

Abstract
Histone methylation is implicated in various biological and pathological processes including cancer development. In this study, we discovered that ectopic expression of KDM5B, a histone H3 lysine 4 (H3K4) demethylase, promoted epithelial-mesenchymal transition (EMT) of cancer cells. KDM5B increased the expression of transcription factors, ZEB1 and ZEB2, followed by downregulation of E-cadherin and upregulation of mesenchymal marker genes. The expression of the microRNA-200 (miR-200) family, which specifically targets ZEB1 and ZEB2, was reduced in the cells with KDM5B overexpression. We found that KDM5B repressed the expression of the miR-200 family by changing histone H3 methylation status of their regulatory regions. The introduction of miR-200 precursor in the cells inhibited EMT induction by KDM5B, suggesting that miR-200 family was a critical downstream mediator of KDM5B-promoted EMT. Furthermore, knockdown of KDM5B was shown to affect the expression of EMT-related genes, indicating the involvement of endogenous KDM5B. Our study demonstrated a novel role of KDM5B histone lysine demethylase in EMT, which may contribute to malignant progression of cancer.
Keywords: tumor progression, histone methylation, transcription, microRNA, epithelial-mesenchymal transition
Introduction
Histone modifications such as acetylation, phosphorylation and methylation can function individually or in combination to elicit specific effects on chromatin structure and gene expression.1 Histone lysine (K) methylation, in particular, has emerged as an important modification due to its central role in transcriptional regulation.2,3 Histone lysine methylation is associated with activated or repressed transcription of individual genes, depending on the methylated residue and the degree of methylation, since lysine residues can be mono-, di- or tri-methylated (me1, me2 or me3).4,5 Moreover, a given methylated mark is often linked to a specific position of the gene, either around the transcription initiation site or in the coding region, regulating transcriptional initiation or elongation. In general, methylation of lysine-4 of histone H3 (H3K4) around TSS and methylation of H3K36 and H3K79 on the coding region are associated with active transcription. Methylation of H3K9 and H3K27 on promoters correlates with transcriptional repression.6,7 The discovery of a large number of site-specific histone methyltransferases and demethylases reveals the dynamic regulation of histone methylation.4,5 Reversible histone lysine methylation is implicated in diverse biological processes, including cellular proliferation, differentiation, DNA repair and recombination. The importance of the tight regulation of histone methylation is demonstrated by emerging links of histone methylation to human disease such as cancer.4
Identification of genes involved in cancer gives us crucial information about the molecular mechanism of cancer development. Genetic screens for mutations contributing to tumor formation in model organisms facilitate the efficient identification of cancer genes in an in vivo setting. Retroviral insertional mutagenesis in mice is one of the potent cancer gene discovery tools.8 Previously we have accomplished high-throughput cloning of retroviral integration sites from the tumors of murine leukemia virus (MuLV)-infected mice. This screen led to the identification of hundreds of candidate cancer genes, including many genes encoding histone lysine methyltransferases and demethylases.9,10 These results provided us with an opportunity to explore the functions of these enzymes in the course of cancer development. Previously, we reported that JMJD2C, a histone H3K9 demethylase, increased the expression of MDM2 oncogene, resulting in the reduction of p53 tumor suppressor in the cells.11 We also showed that UTX H3K27 demethylase enhanced the expression of RB1 and RBL2 genes that play important roles in cell proliferation.12 In addition, we demonstrated that KDM5B/PLU1 H3K4 demethylase repressed the expression of KAT5/TIP60 and CD82/KAI1 genes, thereby increasing the invasive activity of the cancer cells.13 Thus our studies revealed that histone methyl-modifying enzymes were involved not only in tumor initiation, but also in tumor progression such as invasion and metastasis.
The increased motility and invasiveness of malignant tumor cells are often associated with epithelial-mesenchymal transition (EMT) of the cells.14,15 Loss of E-cadherin-mediated cell interaction is the most important step and a well-known marker for EMT. The upregulation of mesenchymal markers such as Fibronectin and N-cadherin also characterizes EMT process. Many studies on the molecular mechanisms for E-cadherin repression have revealed that several transcription factors, including SNAI1, SNAI2, ZEB1, ZEB2 and TWIST, are involved in the complex network that regulates EMT.16-18 EMT is a dynamic and reversible process that primarily occurs at the invasive front of the tumor. When cancer cells distribute to distant sites of the body, they can revert to an epithelial state via mesenchymal-epithelial transition. The plasticity of EMT suggests that epigenetic regulation such as DNA methylation and histone modification is implicated in the EMT process.19,20 Recent reports indicated the connection between EMT and histone methylation. SNAI1 was shown to interact with LSD1 H3K4 demethylase or G9A H3K9 methyltransferase and recruited them to CDH1/E-cadherin promoter for transcriptional repression during EMT.21,22 However, the exploration for the role of histone methyltransferase or demethylase in EMT has just begun.
Histone H3K4 demethylase KDM5B/PLU1/JARID1B has been reported to play important roles in cancer development, since upregulation of KDM5B was observed in many types of malignant tumors.23-26 Previously we reported that KDM5B enhanced the invasive potential of cancer cells, clarifying its function in malignant tumor progression.13 However, the involvement of KDM5B in other processes of cancer progression such as EMT remained unknown.
In this paper we demonstrated that overexpression of KDM5B promoted the EMT process of cancer cells. KDM5B specifically increased the expression of ZEB1 and ZEB2 transcription factors, the critical regulators of EMT induction. Mechanistic investigations indicated that KDM5B regulated the expression of microRNA-200 family targeting ZEB1 and ZEB2 transcripts through the changes of histone H3 methylation on their regulatory regions.
