

Abstract
Aurora kinases play important functions in mitosis. They are overexpressed in many cancers and are targets for anticancer therapy. Inhibition of Aurora B results in cytokinesis failure and polyploidization, leading to activation of the p53 tumor suppressor and its target genes, including p21. The pathways that mediate p21 activation after Aurora B inhibition are not well understood. In this study, we identified a role for the p38 MAP kinase in activation of p21 when Aurora B is inhibited. We show that p38 is required for the acute cell cycle arrest in G1 and to prevent endoreduplication when Aurora B is inhibited. Stabilization of p53 occurs independently of p38, and recruitment of p53 to the p21 promoter also does not require p38. Instead, enrichment of the elongating form of RNA PolII at the distal region of the p21 gene is strongly reduced when p38 is blocked, indicating that p38 acts in transcriptional elongation of p21. Thus, our results identify an unexpected role of p38 in cell cycle regulation in response to Aurora B inhibition, by promoting the transcriptional elongation of the cell cycle inhibitor p21.
Keywords: Aurora B, p53, p38, p21, transcription elongation
Introduction
Aurora kinases are mitotic kinases that play multiple important roles in mitosis.1-3 Three different Aurora kinases, Aurora A, B and C, exist in mammals. Aurora A is involved in mitotic entry and bipolar spindle assembly.4 Aurora B is part of the chromosomal passenger complex (CPC), which also contains the inner centromere protein INCENP, Survivin and Borealin. The CPC shows a characteristic pattern of association with chromatin in prophase and centromeric localization in prometaphase and metaphase. It then transfers to the midzone and midbody in late anaphase and telophase.5 The CPC regulates chromosome condensation, is required for chromosome biorientation and for assembly of the mitotic spindle. One of the key functions of the CPC is to promote the correct attachment of the mitotic spindle by destabilizing defective microtubule-chromosome attachments.6 In addition Aurora B plays a crucial role in cytokinesis, the final process of cell division at the end of mitosis. Aurora C is also a chromosomal passenger protein; however, it is expressed only in testis and its function is not well understood.7
Aurora B is expressed at high levels in various tumors, including lung, breast, colon and pancreatic tumors, and its overexpression correlates with a poor prognosis.8 Several inhibitors of Aurora kinases have been developed as potential anticancer agents.9 Promising results have been obtained in preclinical models with Aurora kinase inhibitors, and several Aurora kinase inhibitors are currently tested in clinical trials.9
Inhibition or depletion of Aurora B causes premature exit from mitosis without cell division as a result of cytokinesis failure. This results in polyploid cells containing DNA contents of 4N or more.10-12 Cells that exit mitosis without cell division arrest as tetraploid cells in the G1 phase of the subsequent cell cycle.13,14 Ultimately, these tetraploid cells are often eliminated by apoptosis. Therefore, prolonged exposure to Aurora B inhibitors has been reported to result in loss of cell viability. Arrest in the G1 phase after failed cytokinesis is dependent on the tumor suppressor p53, an important negative regulator of the cell cycle.13,15,16 Consequently, endoreduplication and polyploidy in response to Aurora B inhibition is enhanced in the absence of p53.10 A recent study provided new insights in the relationship between Aurora B and p53: it was reported that Aurora B directly phosphorylates p53, resulting in its degradation.17 Vice versa, inhibition of Aurora B increased p53 protein levels and inhibited cell cycle progression. However, whether p53-stabilization after inhibition of Aurora B is sufficient to activate p53-target genes is not well understood.
In this study we analyzed the activation of the p53 target gene p21 in response to inhibition of Aurora B. We found that activation of p21 and the block of S-phase entry after inhibition of Aurora B depends on signaling by the p38 MAP kinase. We further show that p38 is not required for stabilization of p53 or for recruitment of p53 to the p21 promoter. Instead, our findings show that p38 it plays a crucial role in transcriptional elongation of p21.
Results
Inhibition of Aurora B results in activation of p53 and p21
We used the chemical inhibitor ZM447439 to investigate the effect of Aurora B inhibition on cell cycle progression. Exposure of U2OS cells to ZM447439 for 24 h resulted in formation of binucleated cells (Fig. 1A). By flow cytometry, an increase in cells with 4N DNA content was detected, indicating failure of cytokinesis, consistent with previous studies (Fig. 1B).11,12 After prolonged exposure to the drug for 48 h, the fraction of cells with a DNA content of > 4N increased, indicating that some cells escaped arrest at 4N and continued through additional cell cycles.12 The percentage of sub-G1 cells remained low, indicating that inhibition of Aurora B does not strongly induce apoptosis in U2OS cells. Instead, after prolonged treatment with ZM447439, cells became senescent, as evidenced by β-galactosidase staining (Fig. 1C).

