubMed] [Google Scholar]
- Williams PA, Dou P, Dudek FE. Epilepsy and synaptic reorganization in a perinatal rat model of hypoxia-ischemia. Epilepsia. 2004;45:1210–1218. doi: 10.1111/j.0013-9580.2004.60403.x. [DOI] [PubMed] [Google Scholar]
- Wills GD, Wesley AL, Moore FR, Sisemore DA. Social interactions among rodent conspecifics: A review of the experimental paradigms. Neuroscience & Biobehavioral Reviews. 1983;7:315–323. doi: 10.1016/0149-7634(83)90035-0. [DOI] [PubMed] [Google Scholar]
- Winter JD, Dorner S, Lukovic J, Fisher JA, St Lawrence KS, Kassner A. Noninvasive MRI measures of microstructural and cerebrovascular changes during normal swine brain development. Pediatric Research. 2011;69:418–424. doi: 10.1203/PDR.0b013e3182110f7e. [DOI] [PubMed] [Google Scholar]
- Wise SP, Jones EG. The organization and postnatal development of the commissural projection of the rat somatic sensory cortex. Journal of Comparative Neurology. 1976;168:313–343. doi: 10.1002/cne.901680302. [DOI] [PubMed] [Google Scholar]
- Wood SL, Beyer BK, Cappon GD. Species comparison of postnatal CNS development: functional measures. Birth Defects Research. 2003;68:391–407. doi: 10.1002/bdrb.10037. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Krach L, Ward E, Mueller BA, Muetzel R, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Archives of Clinical Neuropsychology. 2007;22:555–568. doi: 10.1016/j.acn.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ling EA. Studies of the ultrastructure and permeability of the blood-brain barrier in the developing corpus callosum in postnatal rat brain using electron dense tracers. Journal of Anatomy. 1994;184:227–237. [PMC free article] [PubMed] [Google Scholar]
- Yager JY, Ashwal S. Animal models of perinatal hypoxicischemic brain damage. Pediatric Neurology. 2009;40:156–167. doi: 10.1016/j.pediatrneurol.2008.10.025. [DOI] [PubMed] [Google Scholar]
- Yager JY, Thornhill JA. The effect of age on susceptibility to hypoxicischemic brain damage. Neuroscience & Biobehavioral Reviews. 1997;21:167–174. doi: 10.1016/s0149-7634(96)00006-1. [DOI] [PubMed] [Google Scholar]
- Yuan W, Holland SK, Schmithorst VJ, Walz NC, Cecil KM, et al. AJNR American Journal of Neuroradiology. 2007;28:10. doi: 10.3174/ajnr.A0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BL, Chen X, Tan T, Yang Z, Carlos D, et al. Traumatic brain injury impairs synaptic plasticity in hippocampus in rats. Chinese Medical Journal. 2011;124:740–745. [PubMed] [Google Scholar]
- Zhang ZW. Postnatal development of the mammalian neocortex: Role of activity revisited. Canadian Journal of Neurological Science. 2006;33:158–169. doi: 10.1017/s0317167100004911. [DOI] [PubMed] [Google Scholar]
- Zhong J, Carrozza DP, Williams K, Pritchett DB, Molinoff PB. Expression of mRNAs encoding subunits of the NMDA receptor in developing rat brain. Journal of Neurochemistry. 1995;64:531–539. doi: 10.1046/j.1471-4159.1995.64020531.x. [DOI] [PubMed] [Google Scholar]
- Zhou M, Schools GP, Kimelberg HK. Development of GLAST(+) astrocytes and NG2(+) glia in rat hippocampus CA1: Mature astrocytes are electrophysiologically passive. Journal of Neurophysiology. 2006;95:134–143. doi: 10.1152/jn.00570.2005. [DOI] [PubMed] [Google Scholar]
- Zhu C, Qiu L, Wang X, Xu F, Nilsson M, et al. Age-dependent regenerative responses in the striatum and cortex after hypoxia-ischemia. JCBFM. 2009;29:342–354. doi: 10.1038/jcbfm.2008.124. [DOI] [PubMed] [Google Scholar]
- Zhu C, Wang X, Xu F, Bahr BA, Shibata M, et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differentiation. 2005;12:162–176. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]