Results
Ectopic expression of KDM5B induced morphological changes of the cells
Previously, we demonstrated that KDM5B/PLU1 histone lysine demethylase played an important role in the cell invasion process of cancer progression.13 To investigate the involvement of KDM5B in other processes of malignant progression, we examined whether ectopic expression of KDM5B would affect epithelial-mesenchymal transition (EMT) of cancer cells. We used a lung cancer cell line, A549, because it shows clear morphological changes during EMT caused by the treatment of Transforming Growth Factor-β (TGF-β).29 A549 cells were infected with the retroviruses expressing either without insert or with FLAG-tagged wild-type KDM5B or catalytically inactive KDM5B mutant H499Y,13,25 and the infected cells were selected in the medium containing 800 μg/ml of G418. The cell lysates of the infected cells were prepared and subjected to western blot analysis with anti-FLAG antibody (Fig. S1A). We confirmed that exogenously introduced wild-type and mutant KDM5B proteins were produced at a similar level in A549 cells (Fig. S1A).
As a positive control for EMT induction, we used A549 cells treated with TGF-β for 48 h. After TGF-β treatment, the cells were dispersed, elongated and assumed a fibroblast-like appearance (Fig. 1A, control plus TGF-β). Similar morphological changes of A549 cells were observed with the expression of wild-type KDM5B but not of inactive KDM5B mutant (Fig. 1A, KDM5B WT and Mut), suggesting that overexpression of KDM5B might induce EMT. To confirm this, we performed immunofluorescence assay for A549 cells using an antibody against E-cadherin, an epithelial cell marker. Untreated A549 cells showed heterogeneous E-cadherin staining (Fig. 1B, control), and this staining was almost lost in TGF-β-treated cells (Fig. 1B, control plus TGF-β) as described in the previous reports.29 We discovered that E-cadherin staining significantly reduced in the cells with the expression of wild-type KDM5B (Fig. 1B, KDM5B WT). No significant change was observed after infection with the virus expressing the inactive mutant (Fig. 1B, KDM5B Mut). These results indicated that the epithelial property of the cells might be lost by the expression of wild-type KDM5B. We further examined the status of actin in the cells by TRITC-conjugated phalloidin staining, since actin reorganization occurs during the EMT process.17 In contrast to untreated cells (Fig. 1C, control), treatment of TGF-β dramatically induced actin fiber formation typical of EMT (Fig. 1C, control plus TGF-β). Phalloidin staining revealed a similar actin fiber formation in the cells with wild-type KDM5B, but not with the inactive mutant (Fig. 1C, KDM5B WT and Mut).

Figure 1. Overexpression of KDM5B caused morphological changes of A549 cells. (A) Cell morphological changes of A549 cells induced by KDM5B. A549 cells infected with the control retrovirus, the control retrovirus with TGF-β treatment or the retrovirus expressing FLAG-tagged wild-type (WT) KDM5B or the H499Y mutant (Mut) were stained with crystal violet. (B) Immunofluorescence images of cells showing the localization of E-cadherin. The panels of A549 cells with the same arrangement with (A) were stained with anti-E-cadherin antibody and with DAPI. (C) Fluorescence images of cells showing reorganization of actin cytoskeleton. The cells were stained with TRITC-phalloidin (indicated as Actin) and with DAPI.
We next examined whether KDM5B overexpression would cause similar effects on other EMT models. We used a mouse mammary epithelial cell line, NMuMG, and a human colon cancer cell line, HT29, because these cell lines respond to TGF-β for EMT induction. We first confirmed the expression level of wild-type and mutant KDM5B proteins in NMuMG cells (Fig. S1B) and HT29 cells (Fig. S1C). The expression of wild-type KDM5B but not its mutant induced similar morphological changes of NMuMG cells (Fig. S2A) and HT29 cells (Fig. S3A) as TGF-β treatment did. Moreover, the cells with wild-type KDM5B showed disappearance of E-cadherin staining and formation of actin stress fiber (Figs. S2B and C, S3B and C). Altogether, these results indicated that ectopic expression of KDM5B caused morphological changes and cytoskeletal rearrangement, consistent with EMT in A549 lung cancer cells, NMuMG mammary epithelial cells and HT29 colon cancer cells, dependent on its demethylase activity.
KDM5B affected the expression of EMT related genes
Besides the reduction of E-cadherin, EMT is characterized by upregulation of mesenchymal marker genes.14,15,17 Therefore we analyzed the expression of two representative mesenchymal marker genes, FN1/Fibronectin and CDH2/N-cadherin, as well as an epithelial marker, CDH1/E-cadherin, in the cells with KDM5B expression. Quantitative RT-PCR analysis revealed that TGF-β treatment decreased the expression of CDH1 mRNA in A549 cells (Fig. 2A) as previously reported.29 Expression of wild-type KDM5B similarly reduced the CDH1 expression, but its mutant had no effect (Fig. 2A). On the other hand, for FN1 or CDH2 whose expression was elevated by TGF-β, wild-type KDM5B increased their expression significantly, but the mutant did not change the expression (Fig. 2B and C). These results again indicated that the effect was dependent on the demethylase activity of KDM5B. Judging from the changes in the epithelial and mesenchymal marker gene expression, we could conclude that overexpression of wild-type KDM5B induced EMT of A549 cells.

Figure 2. KDM5B affected the expression of EMT-related genes. Quantitative RT-PCR analysis was performed to detect the expression of CDH1/E-cadherin (A), FN1/fibronectin (B), CDH2/N-cadherin (C), SNAI1 (D), SNAI2 (E), ZEB1 (F) and ZEB2 (G) in A549 cells infected with the control retrovirus, the control retrovirus with TGF-β treatment or the retrovirus expressing wild-type (WT) KDM5B or the mutant (Mut). PCR data were normalized with respect to control human GAPDH expression. The averages from at least three experiments are presented with the standard deviations (*p < 0.001, comparing to control). (H) Western blotting was performed to detect the expression of E-cadherin, ZEB1 and ZEB2 proteins using the corresponding antibodies. As a control, anti-GAPDH antibody was used to show that equal amounts of proteins were loaded on the gel.