Figure 1. Activation of p53 and p21 in response to Aurora B inhibition. (A) U2OS cells were treated with 1 µM ZM447439 for 24 h. Nuclei were stained with Hoechst 33258. p21 was detected by immunostaining. Bar: 50 µm. (B) U2OS cells treated with 1 µM ZM447439 for 24 and 48 h were analyzed by FACS. (C) Cells were treated with 1µM ZM447439 and levels of p53 and p21 were determined by immunoblotting. β-actin served as a control for equal loading (D) U2OS cells were treated with 1 µM ZM447439 for 72 h. Four days later, senescent cells were detected by staining for β-galactosidase. (E) Cells stably expressing a control shRNA or a p53-specific shRNA were treated with ZM447439 for 36 h and p21 protein levels were determined by immunoblotting (F) U2OS cells were treated with AZD1152 as indicated and p21 levels were determined by immunoblotting. β-actin served as a control for equal loading. (G) U2OS cells were transfected with a control siRNA (ctrl) or with an Aurora B-specific siRNA. 48 h later, p21 levels were determined by immunoblotting. Tubulin served as control.
In response to ZM447439 treatment, p53 accumulated, and its downstream target p21, a cyclin-dependent kinase inhibitor, was induced, as reported previously (Fig. 1A and D).10-12 Depletion of p53 by a p53-specific shRNA prevented upregulation of p21, confirming that activation of p21 is p53-dependent (Fig. 1E). Treatment of U2OS cells with AZD1152, a selective Aurora B inhibitor, or RNAi-mediated depletion of Aurora B also resulted in induction of p21, confirming that the effects are indeed due to Aurora B inhibition and not an artifact of pharmacological inhibition (Fig. 1F and G). p21 induction and accumulation of cells with 4N and > 4N DNA content was also observed in HCT116 cells treated either with ZM447439 or with AZD1152, indicating that the effect is not specific to one cell line (Fig. 2A and B).

Figure 2. Polyploidization in response to Aurora B inhibition is increased in the absence of p21. (A) HCT116 cells were treated with ZM447439 or AZD1152 for 24 h or 48 h. p21 levels were determined by immunoblotting. β-actin served as a control. (B) FACS assays of HCT116 cells treated either with 0.5 µM and 1 µM ZM447439 or with 50 nM and 100 nM AZD1152 for 24 h or 48 h. (C) HCT116 wild type and p21−/− cells were treated with 1 µM ZM447439 for 24 or 48 h. Cell cycle distribution was analyzed by FACS.
To directly test the role of p21 in limiting endoreduplication in response to Aurora B inhibition, we used HCT116 p21−/− cells in which both copies of the p21 gene have been disrupted by homologous recombination.18 As a control, isogenic HCT-116 p21+/+ cells were used. The fraction of cells with > 4N after inhibition of Aurora B was significantly increased in p21−/− cells, indicating that p21 limits polyploidization in response to Aurora B inhibition (Fig. 2C).
p38 MAP kinase is required for activation of p21 in response to Aurora B inhibition
The p38 MAP kinase has been implicated recently in cell cycle inhibition in aneuploid cells.19 We therefore next asked whether p38 is involved in p21 activation after Aurora B inhibition. Using an antibody specific for phosphorylated, active p38, we found increased p38 activity in a time-dependent manner after inhibition of Aurora B (Fig. 3A). In contrast, there was no effect on the total p38 level after treatment with ZM447439. The time-course of p38 activation correlates with induction of p53 and p21, suggesting that inhibition of Aurora B could result in activation of a p38-p53-p21 pathway that limits the proliferation of cells.
