

Abstract
Drosophila melanogaster is widely used to study genetic factors causing Parkinson’s disease (PD) due largely to the use of sophisticated genetic approaches and the presence of a high conservation of gene sequence/function between Drosophila and mammals. However, in Drosophila little has been done to study the environmental factors which cause over 90% of PD cases. We used Drosophila primary neuronal culture to study degenerative effects of a well-known PD toxin MPP+. DA neurons were selectively degenerated by MPP+ whereas cholinergic and GABAergic neurons were not affected. This DA neuronal loss was due to post-mitotic degeneration, not by inhibition of DA neuronal differentiation. We also found that MPP+-mediated neurodegeneration was rescued by D2 agonists quinpirole and bromocriptine. This rescue was through activation of Drosophila D2 receptor DD2R, as D2 agonists failed to rescue MPP+-toxicity in neuronal cultures prepared from both a DD2R deficiency line and a transgenic line pan-neuronally expressing DD2R RNAi. Furthermore, DD2R autoreceptors in DA neurons played a critical role in the rescue. When DD2R RNAi was expressed only in DA neurons, MPP+ toxicity was not rescued by D2 agonists. Our study also showed that rescue of DA neurodegeneration by Drosophila DD2R activation was mediated through suppression of action potentials in DA neurons.
Keywords: Drosophila melanogaster, Primary neuronal culture, DD2R, D2 agonist, Parkinson’s disease
Introduction
A cellular hallmark of Parkinson’s disease (PD) is the loss of dopaminergic (DA) neurons in several brain areas, particularly in the substantia nigra. Recent genetic discoveries confirmed that familial forms of PD are directly associated with mutations of certain genes such as α-Synuclein and parkin (Dauer & Przedborski, 2003; Moore et al, 2005; Cookson, 2005). In contrast, the majority of PD cases occur sporadically without clear genetic linkage. Indeed, neurotoxin-based PD models have shown that the loss of DA neurons can be induced by non-genetic, environmental factors (Bove et al, 2005). These findings indicate that a combination of genetic and environmental factors contributes to the pathogenesis of PD.
Among several neurotoxins inducing PD symptoms, a toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is extensively used in a variety of animal models (Bove et al, 2005; Cannon & Greenamyre, 2010). The neurological effects of MPTP were discovered after a small group of drug users developed severe motor dysfunction by administration of a synthetic opioid, 1-methyl-4-phenyl- 4-propionoxypiperidine (MPPP), tainted with MPTP (Langston, 1983). MPTP is converted into the active metabolite 1-methyl-4-phenyl pyridinium (MPP+) by monoamine oxidase in astrocytes. It exerts DA neurotoxicity when up-taken by DA transporter. MPP+ induces oxidative stress leading to mitochondria dysfunction and ultimately causes DA cell death (Przedborski et al, 2004).
Although PD is currently incurable, there are therapeutic drugs such as levadopa which alleviates the PD symptoms (Olanow, 2009; Calabresi et al, 2011). However, an overall lack of clinical treatments that effectively prevent or slow PD development without causing concerns about adverse side effects warrants efforts to understand the molecular and cellular mechanisms underlying PD pathology which may help develop new strategies for PD treatment. Models using toxins to generate PD-like effects allow researchers to study the pathology of PD and develop potential protective treatments from toxin-induced degeneration. One area receiving a great deal of attention is DA receptor signaling (Bozzi & Borrelli, 2006). Indeed, Chen et al (2009) reported that a D2 agonist, ropinirole, prevents rotenone-induced apoptosis of dopaminergic cell line. D2 agonists are also used in PD patient therapy (Schapira & Olanow, 2004). The neuroprotective mechanisms, however, initiated by D2 agonists are poorly understood despite many clinical trials with those drugs.
By utilizing the sophisticated genetic approaches available in Drosophila, research using this animal has been successful in increasing our knowledge about the mechanisms of neurodegenerative disorders (Muqit & Feany, 2002, Bilen & Bonini, 2005). Since the introduction of fly PD models carrying mutated human α-synuclein genes (Feany & Bender, 2000; Auluck et al, 2002), most of known PD-related genes (e.g. parkin, LRRK2, etc) are examined in Drosophila (Whitworth, 2011). The success of Drosophila models makes it a viable option for developing a new system to study environmentally induced PD. Currently, only a handful of studies have examined non-genetic factors (e.g. rotenone, paraquat) causing PD in Drosophila (Coulom & Birman, 2004; Chaudhuri et al, 2007; Lawal et al, 2010). In our study, we developed a cellular model to study a PD toxin MPP+ in primary Drosophila neuronal cultures. Furthermore, we showed that MPP+-mediated neurodegeneration is rescued by D2 receptors through reduced DA neuronal excitability using a combined approach of genetics, pharmacology and electrophysiology.
Methods & Materials
Fly stocks
Flies were raised on a standard cornmeal agar diet at room temperature. A “Cantonized” white eye stock w1118 was used as wild type. TH-Gal4 lines were obtained from Dr. S. Birman at the Dev. Biol. Institute of Marseille, France (Friggi-Grelin et al, 2003). DD2R deficiency line, 1407-Gal4, UAS-DD2R RNAi lines were provided by the Bloomington Drosophila Stock Center.
Drosophila primary neuronal cultures
Mid-gastrula embryos were collected and dechorionated in 50% bleach solution. The embryonic contents were harvested and plated on photoetched or round coverslips (Bellco Glass Inc., Vineland, NJ) as previously described (Park & Lee, 2006; Park et al, 2007). Cultures were then incubated in 5% CO2 at 24 – 25°C and maintained for 9 days in vitro (DIV). Culture medium (DDM1) used in this study was a mixture of high glucose Ham’s F-12/Delbecco’s medium (Irvine Scientific, Santa Ana, CA), L-glutamine (2.5mM; Irvine Scientific), HEPES (20mM), and 4 supplements: 100μM putrescine, 20ng/ml progesterone, 100μg/ml transferrin, and 50μg/ml insulin (Calbiochem, San Diego, CA). MPP+ and D2 agonists (quinpirole and bromocriptine) used in this study were purchased from Sigma (St. Louis, MO).
Pharmacological treatments
All drugs were added to cultures at 3 days in vitro (DIV) except BrdU (Sigma, St Louis, MO) which was added at 0 or 3 DIV. MPP+ iodide (Sigma, St Louis, MO) was stored in darkness. The stock of MPP+ was made freshly in distilled water before use. For all our experiments, MPP+ was prepared and disposed according to the guideline reviewed in Przedborski et al (2001). Quinpirole and bromocriptine were dissolved in distilled water and in DMSO, respectively. The stocks of those chemicals were stored at −20°C until added to the neuronal culture.
Live Imaging
For live imaging, all neurons were cultured on photoetched coverslips which allow us to return to the same field of view (or square). At 3 DIV, squares containing 2–7 DA neurons were selected. These identified squares were re-assessed at days 5 or 9 DIV. In this study, the threshold intensity for GFP(+) neurons was 3x higher than the background (<10% of the maximum pixel intensity) as previously described (Park et al, 2007). More than 50% of the experiments were performed blind with respect to genotype or drug-treated versus control cultures.
Live neuronal culture of TH-GFP (9 DIV) was also used for propidium iodide (PI, In Vitrogen) staining. PI (1μl/ml) was added into culture medium and left for 5 min. After 5 min washing, images of ‘dim (=sub-threshold)’ GFP(+) neurons were acquired. In order to have a clear GFP signal, the brightness of each image was digitally increased. Then individual sub-threshold GFP(+) neurons were examined for overlapping with PI. The average fluorescent intensity of sub-threshold GFP(+) neurons was about 50% of the threshold (3x higher than the background).