During the EMT process, it has been reported that the expression of E-cadherin is regulated by multiple transcriptional repressors.16,17 Thus, we investigated whether overexpression of KDM5B affected the expression of transcription factors such as SNAI1, SNAI2, ZEB1 and ZEB2. Quantitative RT-PCR analysis showed that TGF-β treatment upregulated the expression of all transcription factors we examined in A549 cells (Figs. 2D–G). Expression of wild-type KDM5B did not affect the expression of SNAI1 and SNAI2 significantly (Fig. 2D and E), but resulted in the increased expression of ZEB1 and ZEB2 in A549 cells (Fig. 2F and G). We also confirmed the changes of protein expression for some of the EMT-related gene products in A549 cells. Western blot showed that the expression of wild-type KDM5B resulted in the reduction of E-cadherin protein and the increase of ZEB1 and ZEB2 proteins as TGF-β treatment did (Fig. 2H).
Quantitative PCR and western blot revealed that KDM5B overexpression induced essentially the same changes for the EMT-related gene expression in NMuMG mammary epithelial cells (Fig. S4A–H), although endogenous ZEB2 protein was not detected because of its extremely low expression in NMuMG cells. Moreover, in HT29 colon cancer cells, KDM5B resulted in the similar changes for the EMT-related gene expression (Fig. S5A–E), although endogenous expression of CDH2/N-cadherin, SNAI2 and ZEB2 was extremely low or not detected in HT29 cells. These results together suggested that KDM5B histone lysine demethylase might be specifically involved in the regulation of ZEB family expression and then control the transcriptional network leading to EMT.
KDM5B did not seem to be directly involved in the expression of CDH1/E-cadherin, ZEB1 or ZEB2
Since KDM5B is an enzyme that possesses a demethylase activity of histone H3 to regulate gene expression,23-25 we examined the recruitment of KDM5B and the methylation status of histone H3 on the regulatory regions of CDH1/E-cadherin, ZEB1 or ZEB2 gene. Chromatin immunoprecipitation (ChIP) assay was performed using antibodies that specifically recognize methylated K residues of histone H328 or FLAG epitopes. A549 cells were infected with the control retrovirus or the retrovirus expressing FLAG-tagged wild-type KDM5B, and the cell lysates were prepared. Following immunoprecipitation, total ten different primer sets positioned upstream or around the transcription initiation sites of CDH1, ZEB1 and ZEB2 gene were used in quantitative PCR analysis (Fig. 3A, C and E; Figs. S6A, S7A and S8A). We analyzed the transcriptionally active tri-methylated H3K4 (H3K4me3) status and the repressive H3K27me3 status on the regulatory region of each gene. As a representative result for each gene locus, the data from one primer set (region a) was presented (Fig. 3B, D and F). The data obtained from all other primer sets were shown in Figures S6B, S7B and S8B. In the case of CDH1 gene, H3K4me3 was significantly decreased and H3K27me3 was slightly increased in the regions around the transcription initiation site with the expression of wild-type KDM5B (Fig. 3B; Fig. S6B). Since CDH1 expression was downregulated by KDM5B, this observation was consistent with the previous report showing low levels of H3K4me3 and high levels of H3K27me3 on the regulatory regions linked to transcriptionally repressed status.6 However, the recruitment of KDM5B was not detected on the all regions of CDH1 gene that we have tested (Fig. 3B; Fig. S6B). These results suggested that KDM5B did not seem to be directly involved in the transcriptional regulation of CDH1. On the other hand, we did not detect any significant changes of H3K4me3, H3K27me3 and the recruitment of KDM5B on the all regions of ZEB1 and ZEB2 genes that we have examined (Fig. 3D and F; Figs. S7B and S8B). These results might also exclude the direct involvement of KDM5B in the transcriptional regulation of ZEB1 and ZEB2 genes in the cells with KDM5B overexpression.

Figure 3. Recruitment of KDM5B was not detected on the regulatory regions of CDH1/E-cadherin, ZEB1 and ZEB2 genes. Schematic of the regulatory regions of CDH1/E-cadherin (A), ZEB1 (C) and ZEB2 genes (E) is presented. The boxes shown on the scheme indicate the first and second exons, and the dark area corresponds to the coding region. The arrow points to the transcription initiation site. The regions covered by the primer sets used for ChIP assays are shown as (a). ChIP analyses of H3K4me3, H3K27me3 and FLAG-tagged KDM5B on the regulatory regions of CDH1/E-cadherin (B), ZEB1 (D) and ZEB2 genes (F) are shown. Cross-linked chromatins from A549 cells infected with the control retrovirus or the retrovirus expressing wild-type KDM5B were immunoprecipitated with the specific antibodies. The occupancies of methylated histones or KDM5B protein on the regions were analyzed by quantitative PCR and presented as the percentages of enrichment over input DNA. The averages from at least three experiments are presented with the standard deviations (*p < 0.001, comparing to control; **p < 0.05, comparing to control).
KDM5B regulated the expression of the microRNA-200 family gene through the conversion of histone H3 methylation
The finding that KDM5B increased ZEB1 and ZEB2 expression specifically and indirectly in the EMT process led us to investigate the possibility that the effect could be due to the regulation of the miR-200 family of microRNAs. The miR-200 family has been established as a key regulator of epithelial phenotype and is known to be deeply involved in the EMT process.30,31 The miR-200 family was found to directly target and inhibit ZEB1 and ZEB2 specifically. Genetically, the miR-200 family is grouped in two polycistronic units: miR-200b/200a/429 and miR-200c/141, clustered in chromosome 1 and 12, respectively (see Fig. 4C and E). Therefore we examined whether KDM5B overexpression would affect the expression of two representative microRNAs, miR-200a and miR-200c. Consistent with the previous reports,30 quantitative RT-PCR revealed that TGF-β treatment resulted in a decreased expression of miR-200a and miR-200c in A549 cells (Fig. 4A and B). We could also detect a decrease in the expression of miR-200a and miR-200c with the expression of wild-type KDM5B but not with its mutant (Fig. 4A and B). The expression of miR-302a that belongs to an unrelated miR-302 family was not significantly affected by KDM5B expression (data not shown), suggesting the specific effect of KDM5B on the miR-200 family. We also found the similar downregulation of miR-200a and miR-200c induced by wild-type KDM5B in NMuMG cells and HT29 cells (Fig. S9). These results strongly suggested that KDM5B was implicated in the regulation of the miR-200 family expression and involved in the EMT process through this regulation.

