

One outcome of persistent activation of the DNA damage response (DDR) is cellular senescence; a programmed, permanent, cell cycle exit. DDR activation serves as a block to malignant transformation with senescence as a core feature of this effect. Diverse conditions that induce senescence, for example ionizing radiation, oncogene activation and telomere attrition, themselves induce DDR activation. In virtually all of these instances, senescence can be rescued by ablation of p53 function, thereby allowing the cells to re-enter the cell cycle in the presence of DNA damage. In this issue, Monasor and colleagues provide new evidence suggesting that the well-known tumor suppressor INK4a/ARF enables replicative stress (RS)-induced senescence.1 This is notable, since, in contrast to p53, INK4a/ARF has generally been thought not to play a role in DNA damage-induced senescence. Loss of the INK4a/ARF locus on chromosome 9p21 is one of the most frequent abnormalities observed in human tumors, second only to loss and/or mutation of TP53 (p53). The INK4a/ARF locus encodes two proteins, p16(INK4a) and ARF (p16Ink4A and p19ARF in mice), which regulate the Retinoblastoma and p53 pathways respectively.2 In normal cells, INK4a/ARF levels are usually barely detectable. Monasor and colleagues report that persistent RS is also associated with elevated INK4a/ARF expression, and that INK4a/ARF deficient cells can sustain growth in the presence of substantial levels of RS. Furthermore, upon interrogation of the Cancer Cell Line Encyclopaedia Project (http://www.broadinstitute.org/ccle/home), they found a correlation between copy number variation (CNV) and p16INK4A levels; cell lines with high levels of CNVs (RS has been shown to cause CNV3) exhibited low levels of p16INK4A. The authors consequently propose INK4a/ARF as a bona fide RS-checkpoint activator.1
ATR is an activating protein kinase of the DDR. Persistent ATR activation can drive cells to senescence, although independently of INK4a/ARF.4 Previous work from this team in modeling ATR-defective Seckel syndrome in mice (AtrS/S) showed that impaired ATR function also results in RS-induced DNA damage, senescence and cell death.5 AtrS/S mice exhibit severe embryo-wide cellular attrition with the surviving postnatal animals exhibiting a profound growth restriction, progeria and early death. AtrS/S MEFs fail to cycle, instead undergoing premature senescence. They do not undergo spontaneous immortalization and are even impervious to oncogene-induced immortalization. Genetic ablation of TP53 did not rescue senescence in either the AtrS/S animal or their derived MEFs.5 In fact, synthetic lethality was observed, a serendipitous observation now being actively pursued in the context of ATR kinase inhibitors (ATRi) as an anticancer therapy.6
The characteristic premature senescence seen in AtrS/S MEFs is overcome by reduced INK4a/ARF, a rescue not concomitant with a restoration of ATR expression or a reduction of RS.1 Indeed, ATRS/S + INK4a/ARF−/− cells effectively cycle with persistent RS. In contrast to these MEFs, deletion of INK4a/ARF does not rescue the phenotype of the AtrS/S mouse. The authors argue that INK4a/ARF-dependent senescence is the likely outcome of modest RS, while in the context of high levels of RS (e.g., during AtrS/S embryonic development and/or following high doses of ATRi), significant cell death occurs. This is significant, as it implies that ATR inhibitors would still be toxic toward INK4a/ARF-deficient tumors, as, indeed, demonstrated by the authors here using a mouse pancreatic cancer cell model.
While INK4a/ARF deletion cannot rescue the ATRS/S phenotype, the authors suggest that this does not exclude the fact that the RS-checkpoint defined here could be operative in other contexts associated with more modest levels of RS. One situation where low but persistent RS likely occurs, as discussed by Monasor and colleagues, is oncogene-induced senescence. Oncogene-induced RS, which has long been associated with p16INK4a expression,7 represents a barrier, characterized by DDR activation,8 that must be overcome on the route to frank malignant transformation. The model proposed by Monasor and colleagues is that a key role of the p16 INK4a -Rb pathway would be to promote senescence in cells experiencing chronic RS, thus stopping them on the march toward transformation (Fig. 1). Still, how persistent RS activates INK4a/ARF remains a provocative, open question.
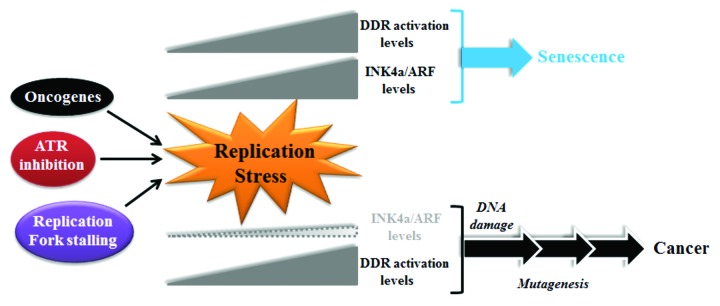
Figure 1. A schematic summarizing the contrasting outcomes of elevated replicative stress in the presence or absence of INK4a/ARF, as proposed by Monasor and colleagues. Prolonged replicative stress, which can occur under various non-mutually exclusive circumstances, results in elevated DDR activation. In the presence of functional INK4a/ARF, its expression is also increased, and this usually drives the cells toward senescence. In the absence of functional INK4a/ARF, cells can replicate even in the context of an elevated DDR, thereby creating the environment for mutation fixation, establishing the pathway to cellular transformation.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25319
References
- 1.Monasor A, Murga M, Lopez-Contreras AJ, Navas C, Gomez G, Pisano DG, et al. INK4a/ARF limits the expansion of cells suffering from replication stress. Cell Cycle. 2013;12 doi: 10.4161/cc.25017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Arlt MF, Wilson TE, Glover TW. Replication stress and mechanisms of CNV formation. Curr Opin Genet Dev. 2012;22:204–10. doi: 10.1016/j.gde.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22:297–302. doi: 10.1101/gad.452308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009;41:891–8. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol Oncol. 2011;5:368–73. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 8.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]