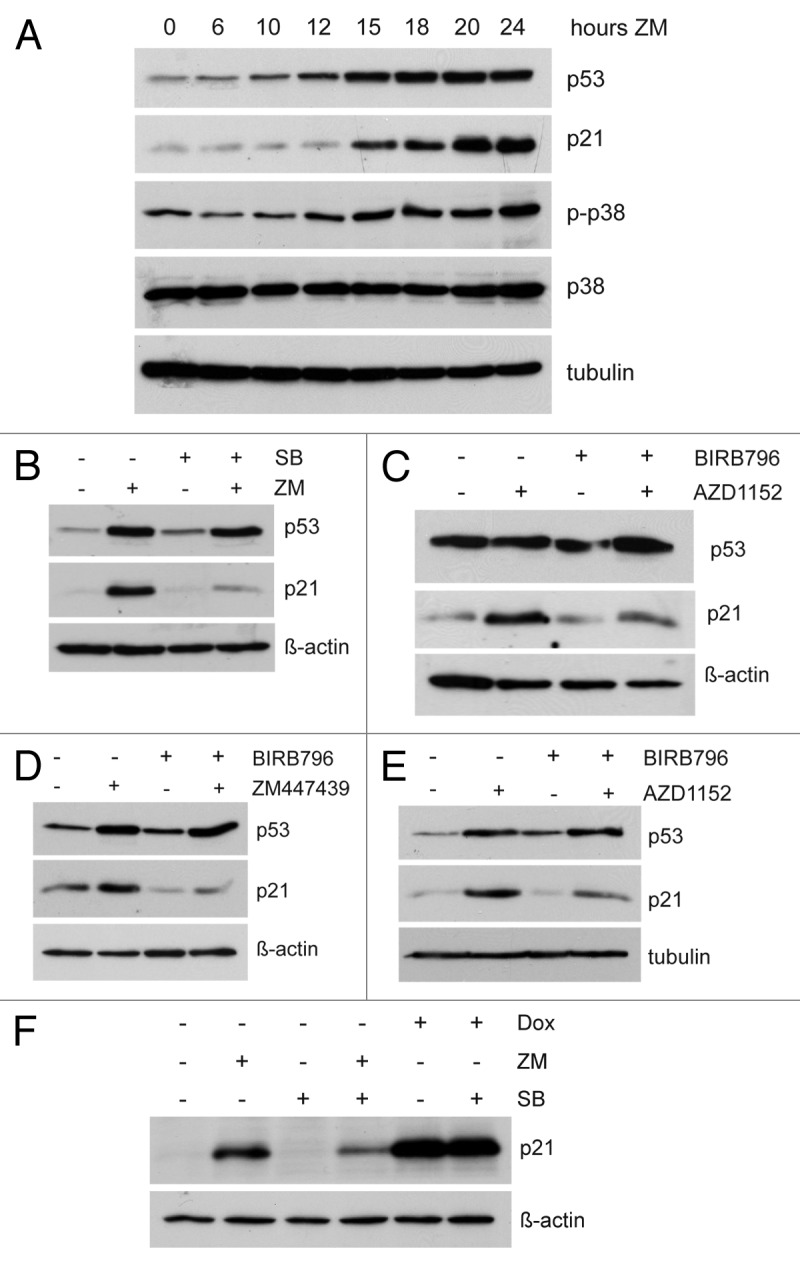
Figure 3. p38 is required for induction of p21 in response to Aurora B inhibition. (A) U2OS cells were treated for the indicated time with 1 µM ZM447439. Levels of p53, p21, phosphorylated p38 (p-p38) and total p38 were determined by immunoblotting. (B) Cells were pretreated with DMSO or 10 µM SB202190 for 2 h and then treated with 1 µM ZM447439 for 24 h. Levels of p21 and p53 were determined by immunoblotting. (C) Cells were pretreated with DMSO or 1 µM BIRB796 for 2 h and then treated with 50 nM AZD1152 for 24 h. Levels of p21 and p53 were determined by immunoblotting. (D) HCT116 cells were pretreated with the p38 MAP kinase inhibitor BIRB796 (1 µM) for 2 h and then treated with ZM447439 (1 µM) for 24 h. p21 and p53 protein levels were determined by immunoblotting. (E) HCT116 cells were treated as in (D), but with a different Aurora B inhibitor, AZD1152 (50 nM). (F) Cells were pre-treated with 10 µM SB202190 for 2 h, as indicated. Cells were than treated either with DMSO, 1 µM ZM447439 or 1 µM doxorubicin (Dox). Levels of p21 were determined by immunoblotting.
Next, we tested whether p38 is required for activation of p53 and p21 in response to inhibition of Aurora B. To do so, we used SB202190, a selective inhibitor of p38α and p38β, but not of other MAP kinases. Co-treatment with SB202190 prevented the activation of p21 by ZM447439, indicating that signaling by p38 contributes to activation of p21 after Aurora B inhibition (Fig. 3B). A different, highly specific pharmacological inhibitor of p38, BIRB796,20 was also able to inhibit the activation of p21 in response to Aurora B inhibition (Fig. 3C), confirming that p38 is required for induction of p21. Furthermore, BIRB796 inhibited the activation of p21 by either ZM447439 or AZD1152 in HCT116 cells (Fig. 3D and E). To investigate whether p38 is also required for induction of p21 following other types of stress, we treated cells with the chemotherapeutic drug doxorubicin to induce DNA damage. DNA damage robustly induced p21, but this p21 accumulation was unaffected by co-treatment SB202190, indicating that it does not depend on p38 (Fig. 3F).
Cell cycle arrest before S-phase in response to Aurora B inhibition requires p38
Next we asked whether activation of p38 is required to limit entry into S-phase in response to Aurora B inhibition. To address this question, we treated U2OS cells with ZM447439 and measured DNA synthesis by BrdU incorporation. DNA synthesis was strongly inhibited after partial inhibition of Aurora B with 0.5 µM ZM447439 (Fig. 4A). Significantly, BrdU incorporation was almost completely restored by co-inhibition of p38 with SB202190, demonstrating that p38 is required for cell cycle arrest in G1 following Aurora B inhibition. Consistently, FACS assays showed that G1 arrest induced by low doses of ZM447439 was overcome by inhibition of p38 (Fig. 4B). Furthermore, polyploidization was increased when cells were co-treated with ZM447439 and SB202190. After prolonged co-treatment with 0.5 µM ZM447439 and SB202190, the number of colonies in a colony-formation assay were reduced when compared with treatment with 0.5 µM ZM447439 alone (Fig. 4C). Furthermore, co-treatment with ZM447439 and SB202190 increased the percentage of cells that stained positive for Annexin V, a marker of apoptosis, (Fig. 4D). Taken together, these data suggest that the simultaneous inhibition of Aurora B and p38 induces apoptosis, which could account for the reduced colony formation.