Figure 4. KDM5B decreased the expression of miR-200a and miR-200c by converting the histone H3 methylation on the regulatory regions of miR-200 gene clusters. Quantitative RT-PCR analysis was performed to detect the expression of miR-200a (A), and miR-200c (B) in A549 cells infected with the control retrovirus, the control retrovirus with TGF-β treatment or the retrovirus expressing wild-type (WT) KDM5B or the mutant (Mut). PCR data were normalized with respect to control human RNU6B expression (*p < 0.001, comparing to control). Schematic representation of the regulatory regions of miR-200b/200a/429 (C) and miR-200c/141 genes (E) is shown. The regions covered by the primer sets used for ChIP assays are shown as (a) and (b). ChIP analyses of H3K4me3, H3K27me3 and FLAG-tagged KDM5B on the regulatory regions of miR-200b/200a/429 (D) and miR-200c/141 genes (F) are shown (*p < 0.001, comparing to control; **p < 0.005, comparing to control).
Next we examined the methylation status of histone H3 on the regulatory regions of the miR-200 family genes by ChIP analysis. As shown in Figure 4C and E, the transcription initiation sites of miR-200b/200a/429 and miR-200c/141 precursor RNAs were reported previously.32,33 Following immunoprecipitation, the two primer sets positioned around the transcription initiation sites of the microRNA clusters were used in quantitative PCR analysis (Fig. 4C and E). On the regulatory regions of both miR-200b/200a/429 and miR-200c/141 genes, the level of H3K4me3 was dramatically decreased by the expression of KDM5B (Fig. 4D and F). Moreover, ChIP assay using anti-FLAG antibody clearly demonstrated the increased occupancies of FLAG-tagged wild-type KDM5B on the regulatory regions (Fig. 4D and F). The level of repressive H3K27me3 mark was significantly increased by KDM5B expression (Fig. 4D and F), which was correlated with the transcriptional repression by KDM5B. These results suggested that KDM5B which was recruited on the regulatory regions of miR-200b/200a/429 and miR-200c/141 genes converted the chromatin structure to a more transcriptionally repressed status by demethylating H3K4. As a control experiment, we performed ChIP assay on the regulatory regions of BRCA1 and HOXA5 genes that were reported to be the target genes regulated by KDM5B25 and also unrelated GAPDH gene (Fig. S10). In the case of BRCA1 and HOXA5 genes, the decrease of H3K4me3 and the increase of H3K27me3 were detected simultaneously with the recruitment of KDM5B on the regulatory regions (Fig. S10B and D). On the other hand, there were no significant changes in H3K4 and H3K27 methylation and KDM5B occupancies on the regulatory regions of GAPDH gene by KDM5B (Fig. S10F). These results indicated that KDM5B-induced changes of histone H3 methylation were specific to the target genes regulated by KDM5B, including miR-200b/200a/429 and miR-200c/141 genes, which was closely correlated with the specificity of transcriptional regulation by KDM5B.
The regulation of miR-200 family expression was important in KDM5B-induced EMT
In order to clarify the role of the miR-200 family downregulated by KDM5B in EMT, we transfected the precursor of miR-200a or miR-200c into KDM5B-expressing A549 cells to see the rescue effects in the EMT process. Figure 5A showed that the expression of wild-type KDM5B decreased CDH1/E-cadherin expression, but simultaneous introduction of miR-200a or miR-200c precursor recovered CDH1 expression. Transfection of miR-200a or miR-200c was also shown to inhibit the enhanced expression of mesenchymal marker CDH2/N-cadherin by KDM5B (Fig. 5B). These results suggested that overexpression of miR-200a or miR-200c inhibited EMT induction by KDM5B expression. Moreover, the expression of miR-200a or miR-200c counteracted the enhanced expression of ZEB1 and ZEB2 induced by KDM5B (Fig. 5C and 5D) but had no effect on the expression of SNAI1 and SNAI2 (data not shown). We found that the introduction of miR-200a or miR-200c precursor resulted in the loss of mesenchymal cell appearance of the KDM5B-expressing cells by the staining of E-cadherin and actin (Fig. S11A and B). We also confirmed that the expression of miR-200a or miR-200c did not affect the expression level of KDM5B proteins (Fig. S12). These results clearly indicated the inhibitory function of these microRNAs in EMT process induced by KDM5B. Taken together, we could conclude that the reduced expression of miR-200a and miR-200c was primarily responsible for the EMT induction by KDM5B overexpression.
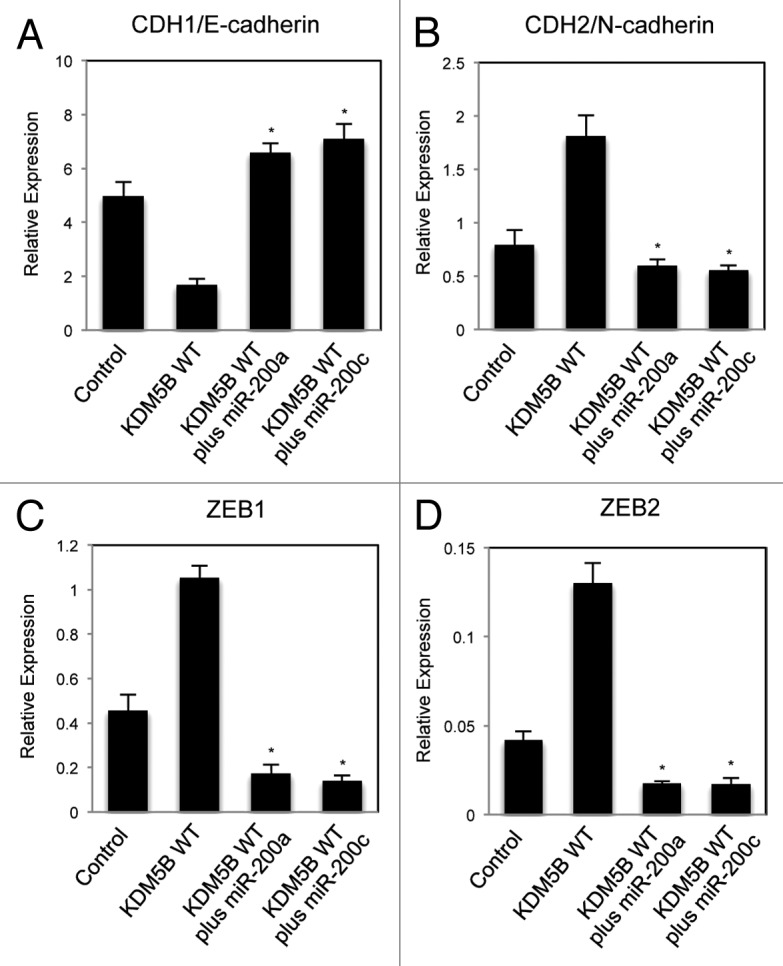
Figure 5. Altered expression of EMT-related gene induced by KDM5B was cancelled with the introduction of exogenous miR-200. Quantitative RT-PCR analysis was performed to detect the expression of CDH1/E-cadherin (A), CDH2/N-cadherin (B), ZEB1 (C) and ZEB2 (D) in A549 cells infected with the control retrovirus, the retrovirus expressing wild-type KDM5B, KDM5B with miR-200a precursor and KDM5B with miR-200c precursor (*p < 0.001, comparing to KDM5B WT).