Immunofluorescence assay
Neurons in culture were fixed with 4% paraformaldehyde for 40 minutes on ice. Then, the cultures were washed 3 times with 10 mM phosphate buffer solution (PBS). Blocking and permeabilization step was done by using 0.1% Triton X-100 and 5% goat serum (Sigma, St. Louis, MO) for 30 minutes on ice. The permeablized cultures were incubated with a primary Ab (e.g., mouse anti-BrDU, mouse anti-tyrosine hydroxylase) overnight at 4°C and then with a secondary Ab (e.g., FITC or TRITC labeled) for 1 hour at 25°C. After staining was completed the cultures were washed 3 times for 10 minutes, then the coverslips were mounted on slide glasses and viewed under a fluorescent microscope (Olympus IX71). Images were taken with a Spot CCD digital camera (Diagnostic Instruments, Sterling Heights, MI).
DAPI-based quantification of TH(+) DA neurons
To count total cells in each image, DAPI (Molecular Probes, Eugene, OR) was used as previously described (Park & Lee, 2006). In order to minimize the subjectivity in manual cell counting, we used Image J software - a public domain image processing and analysis program (http://rsb.info.nih.gov/ij/index.html). A sample area in each image was chosen to calculate the unit pixel intensity (UPI) for each cell. The UPI was obtained by dividing the pixel intensity in a sample area with the manually counted number of cells in that area (typically 30–60 cells). Then the total DAPI signal intensity in an image was measured and divided by the UPI in order to quantify the total number of DAPI(+) cells in that image. Finally, the number of TH(+) DA neurons in that image was normalized to 1,000 DAPI(+) cells.
Action potential (AP) recording
Each coverslip containing Drosophila neuronal cultures was transferred into a recording chamber containing the following external solution (mM): 140 NaCl, 1 CaCl2, 4 MgCl2, 3 KCl, and 5 HEPES, pH 7.2. Action potentials (APs) were extracellularly recorded with patch pipettes (tip resistance 4~6 MΩ) in a cell-attached mode as previously described (Yuan & Lee, 2007). The pipette was filled with internal solutions containing the following ingredients (mM): 120 CsOH, 120 D-gluconic acid, 0.1 CaCl2, 2 MgCl2, 20 NaCl, 1.1 EGTA, and 10 HEPES, pH 7.2. Axopatch 200B amplifier (Axon Instruments Inc, Union City, CA, USA) was used to measure APs.
DA neurons were identified using a live marker GFP, of which expression was driven by a DA specific driver TH-Gal4 (see above). A D2 agonist quinpirole were focally delivered to the patched neurons showing APs by using a Picospritzer III (Parker Hannifin Corp, Fairfield, NJ, USA) for duration of 30 seconds. AP frequency was analyzed using the Minianalysis detection software (Synaptosoft, Decatur, GA, USA).
Results
MPP+ degenerates GFP(+) dopaminergic (DA) neurons in Drosophila primary culture
In a previous study, we developed a live tracking method which allowed quantification of pre-identified dopaminergic (DA) neurons over time in Drosophila primary culture (Park et al, 2007). Using a method similar to this, we studied neurodegeneration induced by a well known PD toxin MPTP. The standard UAS-Gal4 system (Brand & Perrimon, 1993) was used to drive expression of GFP specifically in DA neurons under the control of TH-Gal4, a DA specific driver (Friggi-Grelin et al, 2003). First, cultured neurons were examined with a fluorescent microscope 3 days after plating. Then, we chose squares containing GFP-positive DA neurons (Fig. 1A). These same squares were later re-assessed to count all GFP-positive neurons at 5 and 9 days in vitro (DIV). Neuronal cultures were randomly split into two groups: half control and half for treatments. At 3 DIV, the number of GFP(+) neurons was not different between these two culture groups (Figure 1B inset). We used 3 different concentrations (10, 40, 100μM) of MPP+ (active metabolite of MPTP toxin). DA degeneration by 40μM MPP+ was clearly observed at 9 DIV (Student t-test, p<0.01). 10μM MPP+ caused the number of GFP(+) DA neurons to be significantly lower at 9 DIV but the effect (~13% reduction) was modest compared to 40μM (~37% reduction). We also tested a higher concentration of MPP+ (100μM). The toxic effect on DA neurons by 100μM was similar to that by 40μM MPP+ but we also observed signs of non-specific neurodegeneration in the culture such as morphological alterations in non-DA neuronal processes. The results show that 40μM MPP+ causes degeneration of DA neurons (Figure 1B) and is appropriate for further examinations.
Figure 1. MPP+ reduces number of dopaminergic neurons in Drosophila primary culture.

(A) An overlapped image of bright-field and GFP signals in a marked square (8K in this case). Six GFP(+) dopaminergic (DA) neurons are indicated by arrows. Neuronal cultures were derived from a cross between TH-Gal4 and UAS-GFP lines (=TH-GFP). As shown previously (Park et al, 2007), these cultures were plated onto photoetched coverslips which allow for identifying a square containing GFP(+) neurons that can then be returned to at a later time. Scale bar = 20μm. Inset An enlarged view of the dotted square in (A). showing a GFP(+) neuron is in a neuronal cluster containing GFP(−) neurons. (B) A graph showing percent change in number of GFP(+) DA neurons in the absence and presence of MPP+. Number of GFP(+) neurons at 5 and 9 days in vitro (DIV) was normalized to that at 3 DIV. The control and MPP+ treated cultures have a similar number of GFP(+) DA neurons at 3DIV (Inset, see below). However, at 9 DIV, the number of GFP(+) neurons in the control dish has increased while the number has slightly decreased in the MPP+ treated cultures. Inset A graph showing number of GFP(+) neurons per each square averaged at 3 days in vitro (DIV). Coverslips were randomly split into three groups: control, 10μM MPP+ or 40μM MPP+. Control (n=110), 10μM MPP+ (n=55) & 40μM MPP+ (n=61) from total 6 separate experiments. Student t-test, *P<0.05 & **P<0.01.
Interestingly, in the control group, the number of GFP(+) neurons per square at 9 DIV increased by 142.1+/−6.2% compared to that at 3 DIV. This increase may be explained by newly differentiated DA neurons in culture after 3 DIV. If so, DA neuronal loss by MPP+ may not be due to degeneration, but due to suppressed mitotic division of DA neurons. Thus, we wanted to examine whether DA neurons differentiate after 3 DIV. A mitotic marker BrdU (Boone & Doe, 2008) was added to the neuronal culture as soon as prepared (0 day) or after being incubated for 3 days. Then we stained BrdU signals using anti-BrdU antibody. The vast majority of neurons in culture were anti-BrdU positive at 3 DIV (Fig. 2). In contrast, few neurons (<2%) were anti-BrdU positive when neurons were stained at 6 DIV after 3 day incubation with BrdU. The BrdU signal was also examined in GFP(+) DA neurons with anti-BrdU antibody. Figure 2C shows that the vast majority of DA neurons are also differentiated for the first 3 DIV and then maintained post-mitotically.
Figure 2. Cultured primary neurons including DA neurons are post-mitotic at 3 DIV or older.
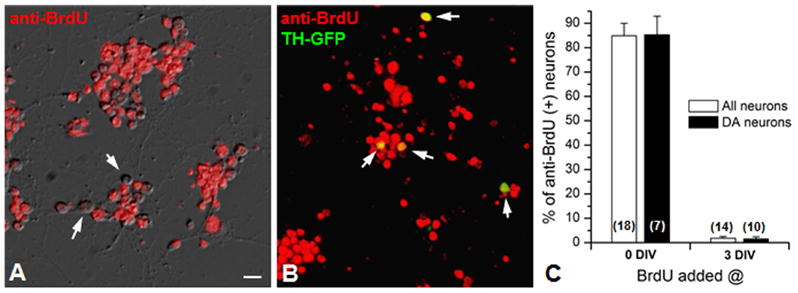
(A) An overlapped image of bright-field and anti-BrdU (red) signals at 3 DIV. In this field of view, all neurons are BrdU(+) except a few neurons indicated by arrows. (B) An overlapped image of anti-BrdU (red) and GFP(+) signals. TH-Gal4 x UAS-GFP (=TH-GFP) line was used to mark DA neurons and thus overlapped signals (yellow, indicated by arrow) are anti-BrdU(+) DA neurons. (C) A bar graph showing percentage of anti-BrdU(+) neurons in culture. BrdU was added at 0 or 3 DIV and neurons were incubated for the following 3 days before immunostaining. Scale bar=20μm.