Figure 4. p38 is required to prevent entry into S-phase when Aurora B is inhibited (A) Cells were pretreated with DMSO or 10 µM SB202190 for 2 h followed by treatment with 0.5 µM ZM447439 for 3 d. Before analysis, cells were pulse labeled with 15 µg/ml BrdU for 2 h. The percentage of BrdU-positive cells was determined by immunofluorescence. Error bars represent standard deviation of three independent experiments. At least 800 cells were counted. (B) Cells were treated with 0.5 µM ZM447439 and 10 µM SB202190 as indicated. The percentage of cells in the different phases of the cell cycle and of polyploid cells (> 4N) was determined by FACS. (C) Cells were plated in 10 cm dishes, pretreated with DMSO or 10 µM SB202190 for 2 h followed by treatment with 0.5 µM ZM447439 for 3 d. Ten days later, colonies were fixed and stained with crystal violet. (D) Cells were treated with 0.5 µM ZM447439 and 10 µM SB202190 for 6 h, as indicated. Apoptotic cells were determined by Annexin V staining. Four independent experiments were performed. The differences between ZM447439- and ZM447439 + SB202190-treated cells were statistically significant (p < 0.0253; Student’s t-test).
p38 MAP kinase promotes transcriptional elongation of p21 when Aurora B is inhibited
Interestingly, while activation of p21 after inhibition of Aurora B was dependent on p38, p53 accumulation was unaffected by treatment with SB202190 or BIRB796 (see Fig. 3B–E). p38 was also not required for localization of p53 to the nucleus (Fig. 5A). It has been reported that phosphorylation of p21 by p38 increases the stability of the p21 protein when cells are arrested in G1 by TGF-β1.21 However, in response to Aurora B inhibition, the half-life of p21 was not affected by inhibition of p38, indicating that p38 does not influence p21 protein stability under these conditions (Fig. 5B). In contrast, p21 mRNA levels increased after inhibition of Aurora B, and this increase required signaling by p38 (Fig. 5C). The stability of the p21 mRNA was not affected by inhibition of p38 (Fig. 5D), indicating that p38 affects transcription of p21.

Figure 5. p38 is required for transcription of p21 but not for nuclear localization of p53 or for p21 protein or mRNA stability (A) U2OS cells were treated with DMSO, 1 µM ZM447439 or 1µM ZM447439 and 10 µM SB202190 for 24 h and p53 localization was determined by immunostaining. Nuclei were counterstained with Hoechst 33258 Scale bar: 25 µM. (B) U2OS cells were treated with ZM447439 or with ZM447439 and SB202190 and then with cycloheximide (CHX) for the indicated time points to block protein synthesis. p21 levels were determined by immunoblotting. Tubulin served as a control for equal loading. (C) U2OS cells were pretreated with DMSO or 10 µM SB202190 for 2 h and then treated with 1 µM ZM447439 for 24 h. Levels of p21 mRNA were determined by RT-qPCR. (D) U2OS cells were treated with ZM447439 or with ZM447439 and SB202190 and then with actinomycin to block transcription. p21 mRNA levels were determined by RT-qPCR.
Having established that p38 regulates p21 expression at the level of transcription, we further analyzed the underlying mechanism. First, we performed chromatin immunoprecipitation (ChIP) assays to determine whether p38 signaling is required for binding of p53 to the p21 promoter. Chromatin isolated from control-treated cells and cells treated with ZM447439 in the presence or absence of SB202190 was immunoprecipitated with a p53-specific antibody (Fig. 6B). As a control, we used nonspecific IgG. Promoter regions were detected by quantitative real-time PCR. Treatment with ZM447439 induced robust binding of p53 to the high-affinity binding site 1 (BS1) in the p21 promoter, consistent with increased p21 expression (Fig. 6B). No significant binding of p53 was observed at the GAPDH2 promoter, which was used as a control, or at other regions of the p21 locus, including the proximal promoter and further downstream regions of the p21 gene locus. p53 binding to BS1 was not affected by co-treatment with SB202190. Thus p38 is necessary for the full expression of p21, and it acts at a step subsequent to p53 binding to the p21 promoter.

Figure 6. p38 is required for transcriptional elongation of p21 in response to Aurora B inhibition. (A) Schematic diagram of the p21 gene locus and position of the amplicons used for ChIP analysis and primary transcript analysis. (B) ChIP assays were performed with chromatin from U2OS cells treated with 1 µM ZM447439 and 10 µM SB202190 or with a combination of both drugs. Chromatin was precipitated either with nonspecific IgG or with antibodies directed at p53. ChIP enriched DNA was amplified by qPCR using the indicated amplicons. The GAPDH2 promoter was analyzed as a control (C) Cells were treated as described in C. ChIP assays were performed with antibodies directed at total RNA polymerase II. (D) Cells were treated as described in (C). ChIP assays were performed with antibodies directed at RNA polymerase II phosphorylated at serine 2. (E) U2OS cells were pretreated with DMSO or 10µM SB202190 for 2 h and then treated with 1 µM ZM447439 for 24 h. Total RNA was isolated and subjected to RT-qPCR using primers specific for different regions of the primary p21 transcript.