Knockdown of KDM5B indicated the involvement of endogenous KDM5B in the transcriptional regulation of EMT-related genes
To extend our understanding for the regulation of EMT by KDM5B, we examined the effects of KDM5B knockdown in the cells. We used the retroviruses expressing small hairpin RNA (shRNA) for efficient and stable knockdown of KDM5B as described previously.13 A549 cells were infected with the control retrovirus or the retrovirus expressing KDM5B shRNA, and we examined the expression of CDH1/E-cadherin, FN1/Fibronectin and CDH2/N-cadherin in the cells with KDM5B knockdown by quantitative RT-PCR. Knockdown of KDM5B increased the CDH1 expression, but decreased the expression of FN1 and CDH2 (Fig. 6A–C). These results indicated that KDM5B knockdown resulted in the opposite effects compared with KDM5B overexpression. By immunofluorescence, KDM5B knockdown induced stronger staining of E-cadherin on the cell membrane compared with control cells (Fig. S13A, KDM5B sh vs. control), but had no effect on actin organization (Fig. S13B). Next we examined the expression of EMT-related transcription factors by quantitative RT-PCR and found that KDM5B knockdown decreased the expression of ZEB1 and ZEB2 (Fig. 6D and E). We also confirmed the corresponding changes of protein expression for E-cadherin, ZEB1 and ZEB2 in A549 cells by KDM5B knockdown (Fig. 6F). Furthermore, we found that the expression of miR-200a and miR-200c was elevated in the KDM5B-knockdown cells (Fig. 6G and H). From the ChIP experiments, we could detect the increase of H3K4me3 and the decrease of H3K27me3 on the regulatory regions of miR-200b/200a/429 and miR-200c/141 genes induced by KDM5B knockdown (Fig. 6I and J). These results indicated that endogenous KDM5B was involved in the transcriptional regulation of EMT-related genes in A549 cells. Next we examined whether KDM5B knockdown could be refractory to TGF-β-induced EMT process. KDM5B knockdown did not affect the loss of E-cadherin staining and actin fiber formation induced by TGF-β (Fig. S13A and B, KDM5B sh plus TGF-β vs. control plus TGF-β). Based on the quantitative PCR analyses for EMT-related gene expression, KDM5B knockdown did not counteract with TGF-β-induced changes of gene expression either (Fig. S14A–E). Finally, it was shown that the level of endogenous KDM5B was slightly but significantly increased after TGF-β treatment in A549 cells (Fig. S15). Taken together, these results suggested that endogenous KDM5B played an important role in the transcriptional regulation of EMT-related genes, possibly through the regulation of the miR-200 family expression, although TGF-β treatment could overcome the depletion of KDM5B for the induction of EMT of the cells.

Figure 6. Knockdown of KDM5B affected the expression of EMT-related genes in A549 cells. Quantitative RT-PCR analysis was performed to detect the expression of CDH1 (A), FN1 (B), CDH2 (C), ZEB1 (D) and ZEB2 (E) in A549 cells expressing control shRNA or KDM5B shRNA (*p < 0.001, comparing to control; **p < 0.005, comparing to control). (F) Western blotting was performed to detect the expression of E-cadherin, ZEB1 and ZEB2 proteins using the corresponding antibodies. Quantitative RT-PCR for miR-200a (G) and miR-200c (H) was performed (*p < 0.001, comparing to control; **p < 0.005, comparing to control). (I and J) ChIP analysis of H3K4me3 and H3K27me3 on the regulatory regions of miR-200b/200a/429 (I) and miR-200c/141 genes (J) in A549 cells expressing control shRNA or KDM5B shRNA (*p < 0.001, comparing to control; **p < 0.005, comparing to control; ***p < 0.01, comparing to control).
Significant association between KDM5B and ZEB family expression in human lung cancer tissues
The finding that KDM5B resulted in the increased expression of ZEB family transcription factors led us to examine the correlation between KDM5B and ZEB family expression in human cancer specimens. Quantitative RT-PCR analysis was performed to detect the expression of KDM5B, ZEB1 and ZEB2 in human lung cancer tissues (n = 24). To determine the possible relationship of KDM5B and ZEB family, we compared the gene expression results using Pearson correlation coefficient. As shown in Figure 7, each dot represented the values of two genes (i.e., x- and y-axis), and the correlation coefficient reflected an association between the two genes. We detected a significant association of KDM5B and ZEB1 expression (Fig. 7A) or KDM5B and ZEB2 expression (Fig. 7B). The associations were positive, with r = 0.442 (p < 0.05) and with r = 0.601 (p < 0.005), respectively. These results suggested that KDM5B might regulate the expression of ZEB family, one of the critical EMT inducers, during the development of human lung cancer.