MPP+ is known to cause DA cell death (Chun et al, 2001) and, therefore, we wondered whether reduced number of GFP(+) DA neurons by MPP+ is due to actual neurodegeneration. A cell death marker propidium iodide (PI, 1μl/ml), which binds to DNA, was used to stain TH-GFP neuronal culture at 9DIV. No GFP (+) neurons above the threshold (refer to Methods and Materials) were co-stained with PI in both control and MPP+-treated cultures. Thus, we focused on ‘dim’ GFP(+) neurons which were below the threshold. These sub-threshold GFP(+) neurons were selected on the basis of GFP intensity regardless of treatments (i.e. control vs. MPP+-treated) and showed about 50% of the threshold intensity on average. A subset (0.52+/−0.3%) of sub-threshold GFP(+) neurons in control was overlapped with PI (Figure 3A). This percent significantly increased (~8 times) when MPP+ was treated (Figure 3B). The results show that MPP+ enhances DA neurodegeneration and cell death.
Figure 3. Increase of propidium iodide (PI)-positive neurons by MPP+.
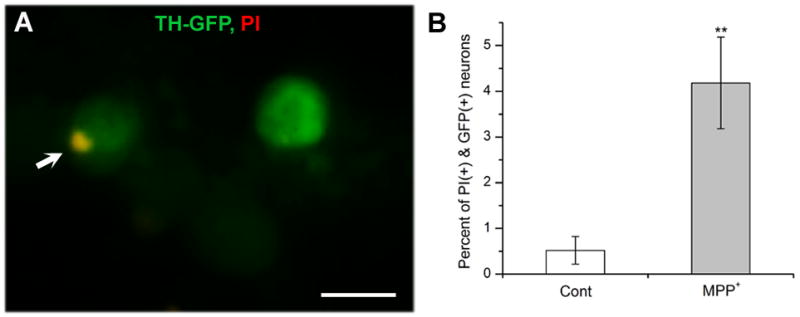
<>br(A) A sub-threshold GFP(+) neuron was overlapped with PI(+) signal (yellow spot indicated by an arrow) while the other ‘brighter’ GFP(+) neuron was not. Scale bar=10μm. (B) A graph showing percent of GFP(+) neurons (9DIV) co-stained with PI in the absence (Cont) and presence of MPP+. 40μM MPP+ was added at 3DIV. In these experiments, total sub-threshold GFP(+) neurons examined were 1,319 neurons for control and1,906 neurons for MPP+ treated from 4 separate experiments, respectively. Average fluorescent intensity of these sub-threshold neurons were about 50% of the threshold cut-off. Student t-test, **P<0.01.
Anti-TH(+) DA neurons are decreased by MPP+
Our results demonstrate that MPP+ induces neurodegeneration. However, Figure 1B shows that the number of GFP(+) neurons did not change significantly at 9 DIV after MPP+ treatment compared to 3 DIV while it was higher in control. Thus, we wanted to confirm whether MPP+ would decrease the number of DA neurons with another well known DA marker tyrosine hydroxylase (TH). Using anti-TH, we stained neurons in cultures derived from two different strains: wild type and TH-GFP. Both strains, considered as controls, showed that the number of TH(+) DA neurons was reduced at 9 DIV compared to that at 3 DIV while there was no difference between these two strains (Fig. 4B). We also examined the number of DA neurons in MPP+ treated culture at 9 DIV. The results showed a clear reduction of DA neurons by MPP+ (Fig. 4C). The results show that MPP+ degenerates anti-TH(+) DA neurons in Drosophila neuronal culture. Taken together, our study demonstrated that both GFP marking and anti-TH staining are very useful and reliable in quantifying DA neurons in primary cultures although two methods showed different patterns in changes of DA neuron number.
Figure 4. Reduction of anti-TH(+) neuron number in culture by MPP+.
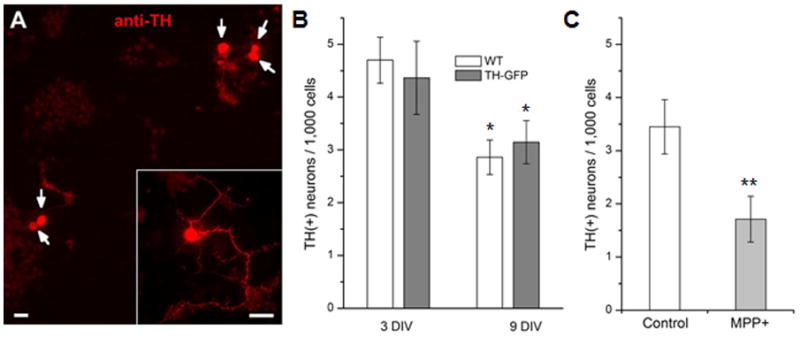
(A) A fluorescent image of cultured neurons strained with anti-TH. Five DA neurons (indicated with arrow) were observed in this field of view. Inset High power image shows TH(+) neuritic processes in addition to the soma. Scale bar=20μm. (B) Anti-TH (+) neurons were quantified at 3 and 9 DIV in wild type (WT) and TH-GFP lines. The number of anti-TH(+) neurons at 9 DIV was significantly reduced compared to that at 3 DIV (Student t-test, *P<0.05). In contrast, there was no difference between two fly lines. In this graph, the number of anti-TH (+) neurons was normalized to 1,000 DAPI (+) cells as described in Materials and Methods section. Data from 6 separate experiments. (C) A graph showing number of anti-TH(+) neurons in the absence and presence of 40μM MPP+. MPP+ was added at 3 DIV and neuronal cultures were stained with anti-TH antibody at 9 DIV. Data from 4 separate experiments. Student t-test, **P<0.01.
MPP+ toxicity is enhanced by overexpression of dopamine transporter (DAT)
MPP+ (40μM) decreases GFP(+) DA neurons in Drosophila primary culture. However, 40μM MPP+ concentration is higher compared to other studies using rodent cells (1μM – 10μM MPP+; Marini et al, 1989; Sanchez-Ramos et al, 1997; Bains et al, 2007). It has been known that MPP+ is taken up by DA transporters (DAT) in DA neurons (Storch et al, 2004). Therefore, we suspected that DAT in Drosophila does not transport MPP+ as effectively as in rodents. DAT was overexpressed in DA neurons by a cross between two fly transgenic lines TH-Gal4 and UAS-DAT. 10μM MPP+ significantly decreased TH(+) neurons and this toxic effect was similar to that of 40 μM (Table 1), strongly indicating that Drosophila DA neurons are less sensitive to MPP+ due to lower activity of DAT.
Table 1.
Overexpression of dopamine transporter (DAT) in DA neurons increases sensitivity to MPP+.
| Treatment | Number of DA neurons per 1,000 cells (+/−SEM) | N (# of images) |
|---|---|---|
| Control | 2.84+/− 0.17 | 3 (68) |
| 10μM MPP+ | 1.88+/− 0.17* | 3 (73) |
| 40μM MPP+ | 1.58+/−0.29* | 3 (59) |
N = number of experiment sets;
p≤0.05 (Student t-test)
Degeneration by MPP+ is not observed in non-DA neurons
In this study, we wanted to examine whether MPP+ toxicity is also observed in nonDA neurons. Cholinergic and GABAergic neurons are two major types of neurons in the intact nervous system as well as the primary neuronal culture (Lee & O’Dowd, 1999; Lee et al, 2003), so we examined the toxic effects of MPP+ on both of those neuronal subtypes. We prepared neuronal cultures on photoetched coverslips derived from a fly strain carrying the following transgenes: Cha-Gal4 x UAS-GFP and Gad1-RFP. The culture is expected to express GFP in cholinergic neurons while red fluorescent protein (RFP) is in GABAergic neurons. The selectivity of 40μM MPP+ was tested by quantifying live cholinergic and GABAergic neurons treated with MPP+ using the same method for GFP(+) DA neuron quantification. Figure 5 shows that neither cholinergic nor GABAergic neurons were affected by 40μM MPP+ at 5 or 9 DIV. There was no statistical difference between the cholinergic neurons in the control and MPP+ treated cultures. Similarly, the percent change of GABAergic neurons was not statistically significant between the control and the 40μM MPP+ treated cultures. This confirms that 40μM MPP+ is selective for DA neurons and thus a proper concentration for the rest of the studies.