It has been reported that expression of p21 can be regulated at the level of transcriptional elongation.22-25 To investigate whether p38 regulates p21 expression at this step, we performed ChIP assays using antibodies specific for RNA polymerase II (RNAPII). We found robust binding of RNAPII at the core promoter region (−20 region) even before inhibition of Aurora B (Fig. 6C). Binding of RNAPII to binding to the transcribed region was low, consistent with polymerase stalling at the promoter.26 After inhibition of Aurora B, levels of RNAPII at the core promoter increased moderately (less than 2-fold), while in the coding region a stronger increase (3–4-fold) was observed, especially toward distal region of the p21 locus (Fig. 6C). Recruitment of RNAPII at the proximal promoter region (−20) was not inhibited but even slightly enhanced after co-inhibition of p38. In stark contrast, binding of RNAPII to the distal region of the gene was prevented when p38 was co-inhibited together with Aurora B. These results are consistent with a role for p38 in transcriptional elongation of p21 after recruitment of RNAPII.
To confirm that p38 regulates the transcriptional elongation of p21 in response to Aurora B inhibition, we performed ChIP assays with antibodies against RNAPII phosphorylated at Ser2 (Fig. 6D). Serine 2 phosphorylation of the CTD of RNAPII is a hallmark of elongating polymerase.27 We found that RNA phosphorylated at Ser2 becomes strongly enriched in the 3′ region of the p21 gene locus after Aurora B is inhibited with ZM447439. Most importantly, the increase in Ser2-phosphorylated RNAPII at the distal regions of the p21 gene was impaired when Aurora B inhibition was combined with p38 inhibition.
We next analyzed nascent, unprocessed p21 mRNA transcripts after treatment with ZM447439 and SB202190 to further confirm that p38 regulates the elongation step of p21 transcription (Fig. 6E). Short p21 transcripts were analyzed by RT-qPCR with primers specific for an amplicon in the first intron of the gene (+507). Long p21 transcripts were analyzed with primers specific for the second intron just before the third exon (+7,011). We found that short transcripts were induced by inhibition of Aurora B and, importantly, their induction was not prevented by co-inhibition of p38. Longer p21 transcripts were also induced after Aurora B inhibition. Importantly, however, accumulation of the longer transcript was significantly reduced when p38 signaling was inhibited (Fig. 6E). Together, these data indicate that p38 is required for transcriptional elongation of the p21 gene in response to Aurora B inhibition.
Discussion
It has been previously been demonstrated that inhibition of Aurora B results in activation of p53 and p21.17,28-30 However, the functional significance of this pathway and the molecular requirements for induction of p21 were not well understood. Here, we report an important role for p38 MAPK in transcriptional elongation of p21 in response to Aurora B inhibition. Our findings are summarized in Figure 7. When Aurora B is inhibited, p38 is activated in a time-dependent manner, and this activation correlates with the induction of p53 and p21. Pharmacological inhibition of p38 with two different specific inhibitors prevented the induction of p21 in response to Aurora B inhibition. These observations suggest that the p38 > p53 > p21 pathway limits DNA replication in response to Aurora B inhibition. Consistent with this notion, polyploidy caused by Aurora B inhibition is exacerbated when p21 is deleted or when p38 is inhibited.

Figure 7. Summary and model for the role of p38 in transcriptional elongation of p21 when Aurora B is inhibited. During normal cell cycle progression Aurora B phosphorylates p53 resulting in its degradation.17 When Aurora B is inhibited, p53 is stabilized and recruited to the p21 promoter in a p38-independent manner. Aurora B inhibition also results in cytokinesis failure and chromosome missegregation triggering p38 activation, which is required for transcriptional elongation of p21.
While p38 was required for the activation of p21 in response to Aurora B inhibition, the stabilization of p53 occurred independently of p38. That p53 is stabilized after inhibition of Aurora B is consistent with recent reports showing that Aurora B associates with p53 and promotes its ubiquitin-dependent degradation.17,28 When Aurora B was inhibited, p53 bound to the p21 promoter, but did not activate transcription without p38 MAPK signaling. This indicates a role for p38 downstream of p53.
The expression of p21 is known to be regulated by multiple mechanisms and, consistent with previous studies, we find that RNA Pol II is paused at the proximal p21 promoter in the basal state.22-25 After inhibition of Aurora B, RNA Pol II binding at the proximal promoter increased moderately, which was not affected by p38 inhibition. In contrast, the failure to activate p21 when p38 is inhibited is linked to a defect in transcriptional elongation, as determined by ChIP assays for RNA Pol II phosphorylated at Ser2. Specifically, enrichment of the elongating form of RNA Pol II in the distal region of the p21 gene was prevented when p38 was inactive, indicating that the transition from stalled RNA Pol II to productive elongation requires p38 signaling. Promoter proximal pausing of RNA Pol II may allow for the rapid induction of p21 in response to stress signals transmitted by p38.