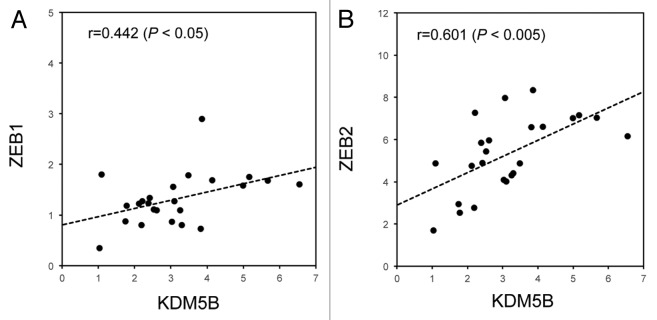
Figure 7. Significant association between KDM5B and ZEB family expressions in human lung cancer samples. Quantitative RT-PCR analysis was performed to detect the expression of KDM5B, ZEB1 and ZEB2 in human lung cancer tissues (n = 24). PCR data were normalized with respect to control human DDX5 expression. Association of KDM5B and ZEB1 expression (A) or KDM5B and ZEB2 expression (B) was estimated using Pearson correlation coefficient. The associations were significantly positive, with r = 0.442 (p < 0.05) and with r = 0.601 (p < 0.005), respectively.
Discussion
In the current study, we found that ectopic expression of KDM5B H3K4 demethylase enhanced the expression of ZEB1 and ZEB2 and resulted in decreased CDH1/E-cadherin expression, thereby promoting EMT. This effect of KDM5B was commonly observed in several EMT model systems, including A549 human lung cancer cells, HT29 colon cancer cells and NMuMG mouse mammary epithelial cells. KDM5B was shown to regulate the expression of the microRNA-200 family, which specifically inhibits ZEB1 and ZEB2 through the conversion of histone H3 methylation on their regulatory regions. We also showed that knockdown of KDM5B affected the expression levels of EMT-related genes, suggesting the involvement of endogenous KDM5B. Moreover, we detected a positive association between KDM5B and ZEB family expression in human lung cancer tissues. Our study uncovers a novel role of KDM5B histone lysine demethylase in EMT, and has important implication in targeting cancer metastasis.
A growing body of evidence indicates that overexpression or mutations of histone methyltransferases and demethylases have been linked to the development of many human cancers.4 A histone H3K27 methytransferase, EZH2, and H3K4 demethylases LSD1 and KDM5B have been thought to play important roles not only in tumor initiation, but also in tumor progression, since overexpression of these genes has been reported in many types of malignant tumors.24,26,34-37 Previous papers revealed that upregulation of EZH2 or LSD1 enhanced cell migration, cell invasion and EMT to promote malignant progression of cancer cells.35,36 During the EMT process, it was shown that EZH2 repressed E-cadherin gene expression,35 and that LSD1 interacted with SNAI1 to repress E-cadherin and other epithelial genes.21 Although we previously reported that overexpression of KDM5B/PLU1 enhanced the invasive potential of cancer cells through repressing KAT5 and CD82 expression,13 the involvement of KDM5B in EMT remained unknown. Here we identified a novel important role of KDM5B that contributed to EMT, another hallmark of cancer aggressiveness. Overexpression of KDM5B was shown to promote the EMT process of the cells by increasing the expression of ZEB1 and ZEB2 transcription factors through the regulation of microRNA-200 family expression. Moreover, we have observed KDM5B-induced EMT both in cancer cells, A549 and HT29, and benign epithelial cells, NMuMG, which might ensure the generality of novel function of KDM5B.
The members of the miR-200 family are important regulators of EMT. Deregulation of miR-200 family expression occurred in multiple types of cancer cells and was linked to tumor progression.30,31 It was proposed that the expression of miR-200 family was controlled by epigenetic mechanisms.33,38 Increased EZH2 activity in cancer was shown to cause repression of numerous microRNAs, including miR-200b and miR-200c, through H3K27me3 regulation at these loci.39 A recent paper also demonstrated that knockdown of JMJD3 H3K27 demethylase decreased the expression of miR-200b and miR-200c for upregulation of EMT inducers such as ZEB1 and ZEB2,40 suggesting the importance of H3K27 methylation in the regulation of miR-200 family expression. Furthermore, a recent genome-wide profiling of histone methylation during EMT revealed strong correlations between the dynamic changes of histone methylations and gene expression.19 For genes with bivalent marks with H3K4me3 and H3K27me3, the level of transcription was shown to be dependent on the relative intensities of active H3K4me3 and repressive H3K27me3 marks. This was closely correlated with our results of ChIP experiments: overexpression of KDM5B induced the decrease of H3K4me3 and the increase of H3K27me3 on the regulatory regions of the miR-200 family, resulting in decreased expression, but KDM5B knockdown caused the opposite changes of histone methylations for the increased expression of the miR-200 family.
KDM5B has been found highly expressed in various types of tumors, such as breast, prostate, bladder and lung cancers.23,24,26 Increased expression of KDM5B caused proliferation in breast cancer, and depletion of KDM5B inhibited tumor growth in a mouse mammary tumor model.25 KDM5B can demethylate H3K4me3/me2 and was shown to regulate the expression of various cellular genes including BRCA1 tumor suppressor, E2F1 and E2F2 genes.25,26 In this study, we identified miR-200 family genes as the novel target genes regulated by KDM5B. Overexpression of KDM5B led to the repression of miR-200 family genes, and KDM5B knockdown increased their expression. The recruitment of KDM5B and the changes of histone H3 methylation could be detected on the regulatory regions of miR-200 family genes, like the cases of BRCA1 and HOXA5, which have already been reported as KDM5B targets. Furthermore, introduction of miR-200 precursor was shown to inhibit EMT phenotype of the cells induced by KDM5B. These findings strongly suggested that miR-200 family was an important downstream mediator of KDM5B-induced EMT, although we could not rule out the possibility that other target genes regulated by KDM5B might be also involved in EMT.