Figure 5. Cholinergic and GABAergic neurons are not affected by 40μM MPP+.
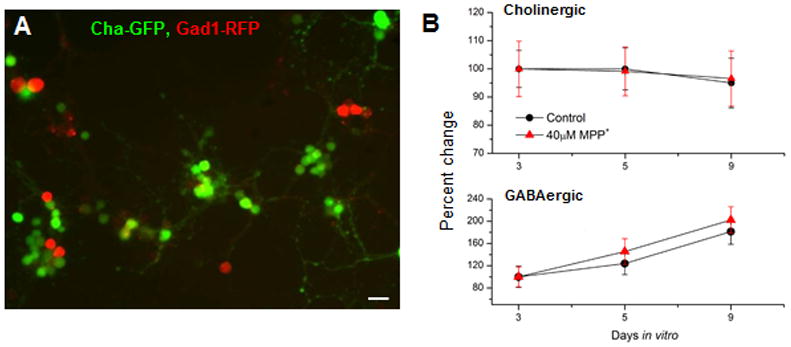
(A) A fluorescent image showing GFP-labeled cholinergic and RFP-labeled GABAergic neurons. Neuronal cultures were prepared from a fly line carrying Cha-Gal4, UAS-GFP and Gad1-RFP transgenes (=Cha-GFP; Gad1-RFP). (B) Top panel Number of GFP-labeled cholinergic neurons was quantified per square at 3, 5 and 9 DIV in the absence and presence of 40μM MPP+. Bottom panel RFP-labeled GABAergic neurons were quantified per square at 3, 5 and 9 DIV in the absence and presence of 40μM MPP+. n number: Cholinergic (n=20) and GABAergic (n=20)
D2 agonists rescue DA neurodegeneration mediated by MPP+
D2 agonists are known to ameliorate PD symptoms (Schapira & Olanow, 2004; Ferrari-Toninelli et al, 2008), so we wanted to test whether these drugs rescue the loss of DA neurons by MPP+. Neuronal cultures were added with D2 agonists in addition to MPP+. We chose two D2 agonists, quinpirole and bromocriptine for this study. The former is known to modify certain motor behaviors in Drosophila (Andretic & Hirsh, 2000; Draper et al, 2007). The latter has been tested for its neuroprotective effects in PD patients (Bozzi & Borrelli, 2006). When 10 μM quinpirole was added to cultures at 3 DIV (Fig. 6A), MPP+ toxicity was significantly rescued although the rescue was not complete. Bromocriptine showed a similar rescue effect. Figure 6B summarizes the rescue effects of both D2 agonists against MPP+ toxicity. In this study, we also examined whether D2 agonists increase DA neuronal survival by themselves. Our results showed that quinpirole or bromocriptine alone did not increase number of GFP(+) neurons (Figure 6C) compared to control, demonstrating that DA neuroprotection by D2 agonists is a direct effect on MPP+ toxicity. In contrast, a D1 agonist SKF38393 (Yuan & Lee, 2007) did not show any rescue effect (data not shown). These results strongly suggest that D2 agonists rescue MPP+ toxicity through D2 receptors. Therefore, we examined whether a D2 antagonist haloperidol can inhibit the rescue effect of quinpirole. When neuronal cultures were treated with 10μM haloperidol at 3DIV in addition to MPP+ and quinpirole, there was no rescue observed (Table 2). Our results show that the rescue of MPP+ toxicity by quinpirole and bromocriptine is D2-specific.
Figure 6. D2 agonists rescue MPP+-induced neurodegeneration.
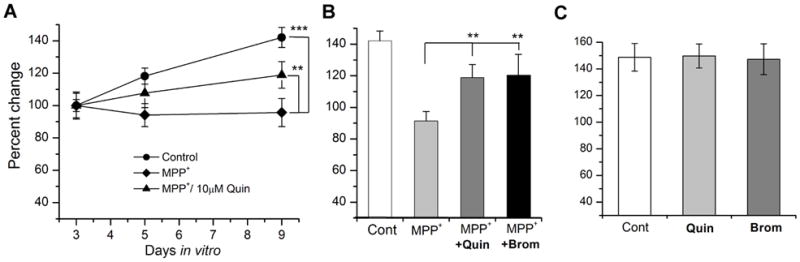
(A) Quinpirole (10μM), added at 3 DIV in addition to MPP+, partially rescued DA neurodegeneration. (B) A graph showing neuroprotective effect of D2 agoinsts quinprirole and bromocriptine against MPP+-induced neurodegeneration. Total GFP(+) neurons in a square was counted at 9DIV and normalized to that at 3 DIV. Student t-test, *p<0.05, **p<0.01. Data from 4 separate experiments: control (n=48), MPP+ (n=41) MPP++Quin (n=31) & MPP++Brom (n=20). (C) Quinpirole or bromocriptine alone without MPP+ did not show any difference in the number of GFP(+) neurons compare to control.
Table 2.
Rescue of MPP+-induced neurotoxicity by quinpirole is significantly suppressed by a D2 antagonist haloperidole (10μM).
| Treatment | Number of DA neurons per 1,000 cells (+/−SEM) | n |
|---|---|---|
| Control | 3.90+/− 0.12 | 55 |
| MPP+ | 2.47+/− 0.06*** | 28 |
| MPP+ + Quin | 3.54+/− 0.27 | 105 |
| MPP+ + Quin + Hal | 2.30+/− 0.12*** | 90 |
p≤0.001 (Student t-test)
Rescue of MPP+ toxicity by quinpirole requires expression of Drosophila D2-like receptor DD2R
Although the rescue effect of D2 agonists are expected to work through D2 receptor signaling, no study has addressed this using molecular means other than pharmacological means. Furthermore, some studies suggest antioxidant effects of D2 agonists (Hara et al, 2006; Lim et al, 2008). Thus, we wanted to examine whether this rescue effect is mediated by D2 receptor activation. In Drosophila, there are four DA receptors (i.e. dDA1, DAMB, DopEcR & DD2R) cloned and characterized (Goetz et al, 1994; Han et al, 1996; Srivastava et al, 2005; Mustard et al, 2005). Among them, DD2R is the only Drosophila D2 receptor that has been identified so far (Hearn et al, 2002). Therefore, we first prepared neuronal cultures from a fly line deficient in the DD2R gene. Since the homozygous DD2R deficient (Df) line mutant is lethal, this Df fly line is maintained over a balancer chromosome - FM6b, act-GFP. Thus, homozygous DD2R Df neuronal cultures were identified by GFP signals. The GFP-negative neuronal cultures were DD2R Df homozygous (refer to Darya et al (2009) for experimental detail). The GFP(−) cultures were treated with MPP+ alone or MPP+ plus quinpirole. As seen in Figure 7A, MPP+ reduced the number of anti-TH(+) neurons but there was no rescue by quinpirole. Although the overall morphology of DD2R Df neuronal cultures was not noticeably altered, we cannot rule out a possibility that this effect is not due to DD2R deficiency, because this line is also deficient in additional gene(s). In Drosophila, genetic approaches allow specific knockdown of D2Rs. Therefore, we also used a fly line carrying RNAi against DD2R (i.e. UAS-DD2R RNAi from Bloomington Drosophila Stock Center). A pan-neuronal driver 1407-Gal4 (Sweeney et al, 1995) was crossed with UAS-DD2R RNAi to drive expression of DD2R RNAi in all neurons. Neuronal cultures prepared from this crossed line were treated with MPP+ plus quinpirole in addition to MPP+ alone. Similar to the DD2R Df experiments, there was no rescue observed in the presence of quinpirole (Figure 7B). The results demonstrate that D2 rescue effect is through the activation of Drosophila DD2R receptor.