In future experiments, it will be important to determine the specific role for p38 in transcriptional elongation of p21. It is possible that p38 itself performs an active role in transcriptional elongation. In support of this possibility, it is known that the yeast p38-related MAP kinase Hog1 associates with the transcribed region of osmoresponsive genes and promotes their transcriptional elongation in response to osmotic stress.31 Although binding of mammalian p38 to chromatin has been demonstrated, for example, during skeletal myogenesis p38 is recruited to the promoter regions of muscle-specific genes Myog and Ckm.32 There is no evidence for direct association of p38 with the elongating RNA polymerase in mammalian cells. Therefore, downstream pathways of p38 may be involved in the control of transcriptional elongation.
How p38 activation is triggered after Aurora B inhibition remains also to be investigated. One possibility is that activation of p38 is mediated by the DNA damage pathway. Chromosome missegregation induced by Aurora B inhibition could result in damage to the chromosomes during cytokinesis.33 Other possible triggers that might result in activation of p38 are damage to the spindle apparatus due to mitotic spindle dysfunction or proteomic stress due to imbalance in protein synthesis for the genes encoded on aneusomic chromosomes.
Aurora kinases are overexpressed in different human cancers and they are being exploited as potential targets for cancer therapeutics.34,35 Although Aurora kinase inhibitors worked well in preclinical studies, they performed relatively poorly in clinical studies.9 The higher proliferation rate of cell lines may make them more susceptible to Aurora B inhibition compared with the relative slow growth rate of human tumors. Because p38 contributes to the inhibition of S-phase entry after Aurora B inhibition, inhibition of p38 may increase proliferation and thus sensitize tumor cells to anticancer therapy with Aurora B inhibitors. Indeed, we find that co-inhibition of p38 and Aurora B reduces colony formation and enhances apoptosis. Therefore, combining the Aurora B inhibitor with a p38 inhibitor may result in a more effective cancer therapy. Lower concentrations of Aurora B inhibitor, which are by themself not efficient, may be sufficient to kill tumor cells when combined with p38 inhibitors. This could not only enhance the effectiveness of Aurora inhibitors, but also result in fewer side effects of Aurora B inhibition.
Materials and Methods
Cell culture
U2OS and HCT116 wild type and HCT116 p21−/− cells were cultured in DMEM (Invitrogen) containing 10% FCS (Invitrogen). Cells were treated with the indicated concentrations of ZM447439 (Enzo), AZD1152 (Selleckchem), SB202190 (Sigma), BIRB796 (Selleckchem), doxyrubicin (Sigma). siRNA oligonucleotides (MWG) were transfected using Lipofectamine RNAi Max (Invitrogen).
SA-β-gal staining
Senescence associated β-galactosidase activity was detected as described.36
Immunofluorescence
Cells grown on coverslips were fixed for 10 min at room temperature with PFA (PBS, 3% paraformaldehyde, 2% sucrose). PFA-fixed cells were permeabilized for 5 min with 0.2% Triton-X-100 in PBS and washed with PBST (0.1% Triton-X-100 in PBS). Slides were blocked for 60 min with 2% BSA in PBS, washed 3 times in PBS and incubated with primary antibodies. Coverslips were washed three times with PBS and incubated with secondary antibody (Invitrogen) in PBST for 30 min. Nuclei were stained with Hoechst 33258 (Sigma).
mRNA and protein stability assays
To determine the half-life of the p21 mRNA, actinomycin D (1 µg/ml) was added to the culture medium and at the indicated time-points total RNA was isolated and used for RT-qPCR. p21 mRNA levels were normalized to GAPDH mRNA levels. To determine the p21 protein stability, cycloheximide (20 µg/ml) was added to the culture medium treatment with ZM447439 and SB202190. At the indicated time points, total lysates were analyzed by immunoblotting.
Retroviral shRNA
Retroviral infections with a p53-specific shRNA were performed as described before.37
Colony-formation assay
For colony-formation assays, 6,000 cells were plated on 10 cm dishes and treated with the indicated drugs for 3 d. Ten days later, cells were fixed and stained with crystal violet.
FACS analysis and BrdU incorporation
For FACS analysis cells were stained with propidium iodide and analyzed on a Beckman Coulter FC500. To detect apoptotic cells, cells were stained with annexin V-FITC (PharMingen) and analyzed by flow cytometry. To determine the fraction of cells in S-phase, cells were pulsed with 15 µM BrdU for 2 h. BrdU incorporation was analyzed by immunostaining.
siRNA
The following siRNA sequence specific for Aurora B was used: 5′ AACGCGGCACUUCACAAUUGA 3′38
Antibodies
The following primary antibodies were used: p21 (Santa Cruz), p38 and phospho-p38 (Thr 180/Tyr 182) (Cell Signaling), p53 (DO-1, Santa Cruz), BrdU-FITC (BD Bioscience), β-actin (Santa Cruz), tubulin (Sigma), RNA PolII (Santa Cruz), Ser2-phosphorylated RNA PolII (Abcam).