We have also detected that ectopic expression of KDM5B increased the cell invasion activity of A549 lung cancer cells and repressed the expression of other KDM5B target genes, KAT5 and CD82, that we previously described (ref. 13 and unpublished results). Since CD82 is involved in integrin-mediated cell migration, these results suggested that KDM5B might affect both EMT and integrin-mediated pathway, leading to cell invasion in A549 cells. However, in MCF10A immortalized breast epithelial cells, KDM5B overexpression enhanced cell invasion but had no effect on CDH1 expression,13 suggesting that EMT might not be necessary for cell invasion activity of MCF10A cells. Therefore, epigenetic regulation by KDM5B might be differently involved in EMT or the cell invasion process in different type of cell lines. Further studies using various cancer cell lines would be required to extend our understanding of KDM5B function in malignant progression.
By the knockdown experiments for KDM5B, we provided evidence supporting the role of endogenous KDM5B in the transcriptional regulation of EMT-related genes. KDM5B knockdown increased CDH1 expression and decreased the expression of ZEB1 and ZEB2 through upregulation of the microRNA-200 family. Alteration of histone H3 methylation on the regulatory regions of the microRNA clusters was suggested to be the underlying mechanism, although the recruitment of endogenous KDM5B on the regulatory regions was not detected, probably due to the low level of protein expression or low sensitivity of the available antibodies. We found that endogenous KDM5B was slightly but significantly induced by TGF-β treatment in A549 cells, but KDM5B knockdown could not counteract with TGF-β-induced EMT process. However, this result did not weaken the importance of endogenous KDM5B function in EMT process. Since TGF-β operated at the upstream of signaling pathway and activated various signaling molecules, transcription factors and epigenetic regulators in the cells for EMT induction, it was possible that KDM5B knockdown could not interrupt the profound effects caused by TGF-β. Further studies would be essential to delineate the precise role of endogenous KDM5B in the process of EMT.
In this paper, we have shown the important function of KDM5B histone demethylase in EMT. The regulation of the miR-200 family expression through histone H3 methylation was shown to be a novel mechanism for KDM5B action. These findings strongly suggest that deregulation of histone methyl-modifying enzymes and microRNA expression contributes to a critical step for malignant progression of cancer. Our study reveals a novel epigenetic mechanism regulating EMT and has important implications in devising novel targets for therapeutic interventions in the treatment of aggressive cancers.
Materials and Methods
Plasmids, cell culture and transfection
The construction of the retrovirus vectors expressing mouse KDM5B or its inactive mutant H499Y was described previously.13 To produce pantropic retroviruses for the infection of human cells, the retrovirus vectors were co-transfected into GP2–293 packaging cells (Takara) with pVSV-G plasmid (Takara) using FuGENE HD reagent (Roche). To produce ecotropic retroviruses for the infection of mouse cells, the retrovirus vectors were transfected into Plat-E packaging cells.11 Forty-eight hours after transfection, the supernatants were collected. The retroviral stock was added into the medium with 6 μg/ml polybrene (Sigma) for infection. The infected cells were selected in the medium containing 800 μg/ml G418 (nacalai) for 2–5 d and used for the experiments.
A549 human lung cancer cell line, HT29 human colon cancer cell line and NMuMG mouse mammary epithelial cell line were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS, 2 mM glutamine and penicillin/streptomycin (Sigma) at 37°C in 5% CO2. For EMT induction, the cells were treated with 1–5 ng/ml of transforming growth factor-β (TGF-β) (R&D Systems) for 24–48 h.
The construction of shRNA-expressing retrovirus vectors expressing KDM5B shRNA#1, #2 and control shRNA was described previously.13 To produce shRNA-expressing retroviruses, the retrovirus vectors were co-transfected into HEK293T cells with MISSION Lentiviral Packaging Mix (Sigma-Aldrich). The infected cells were selected in the medium containing 500 ng/ml puromycin (Nacalai). As described previously,13 the knockdown efficiencies of KDM5B shRNA#1 and #2 were 82 and 77% reduction compared with the control, respectively. We used these two effective shRNA-expressing retroviruses in this study and essentially obtained the same effects of both. Therefore we presented the data of KDM5B shRNA#1 as a representative result.
Transfections of miRNA precursors (Ambion) were performed with Lipofectamine RNAiMAX (Invitrogen) by reverse transfection procedure. Cells were transfected with pre-miR-200a (#PM10991), pre-miR-200c (#PM11714) or negative control #1 (#AM17110) miRNA at a final concentration of 10 nM, and further incubated for 3 d and used for the experiments.
Patient samples
For gene expression studies, 24 tumor specimens were obtained from lung adenocarcinoma patients with written informed consent at Kanazawa University Hospital in Japan. RNA was obtained from the OCT-compound-embedded frozen tissues (Tissue-Tek). The association between mRNA expressions was analyzed by Pearson correlation coefficient, and the results were summarized by scatter plot. All tests were two-sided, and p values < 0.05 were considered statistically significant. The experiment was approved by the Ethics Committee of Kanazawa University School of Medicine.
Quantitative PCR
RNA preparation and quantitative RT-PCR analysis were performed as described previously.11 PCR data were normalized with respect to control human GAPDH, human DDX5 or mouse Actb expression. The averages from at least three independent experiments are shown with the standard deviations. In the quantitative PCR experiments, Student’s t-tests were used to determine the statistical significance of differences between samples treated under different conditions. Differences were considered statistically significant when p < 0.05. Primers used for the quantitative PCR were described previously13 and are listed in Table S1.
For microRNA quantification, TaqMan MicroRNA Assays (Applied Biosystems) for miR-200a (#000502), miR-200c (#002300) and miR-302a (#000529) were used according to the manufacturer’s protocol. The relative miRNA levels were calculated using the equation 2−ΔΔCt. All data were normalized with respect to the expression of RNU6B (#001093) for human or snoRNA202 (#001232) for mouse.
Immunoblotting
Cells were lysed in RIPA buffer as described previously.27 The lysates were separated on SuperSep Ace 10% running gel (Wako) and transferred to Hybond™-LFP membrane (GE Healthcare). Anti-E-cadherin (#610181, BD Transduction Lab), anti-ZEB1 (#3396, Cell Signaling), anti-ZEB2 (#61096, Active Motif) and anti-GAPDH (6C5, Millipore) antibodies were used.