Figure 7. Genetic lesions of Drosophila D2 receptor DD2R causes no rescue of MPP+ toxicity by quinpirole.
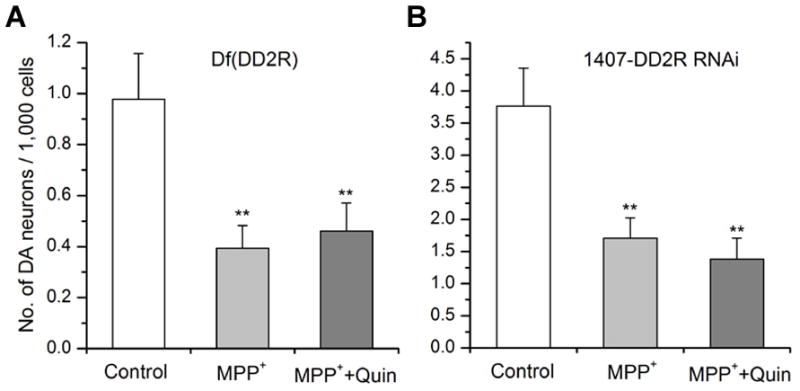
(A) A DD2R deficiency line was used to prepare primary neuronal culture. Homozygous Df(DD2R) was identified using GFP de-selection marking (refer to Darya et al, 2009). Subset of homozygous cultures were treated with MPP+ alone or MPP+ plus quinpirole. Control was not treated with any drug. At 9 DIV, neuronal cultures were fixed and stained with anti-TH in order to quantify DA neurons in culture as described in Materials and Methods section. The number of antiTH(+) neurons was reduced by MPP+ while no rescue was observed in neuronal culture treated with quinpirole (Student t-test, **p<0.01). Data from 3 separate experiments: control (n=81), MPP+ (n=50) & MPP++Quin (n=59). (B) A D2 agonist quinpirole (10μM) did not rescue MPP+-mediated degeneration of DA neurons in Drosophila culture expressing DD2R-RNAi transgene driven by a pan-neuronal driver 1407-Gal4. Neuronal cultures were prepared from a cross of 1407-Gal4 and UAS-DD2R-RNAi lines. A subset of cultures were treated with MPP+ alone or MPP+ plus quinpirole. No rescue was observed in neuronal cultures (9 DIV) treated with quinpirole (Student t-test, **p<0.01). Data from 4 separate experiments: control (n=49), MPP+ (n=59) & MPP++Quin (n=35). Anti-TH antibody was used to identify DA neurons in these experiments.
Down-regulation of Drosophila D2 autoreceptors in DA neurons results in no rescue of MPP+ toxicity by D2 agonists
Since DD2Rs are widely expressed in the central nervous system including DA and non-DA neurons (Hearn et al, 2002), it is possible that DA neuroprotection can be mediated through DD2Rs expressed in DA (called autoreceptors), non-DA neurons or both. In this study, therefore, we used a DA-specific driver TH-Gal4 to induce DD2R RNAi only in DA neurons. Neurons were cultured from a transgenic line carrying UAS-GFP and UAS-DD2R RNAi in addition to a DA driver TH-Gal4. GFP(+) neurons were quantified in the presence or absence of drugs (e.g., MPP+ and/or quinpirole). Figure 8 showed that quinpirole failed to rescue MPP+-mediated neurodegeneration. Interestingly, GFP(+) neurons in the control group were not increased at 9 DIV (close to 100%) compared to 3 DIV unlike in Figure 1. This is perhaps because of the gene-dosage effect as the transgenic fly (heterozygous due to a cross of two line) used in this study carries only one copy of each GFP and TH-Gal4 gene compared to two copies in the fly used in Figure 1 (homozygous and thus two copies of each transgene). The results demonstrated that activation of DD2R autoreceptors is required for the rescue of MPP+ topxicity mediated by D2 agonists.
Figure 8. Down-regulation of D2 autoreceptors by a DD2R RNAi transgene fails to protect MPP+-induced DA degeneration.
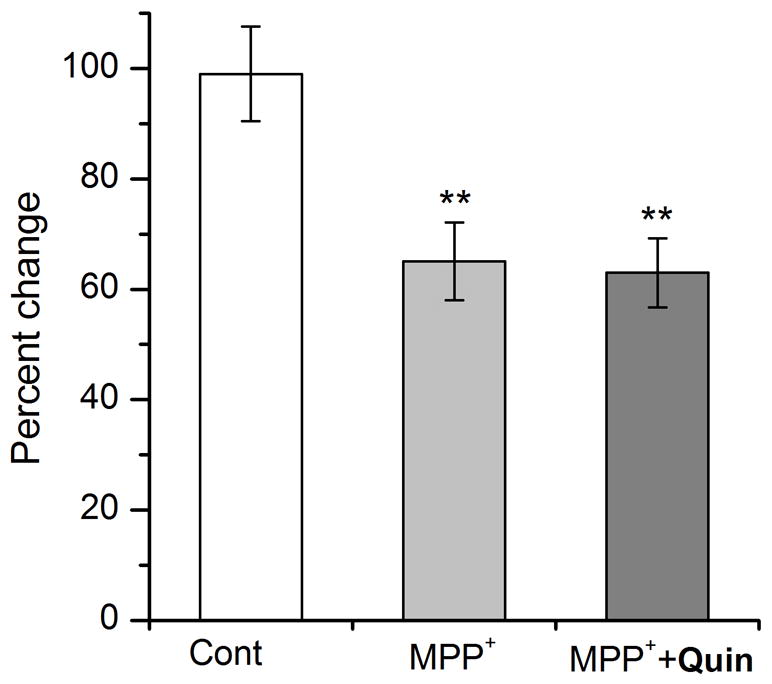
A DA specific driver TH-Gal4 was used to induce expression of DD2R RNAi transgene. A subset of cultures were treated with MPP+ alone or both MPP+ and quinpirole (MPP++Quin). At 9 DIV, DA(+) neurons were quantified using a live marker GFP and normalized that at 3 DIV. Neuronal culture was prepared on photoetched coverslips. No rescue was observed in neuronal cultures treated with quinpirole (Student t-test, **p<0.01). Data from 3 separate experiments: control (n=48), MPP+ (n=35) & MPP++Quin (n=45).
DA neuroprotection is mediated by activation of D2 autoreceptors and a sodium channel blocker TTX through the reduction of DA neuronal excitability
Our next attempt was to understand the mechanisms underlying neuroprotection by DD2R. It is known that D2R regulates excitability of DA neurons, and thus reduce excitotoxic cell death (Piallat et al, 1996; Schapira & Olanow, 2003). Therefore, we examined whether Drosophila DD2R regulates DA cell excitability in the primary neuronal culture. As previously described (Yuan & Lee, 2007), the patch clamping technique was used to measure action potentials (APs) in DA neurons, which were identified with a GFP marker (refer to Fig. 1A). Quinpirole (10μM) was focally applied to a DA neuron showing APs in order to examine the effect of DD2R activation. Figure 9A showed that DD2R activation by quinpirole significantly reduces the frequency of APs in DA neurons. This quinpirole effect was also observed in the presence of nAChR blocker curare and/or GABAA-R blocker picrotoxin (data not shown). Frequency of APs in DA neurons was reduced to 45+/−10.3% to the control (Fig. 8B), demonstrating that quinpirole-mediated activation of DD2R suppresses the excitability of DA neurons. In the next experiment, we wanted to further test whether the reduction of DA neuronal excitability is enough to show neuroprotective actions, independent of DD2R activation. A powerful AP blocker tetrodotoxin (TTX) was added in the neuronal culture in addition to MPP+. The number of DA neurons was increased by TTX, compared to that by MPP+ alone. All these results demonstrate that activation of DD2R autoreceptors mediates DA neuroprotection against MPP+-induced neurodegeneration via suppression of DA neuronal firing.