Oligonucleotide sequences
RT-qPCR
GAPDH
5′GCCCAATACGACCAAATCC3′ and 5′AGCCACATCGCTCAGACAC3′
p21
5′TCACTGTCTTGTACCCTTGTGC3′ and 5′GGCGTTTGGAGTGGTAGAAA3′
ChIP
GAPDH2 promoter
5′ GGCAGCAAGAGTCACTCCA 3′
5′ TGTCTCTTGAAGCACACAGGTT 3′
p21 (−2285, p53BS1)
5′ CTGTGGCTCTGATTGGCTTT 3′
5′ CTCCTACCATCCCCTTCCTC 3′
p21 (−20)
5′ TATATCAGGGCCGCGCTG 3′
5′ GGCTCCACAAGGAACTGACTTC 3′
p21 (+182)
5′ CGTGTTCGCGGGTGTGT 3′
5′ CATTCACCTGCCGCAGAAA 3′
p21 (+507)
5′ CCAGGAAGGGCGAGGAAA 3′
5′ GGGACCGATCCTAGACGAACTT 3′
p21 (+7011)
5′ CCTGGCTGACTTCTGCTGTCT 3′
5′ CGGCGTTTGGAGTGGTAGA 3′
p21 (+8566)
5′ CCTCCCACAATGCTGAATATACAG 3′
5′ AGTCACTAAGAATCATTTATTGAGCACC 3′
RT-PCR
Total RNA was isolated with Trizol (Invitrogen), reverse transcribed with 0.5 units M-MLV-RT Transcriptase (Thermo Scientific) and analyzed with quantitative real-time PCR with SYBR green reagents from Thermo Scientific using the Mx3000 (Agilent technologies) detection system. Expression differences were calculated relative to GAPDH as described before.39
Immunoblotting
Cells were lysed in TNN [50 mM Tris (pH 7.5), 120 mM NaCl, 5 mM EDTA, 0.5% NP40, 10 mM Na4P2O7, 2 mM Na3VO4, 100 mM NaF, 10 mg/mL phenylmethylsulfonyl fluoride, protease inhibitors (Sigma)]. Proteins were separated by SDS-PAGE, transferred to PVDF membrane and detected by immunoblotting
ChIP
Cells were cross-linked with 1% formaldehyde for 10 min at room temperature. The reaction was stopped by adding 125 mM glycine. Cells were lysed for 10 min in lysis buffer [5 mM PIPES (pH 8.0), 85 mM KCl, 0.5% NP40, protease inhibitors (Sigma)]. Nuclei were lysed in nuclei lysis buffer [50 mM Tris (pH 8.1), 10 mM EDTA, 1% SDS, protease inhibitors (Sigma)]. Chromatin was sonicated to an approximate length of 250–500 bp, diluted 1:10 with dilution buffer [0.01% SDS, 1.1% Triton, 1.2 mM EDTA, 16.7 mM Tris (pH 8.2), 167 mM NaCl, protease inhibitors (Sigma)] and used for immunoprecipitation overnight. Immmunoprecipitates were collected with protein G-dynabeads (Life Technologies) for 1 h (blocked with 1 mg/mL BSA and 0.3 mg/ml ssDNA). Beads were washed seven times with LiCl washing buffer [0.25 mM LiCl, 0.5% NP40, 0.5% sodium deoxycholate, 1 mM EDTA, 10 mM Tris (pH 8.0), protease inhibitors (Sigma)] and eluted with elution buffer [50 mM Tris (pH 8.0), 1% SDS, 10 mM EDTA]. The cross-link was reversed overnight with 0.2 mM NaCl at 65°C. After proteinase K incubation for 2 h at 55°C, chromatin was purified using Qiagen DNA purification spin columns. Chromatin (1 µl) was used as template for quantitative real-time PCR.