Chromatin immunoprecipitation (ChIP) assays
ChIP experiments were performed as previously described.13,28 The cross-linked chromatins were immunopreciptated with mouse antibody [anti-H3K27me3, anti-H3K4me3, anti-FLAG (M2, Sigma) and normal IgG], and the complexes were collected by Dynabeads M-280 sheep anti-mouse IgG (Invitrogen). The enrichment of the specific amplified region was analyzed by quantitative PCR, and percentage enrichment of each histone modification over input chromatin DNA was shown. The averages from at least three experiments are presented with the standard deviations.
Cell staining and immunofluorescence assay
To detect the morphological changes of the cells, A549 or NMuMG cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and stained with 0.4% crystal violet (Waldeck). To allow direct fluorescence of actin cytoskeleton, A549 cells were fixed, permeabilized with 0.2% Triton X-100 in PBS for 2 min and subsequently stained with 0.25 μM tetramethylrhodamine isothiocyanate (TRITC)-conjugated phalloidin (Sigma). For indirect immunofluorescence, the specimens were incubated with anti-E-cadherin antibody (BD Biosciences) and treated with Alexa546-conjugated anti-mouse IgG antibody (Invitrogen). Nuclei were visualized with 4’,6-diamidino-2-phenylindole (DAPI).
Supplementary Material
Acknowledgments
We thank Drs Y. Endo, Y. Kido, T. Takino and H. Sato (Cancer Research Institute, Kanazawa University) for providing the cell lines and the protocol for EMT, microRNA experiments. We also thank Dr H. Kimura (Osaka University) and Dr N. Nozaki (Hokkaido University) for providing the antibodies that specifically recognize methylated H3K residues. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Glossary
Abbreviations:
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- H3K4me3
histone H3 tri-methylated Lys4
- HEK293T cells
human embryonic kidney cells expressing the large T-antigen of simian virus 40
- IgG
immunoglobulin G
- miRNA
microRNA
- shRNA
small hairpin RNA
- EMT
epithelial-mesenchymal transition
- TGF-β
transforming growth factor-beta
- ChIP
chromatin immunoprecipitation
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/25135
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25142
References
- 1.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–40. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem. 2010;79:155–79. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 4.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656–72. doi: 10.1038/sj.onc.1209043. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, et al. New genes involved in cancer identified by retroviral tagging. Nat Genet. 2002;32:166–74. doi: 10.1038/ng949. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki T, Minehata K, Akagi K, Jenkins NA, Copeland NG. Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm-deficient mice. EMBO J. 2006;25:3422–31. doi: 10.1038/sj.emboj.7601215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishimura A, Terashima M, Kimura H, Akagi K, Suzuki Y, Sugano S, et al. Jmjd2c histone demethylase enhances the expression of Mdm2 oncogene. Biochem Biophys Res Commun. 2009;389:366–71. doi: 10.1016/j.bbrc.2009.08.155. [DOI] [PubMed] [Google Scholar]
- 12.Terashima M, Ishimura A, Yoshida M, Suzuki Y, Sugano S, Suzuki T. The tumor suppressor Rb and its related Rbl2 genes are regulated by Utx histone demethylase. Biochem Biophys Res Commun. 2010;399:238–44. doi: 10.1016/j.bbrc.2010.07.061. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida M, Ishimura A, Terashima M, Enkhbaatar Z, Nozaki N, Satou K, et al. PLU1 histone demethylase decreases the expression of KAT5 and enhances the invasive activity of the cells. Biochem J. 2011;437:555–64. doi: 10.1042/BJ20110343. [DOI] [PubMed] [Google Scholar]
- 14.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 17.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–37. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyazono K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:314–23. doi: 10.2183/pjab.85.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ke XS, Qu Y, Cheng Y, Li WC, Rotter V, Øyan AM, et al. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genomics. 2010;11:669. doi: 10.1186/1471-2164-11-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu CY, Tsai YP, Wu MZ, Teng SC, Wu KJ. Epigenetic reprogramming and post-transcriptional regulation during the epithelial-mesenchymal transition. Trends Genet. 2012;28:454–63. doi: 10.1016/j.tig.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. 2010;29:4896–904. doi: 10.1038/onc.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122:1469–86. doi: 10.1172/JCI57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, Meier-Ewert S, et al. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274:15633–45. doi: 10.1074/jbc.274.22.15633. [DOI] [PubMed] [Google Scholar]
- 24.Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci USA. 2007;104:19226–31. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007;25:801–12. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 26.Hayami S, Yoshimatsu M, Veerakumarasivam A, Unoki M, Iwai Y, Tsunoda T, et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer. 2010;9:59. doi: 10.1186/1476-4598-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishimura A, Minehata K, Terashima M, Kondoh G, Hara T, Suzuki T. Jmjd5, an H3K36me2 histone demethylase, modulates embryonic cell proliferation through the regulation of Cdkn1a expression. Development. 2012;139:749–59. doi: 10.1242/dev.074138. [DOI] [PubMed] [Google Scholar]
- 28.Kimura H, Hayashi-Takanaka Y, Goto Y, Takizawa N, Nozaki N. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct. 2008;33:61–73. doi: 10.1247/csf.07035. [DOI] [PubMed] [Google Scholar]
- 29.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 31.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–54. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 33.Davalos V, Moutinho C, Villanueva A, Boque R, Silva P, Carneiro F, et al. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene. 2012;31:2062–74. doi: 10.1038/onc.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–7. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- 35.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17:2613–8. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 36.Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, et al. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One. 2012;7:e35065. doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011;128:574–86. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- 38.Vrba L, Jensen TJ, Garbe JC, Heimark RL, Cress AE, Dickinson S, et al. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS One. 2010;5:e8697. doi: 10.1371/journal.pone.0008697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Q, Mani RS, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR, et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011;20:187–99. doi: 10.1016/j.ccr.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pereira F, Barbáchano A, Singh PK, Campbell MJ, Muñoz A, Larriba MJ. Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle. 2012;11:1081–9. doi: 10.4161/cc.11.6.19508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.