Figure 9. Quinpirole and TTX suppress DA neuronal firing and mediate DA neuroprotection.
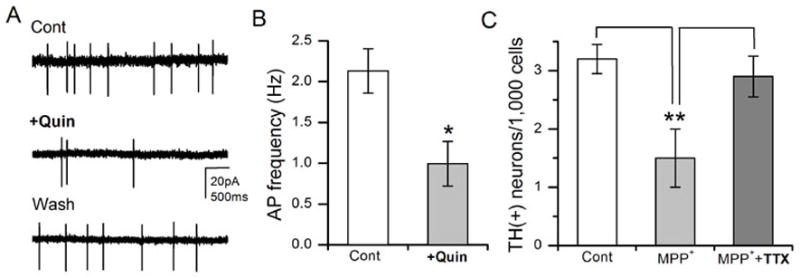
(A) Example traces of extracellular APs recorded in a GFP(+) DA neuron (3 DIV). The APs in DA neurons were inhibited by D2 agonist quinpirole (10μM) indicating functional expression of DA autoreceptors. (B) A graph summarizing effects of quinpirole on APs in DA neurons (n=5). (C) A powerful AP blocker TTX (1μM) rescued DA neurodegeneration by MPP+. Drosophila neuronal cultures were divided into three groups: Control, MPP+ only, and MPP++TTX. MPP+ and TTX were added at 3DIV and neuronal cultures were stained with anti-TH at 9 DIV. Student t-test, *P<0.05 & **P<0.01.
Discussion
The present study showed that MPP+, active metabolite of MPTP, causes selective degeneration of dopaminergic (DA) neurons in a Drosophila primary culture system where the vast majority of neurons are post-mitotic at 3 DIV or older. The number of DA neurons marked with GFP was reduced in the presence of MPP+ compared to control. This finding was further supported by using another specific DA marker anti-TH antibody. Interestingly, DA neurodegeneration by MPP+ was rescued when D2 agonists (quinpirole or bromocriptine) were added. This rescue was not observed in neuronal cultures prepared from Drosophila D2 receptor DD2R deficiency line, or in a line carrying DD2R RNAi transgene. We conclude that this D2 agonist effect is mediated by D2 receptors. Furthermore, our results showed for the first time that activation of D2 autoreceptors in DA neurons is sufficient to rescue MPP+ toxicity and also that this D2-dependant rescue is mediated through suppression of DA neuronal excitability.
PD is a complicated neurodegenerative disease and develops in such a slow fashion that we cannot easily use human subjects to study the mechanisms underlying its pathogenesis. An alternative is to use model systems suitable for molecular and cellular manipulations. Over the last decade, studies with the fruit fly Drosophila melanogaster have contributed to pivotal advances in our understanding of human health and disease (Muqit & Feany, 2002; Bilen & Bonini, 2005). Perhaps the important reason Drosophila continues to afford new insights into human neurodegenerative diseases is that gene sequence and function are highly conserved between flies and humans (Rubin et al, 2000). Of particular relevance to PD, the Drosophila genome encodes orthologs of most of the genes that have thus far been implicated in PD (Whitworth, 2011). Over the last few years, we have developed a cellular model to study PD genes using Drosophila primary neuronal cultures which showed all expected cellular symptoms when human α-Syn was transgenically expressed (e.g., DA cell loss, LB formation; Park & Lee, 2006).
Compared to the intact fruit fly model, the strengths of this in vitro model are twofold. First, the neuronal culture is a system readily available for pharmacological and biochemical manipulations in order to further dissect pathophysiological mechanisms of PD at the cellular and sub-cellular levels (e.g. mitochondria, DA synapse) in a time-dependent manner. As shown in Figure 6, this culture system provides an easy way to study effects of various combinations of pharmacological agents on DA neurodegeneration and protection, which would be very difficult in vivo. This system also affords rapid access to drug screening for possible neuroprotective/PD therapeutic agents due to the short turn over time for cellular PD symptoms - less than 6 days. Second, this neuronal culture is suitable for functional assays, in particular, electrophysiological studies that are difficult to accomplish in intact fly brain (refer to Figure 9, AP experiments). In addition, this neuronal culture appears to contain molecular components essential for DA neuronal function and synaptic DA release (Lee, unpublished data). Furthermore, our current study showed that a Drosophila in vitro model to study PD toxins (e.g. MPP+), in addition to PD genes (e.g. α-Syn), has been successfully developed. Given the availability of sophisticated genetic tools and a live quantification method used in this study, this model will be very useful to study the interaction between genetic and environmental factors causing PD, and also complimentary to a well-established adult fly brain model of PD.
In this study, we used two methods to identify DA neurons: (1) immunocytochemistry by using anti-TH, and (2) green fluorescence protein (GFP) marker specifically expressed in DA neurons. The majority of TH(+) neurons overlapped with GFP(+) neurons at 3DIV. A typical frequency of GFP (TH(+) & GFP(+)/total TH(+); refer to Matsushita et al, 2002) was 81% (89/106) at 3 DIV while it was decreased to 57% (82/145) at 9DIV. Given that the number of GFP(+) DA neurons increases over time and is higher than TH(+) neurons (e.g., ~1.6 fold higher at 9 DIV; also refer to Park & Lee, 2006), it is consistent with our expectation that fewer TH(+) neurons are overlapped with GFP(+) neurons at 9 DIV and also that the number of GFP(+) neurons with no anti-TH signal increases over time. Observation of those GFP(+)/TH(−) neurons would be related to transcriptional (e.g., cis-regulation) and/or posttranslational (e.g. GFP maturation) regulations. Activity of TH promoter (~11kb) used in this study can be differently regulated in some cells and thus GFP can still be expressed, but TH enzyme synthesis can be reduced (or stopped) below the detection level. This discrepancy can also be due to the biochemical properties (e.g. maturation, half-life, etc) of each protein. GFP is known to be very stable in the cell (Tsien, 1998; Day & Davidson, 2009) and thus GFP(+) neurons can be detected even if TH expression is significantly diminished by MPP+. Additionally, GFP(+)/TH(−) neurons would be related to the sensitivity of the TH-antibody used in this study. Since the antibody used in our study has been originally raised against rat TH, it does not necessarily show optimal sensitivity against Drosophila TH and thus cannot detect lower levels of Drosophila TH expressed. Further, GFP fluorescent detection is generally considered more sensitive than immunocytochemistry.
Although we observed a significant number of GFP(+)/TH(−) cells in this study (also refer to Lee & Park, 2006), it is not a unique phenomenon in our culture system. Pesah et al (2005) reported that only 68.6% of TH(+) neurons were overlapped with GFP(+) in the adult fly brain. Friggi-Grelin et al (2003) also reported that a small portion of GFP(+) neurons are not TH(+) in adult fly brain, confirming that this observation in the primary neuronal culture is consistent with those in vivo findings. In addition, this mismatch between GFP and TH signals was reported in rodent and other vertebrate animals. Matsushita et al (2002) reported that a typical frequency of GFP expression (see above) in adult TH-GFP mice varies from 60.3 to 94.1% depending on strains (TH-GFP/6–7 vs. TH-GFP/21–31) or brain areas (SNc vs. VTN). When GFP was expressed by a TH promoter in zebra fish, this overlap was just about 30% (Meng et al, 2008). All of these findings demonstrate that these two methods (anti-TH, TH-GFP) are very popular and powerful, but both have a limitation as they cannot simultaneously detect a portion of the neurons. Nonetheless, the more important observation in our study was that the DA neurons identified by GFP or anti-TH were degenerated by the PD toxin MPP+, although the magnitude of the toxicity was larger with anti-TH detection. However, GFP marking can be a more useful and reliable means to quantify DA neuronal degeneration for future PD research, especially for rapid drug screening.