Acknowledgments
We thank all members of the laboratory for their suggestions and critical reading of the manuscript. This work was supported by grants from the DFG (575/6-1 and TR17-B1) toward S.G.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25100
References
- 1.Carmena M, Ruchaud S, Earnshaw WC. Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol. 2009;21:796–805. doi: 10.1016/j.ceb.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vader G, Lens SMA. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 4.Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278:51786–95. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 5.Earnshaw WC, Cooke CA. Analysis of the distribution of the INCENPs throughout mitosis reveals the existence of a pathway of structural changes in the chromosomes during metaphase and early events in cleavage furrow formation. J Cell Sci. 1991;98:443–61. doi: 10.1242/jcs.98.4.443. [DOI] [PubMed] [Google Scholar]
- 6.Vader G, Maia AF, Lens SM. The chromosomal passenger complex and the spindle assembly checkpoint: kinetochore-microtubule error correction and beyond. Cell Div. 2008;3:10. doi: 10.1186/1747-1028-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Sakashita G, Matsuzaki H, Sugimoto K, Kimura K, Hanaoka F, et al. Direct association with inner centromere protein (INCENP) activates the novel chromosomal passenger protein, Aurora-C. J Biol Chem. 2004;279:47201–11. doi: 10.1074/jbc.M403029200. [DOI] [PubMed] [Google Scholar]
- 8.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Jr., Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–48. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 9.Boss DS, Beijnen JH, Schellens JHM. Clinical experience with aurora kinase inhibitors: a review. Oncologist. 2009;14:780–93. doi: 10.1634/theoncologist.2009-0019. [DOI] [PubMed] [Google Scholar]
- 10.Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–80. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreier MR, Grabovich AZ, Katusin JD, Taylor WR. Short and long-term tumor cell responses to Aurora kinase inhibitors. Exp Cell Res. 2009;315:1085–99. doi: 10.1016/j.yexcr.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–77. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- 13.Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell. 2001;12:1315–28. doi: 10.1091/mbc.12.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senovilla L, Vitale I, Galluzzi L, Vivet S, Joza N, Younes AB, et al. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle. 2009;8:1380–5. doi: 10.4161/cc.8.9.8305. [DOI] [PubMed] [Google Scholar]
- 15.Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol. 1998;18:1055–64. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minn AJ, Boise LH, Thompson CB. Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes Dev. 1996;10:2621–31. doi: 10.1101/gad.10.20.2621. [DOI] [PubMed] [Google Scholar]
- 17.Gully CP, Velazquez-Torres G, Shin J-H, Fuentes-Mattei E, Wang E, Carlock C, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci USA. 2012;109:E1513–22. doi: 10.1073/pnas.1110287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–90. [PubMed] [Google Scholar]
- 19.Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188:369–81. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuma Y, Sabio G, Bain J, Shpiro N, Márquez R, Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J Biol Chem. 2005;280:19472–9. doi: 10.1074/jbc.M414221200. [DOI] [PubMed] [Google Scholar]
- 21.Kim G-Y, Mercer SE, Ewton DZ, Yan Z, Jin K, Friedman E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J Biol Chem. 2002;277:29792–802. doi: 10.1074/jbc.M201299200. [DOI] [PubMed] [Google Scholar]
- 22.Espinosa JM, Verdun RE, Emerson BM. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell. 2003;12:1015–27. doi: 10.1016/S1097-2765(03)00359-9. [DOI] [PubMed] [Google Scholar]
- 23.Beckerman R, Donner AJ, Mattia M, Peart MJ, Manley JL, Espinosa JM, et al. A role for Chk1 in blocking transcriptional elongation of p21 RNA during the S-phase checkpoint. Genes Dev. 2009;23:1364–77. doi: 10.1101/gad.1795709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomes NP, Bjerke G, Llorente B, Szostek SA, Emerson BM, Espinosa JM. Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev. 2006;20:601–12. doi: 10.1101/gad.1398206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valin A, Ouyang J, Gill G. Transcription factor Sp3 represses expression of p21CIP¹ via inhibition of productive elongation by RNA polymerase II. Mol Cell Biol. 2013;33:1582–93. doi: 10.1128/MCB.00323-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price DH. Poised polymerases: on your mark...get set...go! Mol Cell. 2008;30:7–10. doi: 10.1016/j.molcel.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:280–8. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Wu L, Ma CA, Zhao Y, Jain A. Aurora B interacts with NIR-p53, leading to p53 phosphorylation in its DNA binding domain and subsequent functional suppression. J Biol Chem. 2011;286:2236–44. doi: 10.1074/jbc.M110.174755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trakala M, Fernández-Miranda G, Pérez de Castro I, Heeschen C, Malumbres M. Aurora B prevents delayed DNA replication and premature mitotic exit by repressing p21 (Cip1) Cell Cycle. 2013;12:1030–41. doi: 10.4161/cc.24004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernández-Miranda G, Trakala M, Martín J, Escobar B, González A, Ghyselinck NB, et al. Genetic disruption of aurora B uncovers an essential role for aurora C during early mammalian development. Development. 2011;138:2661–72. doi: 10.1242/dev.066381. [DOI] [PubMed] [Google Scholar]
- 31.Proft M, Mas G, de Nadal E, Vendrell A, Noriega N, Struhl K, et al. The stress-activated Hog1 kinase is a selective transcriptional elongation factor for genes responding to osmotic stress. Mol Cell. 2006;23:241–50. doi: 10.1016/j.molcel.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 32.Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet. 2004;36:738–43. doi: 10.1038/ng1378. [DOI] [PubMed] [Google Scholar]
- 33.Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–8. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 34.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–36. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 35.Katayama H, Sen S. Aurora kinase inhibitors as anticancer molecules. Biochim Biophys Acta. 2010;1799:829–39. doi: 10.1016/j.bbagrm.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gagrica S, Hauser S, Kolfschoten I, Osterloh L, Agami R, Gaubatz S. Inhibition of oncogenic transformation by mammalian Lin-9, a pRB-associated protein. EMBO J. 2004;23:4627–38. doi: 10.1038/sj.emboj.7600470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lampson MA, Kapoor TM. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol. 2005;7:93–8. doi: 10.1038/ncb1208. [DOI] [PubMed] [Google Scholar]
- 39.Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, et al. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle. 2007;6:1903–13. doi: 10.4161/cc.6.15.4512. [DOI] [PubMed] [Google Scholar]