Some D2 agonists are experimentally used in studies of neuroprotection. Previously, Chen et al (2009) reported that the D2 agonist ropinirole prevents rotenone-induced apoptosis of dopaminergic cell line. In addition, Kitamura & colleagues (1998) showed that the D2R agonist bromocriptine inhibits MPP+-mediated DA cell death. However, the mechanism of how D2 agonists mediate neuroprotection is not well understood. A few studies have shown that D2 agonists (e.g. bromocriptine, apomorphine) exert D2R-independent neuroprotection against 6-OHDA (Hara et al, 2006) or oxidative stress (Lim et al, 2008). Our results clearly demonstrated neuroprotection by D2 agonists through activation of D2-like receptors. In addition, the concentration of D2 agonists used in this study is considered too low to have scavenging effects on reactive oxygen species (Schapira & Olanow, 2003). Therefore, neuroprotection against MPP+ neurotoxicity is mediated through activation of D2 receptors. D2Rs are widely expressed in the central nervous system including DA and non-DA neurons, and thus it is possible that DA neuroprotection can be mediated through either D2Rs expressed in DA or non-DA neurons. We showed for the first time that activation of D2R autoreceptors mediate DA neuroprotection using Drosophila genetic approaches allowing specific knock-down of D2Rs in DA neurons using DD2R RNAi transgenic lines (Fig. 8).
Dopamine D2 autoreceptors in mammals are known to regulate excitability of DA neurons, and hence they reduce excitotoxic cell death (Piallat et al, 1996; Schapira & Olanow, 2003). In our study, we showed that Drosophila D2 autoreceptors also suppress DA neuronal excitability. Further, our data demonstrated that reduced DA neuronal excitability is neuroprotective against MPP+ toxicity (Fig. 9). It is known that D2 autoreceptors suppress DA synthesis through a negative feedback mechanism, and thus reduce oxidative stress caused by a high level of cytoplasmic DA (Carter & Muller, 1991; Schapira & Olanow, 2003; Park et al, 2007). Recently, it was reported that D2R agonists exert their neuroprotective effects through the blockade of Ca2+-triggered permeability transition pore in mitochondria (Parvez et al, 2010). Therefore, our results strongly support these findings in that activation of Drosophila DD2R autoreceptors reduces DA neuronal excitability, cytoplasmic dopamine/Ca2+ levels, oxidative stress and thus mediates DA neuroprotection. However, it should be noted that D2-dependent rescue can be achieved through other mechanisms such as Akt/GSK-3β pathway (Nair & Olanow, 2008), which do not directly modulate DA neuronal excitability.
Given that D2Rs mediate neuroprotection against PD toxins by suppressing DA neuronal excitability, an important next question is to understand mechanisms mediating this suppression. Several possible mechanisms are proposed to explain D2R-mediated neuroprotection. Then, what are the molecular mechanisms underlying this neuroprotection? The prime suspects are related to the known players downstream of D2R signaling. Since D2R actions are mediated through G-protein signaling, it is highly possible that genetic and pharmacological inhibition of G-protein signaling will eliminate D2R-mediated neuroprotection. Hearn et al (2002) showed that Drosophila D2Rs activate Gαi when they were expressed in a heterologous expression system. Therefore, we suspect that Gαi is involved in D2R-mediated protection. However, it is possible that DD2Rs activate other Gα subunits. Because there are mutant and RNAi transgenic lines available for all five Drosophila Gα subunits (i.e. Gαs, Gαi, Gαo, Gαq, Gαf), future studies should be able to identify a Gα subunit mediating reduced DA neuronal excitability and hence neuroprotection, and will further shed light on molecular components downstream of D2R and G-protein signaling such as an ion channel or a neurotransmitter receptor.
Acknowledgments
This work was partially supported by NIH grant (NS050260) and Korea Institute of Science & Technology (Brain Science Institute), Seoul, Korea. We thank Dr. R.A. Colvin for valuable inputs on previous versions of this manuscript. BS was a recipient of Ohio University Provost’s Undergraduate Research Fund.
Footnotes
Authorship credit: LW & BS conducted all experiments except electrophysiology. They were also involved in designing experiments, analysis of data and writing the manuscript. AG performed the electrophysiology experiment. DL supervised the project and wrote the final draft of the manuscript. All authors have read and approved the manuscript.
Conflict of Interest: Authors declare that they have no conflicts of interest.
References
- Andretic R, Hirsh J. Circadian modulation of dopamine receptor responsiveness in Drosophila melanogaster. Proc Natl Acad Sci USA. 2000;97(4):1873–8. doi: 10.1073/pnas.97.4.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295(5556):865–8. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Bains M, Cousins JC, Roberts JL. Neuron protection by estrogen against MPP+-induced dopamine neuron death is mediated by ERα in primary cultures of mouse mesencephalon. Experimental Neurology. 2007;204:767–776. doi: 10.1016/j.expneurol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barolo S, Castro B, Posakony JW. New Drosophila transgenic reporters: insulated P-element vectors expressing fast-maturing RFP. Biotechniques. 2004;36:436–42. doi: 10.2144/04363ST03. [DOI] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–71. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- Boone J, Doe C. Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev Neurobiol. 2008;68(9):1185–1195. doi: 10.1002/dneu.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bové J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx. 2005;2(3):484–94. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi Y, Borrelli E. Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? TINS. 2006;29:167–74. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 2011;9(11):1106–17. doi: 10.1016/S1474-4422(10)70218-0. [DOI] [PubMed] [Google Scholar]
- Cannon JR, Greenamyre JT. Neurotoxic in vivo models of Parkinson’s disease recent advances. Prog Brain Res. 2010;184:17–33. doi: 10.1016/S0079-6123(10)84002-6. [DOI] [PubMed] [Google Scholar]
- Carter AJ, Müller RE. Pramipexole, a dopamine D2 autoreceptor agonist, decreases the extracellular concentration of dopamine in vivo. Eur J Pharmacol. 1991;200(1):65–72. doi: 10.1016/0014-2999(91)90666-e. [DOI] [PubMed] [Google Scholar]
- Chaudhuri A, Bowling K, Funderburk C, Lawal H, Inamdar A, Wang Z, O’Donnell JM. Interaction of genetic and environmental factors in a Drosophila parkinsonism model. J Neurosci. 2007;27(10):2457–67. doi: 10.1523/JNEUROSCI.4239-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang X, Yang D, Du Y, Li L, Li X, Ming M, Le W. D2/D3 receptor agonist ropinirole protects dopaminergic cell line against rotenone-induced apoptosis through inhibition of caspase- and JNK-dependent pathways. FEBS Lett. 2009;582(5):603–10. doi: 10.1016/j.febslet.2008.01.028. [DOI] [PubMed] [Google Scholar]
- Chun HS, Gibson GE, DeGiorgio LA, Zhang H, Kidd VJ, Son JH. Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism. J Neurochem. 2001;76(4):1010–21. doi: 10.1046/j.1471-4159.2001.00096.x. [DOI] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson’s disease. Ann Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson’s disease in Drosophila melanogaster. J Neurosci. 2004;24:10993–8. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darya K, Ganguly A, Lee D. Quantitative analysis of synaptic boutons in cultured Drosophila neurons. Brain Research. 2009;1280:1–12. doi: 10.1016/j.brainres.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Day RN, Davidson MW. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 2009;38(10):2887–921. doi: 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper I, Kurshan PT, McBride E, Jackson FR, Kopin AS. Locomotor activity is regulated by D2-like receptors in Drosophila: an anatomic and functional analysis. Dev Neurobiol. 2007;67(3):378–93. doi: 10.1002/dneu.20355. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Ferrari-Toninelli G, Bonini SA, Cenini G, Maccarinelli G, Grilli M, Uberti D, Memo M. Dopamine receptor agonists for protection and repair in Parkinson’s disease. Curr Top Med Chem. 2008;8:1089–99. doi: 10.2174/156802608785161402. [DOI] [PubMed] [Google Scholar]
- Friggi-Grelin F, Coulom H, Meller M, Gomez D, Hirsh J, Birman S. Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J Neurobiol. 2003;54:618–27. doi: 10.1002/neu.10185. [DOI] [PubMed] [Google Scholar]
- Han KA, Millar NS, Grotewiel MS, Davis RL. DAMB, a novel dopamine receptor expressed specifically in Drosophila mushroom bodies. Neuron. 1996;16:1127–1135. doi: 10.1016/s0896-6273(00)80139-7. [DOI] [PubMed] [Google Scholar]
- Hara H, Ohta M, Adachi T. Apomorphine protects against 6-hydroxydopamine-induced neuronal cell death through activation of the Nrf2-ARE pathway. J Neurosci Res. 2006;84:860–6. doi: 10.1002/jnr.20974. [DOI] [PubMed] [Google Scholar]
- Hearn MG, Ren Y, McBride EW, Reveillaud I, Beinborn M, Kopin AS. A Drosophila dopamine 2-like receptor: Molecular characterization and identification of multiple alternatively spliced variants. Proc Natl Acad Sci USA. 2002;99:14554–9. doi: 10.1073/pnas.202498299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Taniguchi T, Shimohama S, Akaike A, Nomura Y. Neuroprotective mechanisms of antiparkinsonian dopamine D2-receptor subfamily agonists. Neurochemical Research. 2003;28:1035–1040. doi: 10.1023/a:1023207222944. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):979–80. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lawal HO, Chang HY, Terrell AN, Brooks ES, Pulido D, Simon AF, Krantz DE. The Drosophila vesicular monoamine transporter reduces pesticide-induced loss of dopaminergic neurons. Neurobiol Dis. 2010;40:102–12. doi: 10.1016/j.nbd.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, O’Dowd DK. Excitatory synaptic transmission mediated by nicotinic acetylcholine receptors in Drosophila neurons. J Neurosci. 1999;19:5311–21. doi: 10.1523/JNEUROSCI.19-13-05311.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Su H, O’Dowd DK. GABA receptors containing Rdl subunits mediate fast inhibitory synaptic transmission in Drosophila neurons. J Neurosci. 2003;23:4625–34. doi: 10.1523/JNEUROSCI.23-11-04625.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Kim KM, Kim SW, Hwang O, Choi HJ. Bromocriptine activates NQO1 via Nrf2-PI3K/Akt signaling: Novel cytoprotective mechanism against oxidative damage. Pharmacol. 2008;57:325–31. doi: 10.1016/j.phrs.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Marini AM, Schwartz JP, Kopin IJ. The neurotoxicity of 1-methyl-4-phenylpyridinium in cultured cerebellar granule cells. J Neurosci. 1989;9(10):3665–72. doi: 10.1523/JNEUROSCI.09-10-03665.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita N, Okada H, Yasoshima Y, Takahashi K, Kiuchi K, Kobayashi K. Dynamics of tyrosine hydroxylase promoter activity during midbrain dopaminergic neuron development. J Neurochem. 2002;82(2):295–304. doi: 10.1046/j.1471-4159.2002.00972.x. [DOI] [PubMed] [Google Scholar]
- Meng S, Ryu S, Zhao B, Zhang DQ, Driever W, McMahon DG. Targeting retinal dopaminergic neurons in tyrosine hydroxylase-driven green fluorescent protein transgenic zebrafish. Mol Vis. 2008;14:2475–83. [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Ann Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Feany MB. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci. 2002;3:237–43. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- Mustard JA, Beggs KT, Mercer AR. Molecular biology of the invertebrate dopamine receptors. Arch Insect Biochem Physiol. 2005;59:103–117. doi: 10.1002/arch.20065. [DOI] [PubMed] [Google Scholar]
- Nair VD, Olanow CW. Differential modulation of Akt/glycogen synthase kinase-3beta pathway regulates apoptotic and cytoprotective signaling responses. J Biol Chem. 2008;283(22):15469–78. doi: 10.1074/jbc.M707238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW. Can we achieve neuroprotection with currently available anti-parkinsonian interventions? Neurology. 2009;72(7 Suppl):S59–64. doi: 10.1212/WNL.0b013e318199068b. [DOI] [PubMed] [Google Scholar]
- Park SS, Lee D. Selective loss of dopaminergic neurons and formation of Lewy body-like aggregations in alpha-synuclein transgenic fly neuronal cultures. Eur J Neurosci. 2006;23:2908–14. doi: 10.1111/j.1460-9568.2006.04844.x. [DOI] [PubMed] [Google Scholar]
- Park SS, Schulz EM, Lee D. Disruption of dopamine homeostasis underlies selective neurodegeneration mediated by alpha-synuclein. Eur J Neurosci. 2007;26:3104–12. doi: 10.1111/j.1460-9568.2007.05929.x. [DOI] [PubMed] [Google Scholar]
- Parvez S, Winkler-Stuck K, Hertel S, Schönfeld P, Siemen D. The dopamine-D2-receptor agonist ropinirole dose-dependently blocks the Ca2+-triggered permeability transition of mitochondria. Biochim Biophys Acta. 2010;1797(6–7):1245–50. doi: 10.1016/j.bbabio.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Pesah Y, Burgess H, Middlebrooks B, Ronningen K, Prosser J, Tirunagaru V, Zysk J, Mardon G. Whole-mount analysis reveals normal numbers of dopaminergic neurons following misexpression of alpha-Synuclein in Drosophila. Genesis. 2005;41:154–9. doi: 10.1002/gene.20106. [DOI] [PubMed] [Google Scholar]
- Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci. 1996;8:1408–14. doi: 10.1111/j.1460-9568.1996.tb01603.x. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem. 2001;76(5):1265–74. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J Bioenerg Biomembr. 2004;36(4):375–9. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ramos JR, Song S, Facca A, Basit A, Epstein CJ. Transgenic murine dopaminergic neurons expressing human Cu/Zn superoxide dismutase exhibit increased density in culture, but no resistance to methylphenylpyridinium-induced degeneration. J Neurochem. 1997;68(1):58–67. doi: 10.1046/j.1471-4159.1997.68010058.x. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Olanow CW. Rationale for the use of dopamine agonists as neuroprotective agents in Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S149–57. doi: 10.1002/ana.10514. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Olanow CW. Neuroprotection in Parkinson disease: mysteries, myths, and misconceptions. JAMA. 2004;291(3):358–64. doi: 10.1001/jama.291.3.358. [DOI] [PubMed] [Google Scholar]
- Srivastava DP, Yu EJ, Kennedy K, Chatwin H, Reale V, Hamon M, Smith T, Evans PD. Rapid, nongenomic responses to ecdysteroids and catecholamines mediated by a novel Drosophila G-protein-coupled receptor. J Neurosci. 2005;25:6145–6155. doi: 10.1523/JNEUROSCI.1005-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch A, Ludolph AC, Schwarz J. Dopamine transporter: involvement in selective dopaminergic neurotoxicity and degeneration. J Neural Transm. 2004;111:1267–1286. doi: 10.1007/s00702-004-0203-2. [DOI] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, O’Kane CJ. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–351. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- Takashima H, Tsujihata M, Kishikawa M, Freed WJ. Bromocriptine protects dopaminergic neurons from levodopa-induced toxicity by stimulating D(2)receptors. Exp Neurol. 1999;159:98–104. doi: 10.1006/exnr.1999.7122. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–44. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Whitworth AJ. Drosophila models of Parkinson’s disease. Adv Genet. 2011;73:1–50. doi: 10.1016/B978-0-12-380860-8.00001-X. [DOI] [PubMed] [Google Scholar]
- Yuan N, Lee D. Suppression of excitatory cholinergic synaptic transmission by Drosophila dopamine D1-like receptors. Eur J Neurosci. 2007;26(9):2417–27. doi: 10.1111/j.1460-9568.2007.05870.x. [DOI] [PubMed] [Google Scholar]