

Abstract
Dexamethasone is a key front-line chemotherapeutic for B-cell malignant multiple myeloma (MM). Dexamethasone modulates MM cell survival signaling but fails to induce marked cytotoxicity when used as a monotherapy. We demonstrate here the mechanism behind this insufficient responsiveness of MM cells toward dexamethasone, revealing in MM a dramatic anti-apoptotic role for microRNA (miRNA)-125b in the insensitivity toward dexamethasone-induced apoptosis. MM cells responding to dexamethasone exhibited enhanced expression of oncogenic miR-125b. Dexamethasone also induced expression of miR-34a, which acts to suppress SIRT1 deacetylase, and thus allows maintained acetylation and inactivation of p53. p53 mRNA is also suppressed by miR-125b targeting. Reporter assays showed that both these dexamethasone-induced miRNAs act downstream of their target genes to prevent p53 tumor suppressor actions and, ultimately, resist cytotoxic responses in MM. Use of antisense miR-125b transcripts enhanced expression of pro-apoptotic p53, repressed expression of anti-apoptotic SIRT1 and, importantly, significantly enhanced dexamethasone-induced cell death responses in MM. Pharmacological manipulations showed that the key regulation enabling complete dexamethasone sensitivity in MM cells lies with miR-125b. In summary, dexamethasone-induced miR-125b induces cell death resistance mechanisms in MM cells via the p53/miR-34a/SIRT1 signaling network and provides these cells with an enhanced level of resistance to cytotoxic chemotherapeutics. Clearly, such anti-apoptotic mechanisms will need to be overcome to more effectively treat nascent, refractory and relapsed MM patients. These mechanisms provide insight into the role of miRNA regulation of apoptosis and their promotion of MM cell proliferative mechanisms.
Keywords: NFκB, SIRT1, mir-125b, mir-34a, p53
Introduction
MicroRNAs (miRNAs) are short (~22 nucleotide) single stranded non-coding RNA molecules that regulate translation and protein production by interfering with complementary seed sequences in target mRNA 3′ untranslated regions (UTRs). The role of miRNAs in the pathogenesis of neoplasms has become increasingly appreciated in recent years, particularly as they show potential as biomarkers and drug targets in the search for new cancer therapeutics.1 For example, studies have shown miRNA expression signatures to surpass those of mRNA in predicting tissue of origin and cancer type in both solid tumors and hematological malignancies.2-4
miRNA (miR)-125b is one such miRNA that provides an attractive focus for further research, emerging as a key player in the pathology of numerous cancers, in particular hematological malignancies.5 Several putative targets have been identified, including tumor suppressor p536 and pro-apoptotic Bcl-2 antagonist killer 1 (Bak1),7 suggesting that miR-125b acts as an oncogenic miRNA, or “oncomiR.” Further to this, miR-125b appears to be frequently implicated in drug resistance8 and thus presents an intriguing parallel to the function of another key miRNA, miR-34a.
miR-34a has shown deregulation in a diverse range of cancers through its role as a tumor suppressor.9,10 This activity appears, in part, to be due to the direct transactivation of miR-34a by pro-apoptotic p53.11 In turn, miR-34a targets the 3′UTR of Sirtuin (SIRT)1, an anti-apoptotic histone deacetylase that itself binds to and deacetylates the C terminus of the p53 protein. Activation of this pro-apoptotic cell signaling loop causes disruption of SIRT1 translation, promoting cell cycle arrest and ultimately apoptosis.12 The p53/miR-34a/SIRT1 network has now been well characterized in solid tumors, including breast and neuroblastoma,13 with emerging evidence of a role in leukemias, particularly chronic lymphocytic leukemia (CLL).14 Intriguingly, miR-34a also appears to confer a level of protection against drug resistance in various solid tumors, further highlighting its importance as a tumor suppressor.15,16
As yet, however, there has been little research into the role of either miR-125b or miR-34a in multiple myeloma (MM), despite this pathology sharing numerous characteristics with both solid and blood-borne malignancies. MM is characterized by a clonal expansion of plasma cells in the bone marrow and accounts for approximately 1% of all cancer diagnosis.17 There is currently no cure for MM and, despite the recent addition of thalidomide derivatives, frontline induction therapy continues to follow a similar regime to that seen in clinics over 50 y ago, including synthetic glucocorticoids and potential bone marrow transplant.18 Current treatment regimens favor the synthetic glucocorticoid dexamethasone (dex), which acts as an anti-inflammatory and immunosuppressant via the inhibition of NFκB.19,20 The exact mode of action for dexamethasone in MM is not fully understood; however, it is thought to “prime” malignant plasma cells for apoptosis in response to induction chemotherapies, such as Velcade (bortezomib) or lenalidomide (Revlamid), through its anti-inflammatory properties. Dexamethasone is known to stimulate plasma cell apoptosis in vivo and in vitro via pathways mediated by anti-apoptotic Bcl2,21 while further in vitro studies have demonstrated a role for transcription factors NFκB and p53.20 In patients, however, resistance to dexamethasone is a common problem, signifying a need to elucidate the cellular mechanisms of plasma cell drug resistance.22
Recently interest in hematopoietic cell miRNA expression in response to dexamethasone has increased, and there is evidence that miRNA can control sensitivity and resistance to dexamethasone in leukemic cell lines.23 Given the emerging data on miRNA regulation in response to dexamethasone, we took a microarray approach to investigate dexamethasone-induced miRNA in the dexamethasone-sensitive MM/B-lymphoblast cell line, MM.1S. Our findings led us to focus on miR-125b and the potential to exploit the p53/miR-34a/SIRT1 network to manipulate B-cell apoptosis to improve MM therapy.
Results
Dexamethasone fails to induce net cell death in human MM cells
Dexamethasone is well characterized as an anti-inflammatory and immunosuppressant through its repression of pro-survival transcription factor NFκB expression.19,24 We found, however, that dexamethasone failed to induce greater than approximately 20% cell death over a range of concentrations and time points in the dexamethasone-sensitive MM/B-lymphoblast cell line, MM.1S, compared with the dexamethasone-resistant MM.1R cell line (Fig. 1A). To further investigate the effect of dexamethasone on cell death we analyzed expression of key anti-apoptotic genes Bcl2 and c-FLIP,25 both of which showed a significant (p ≤ 0.001) reduction in mRNA expression over 24 h, compared with MM.1R (Fig. 1B). Repression of Bcl2 and c-FLIP protein in response to dexamethasone was confirmed between 24–48 h, along with repression of phosphorylated NFκB subunit p65 (p-p65) between 4–48 h (Fig. 1C). These apparent contradictions in gene expression and cell-death response led us to further investigate the mechanisms controlling dexamethasone-induced MM cell death.
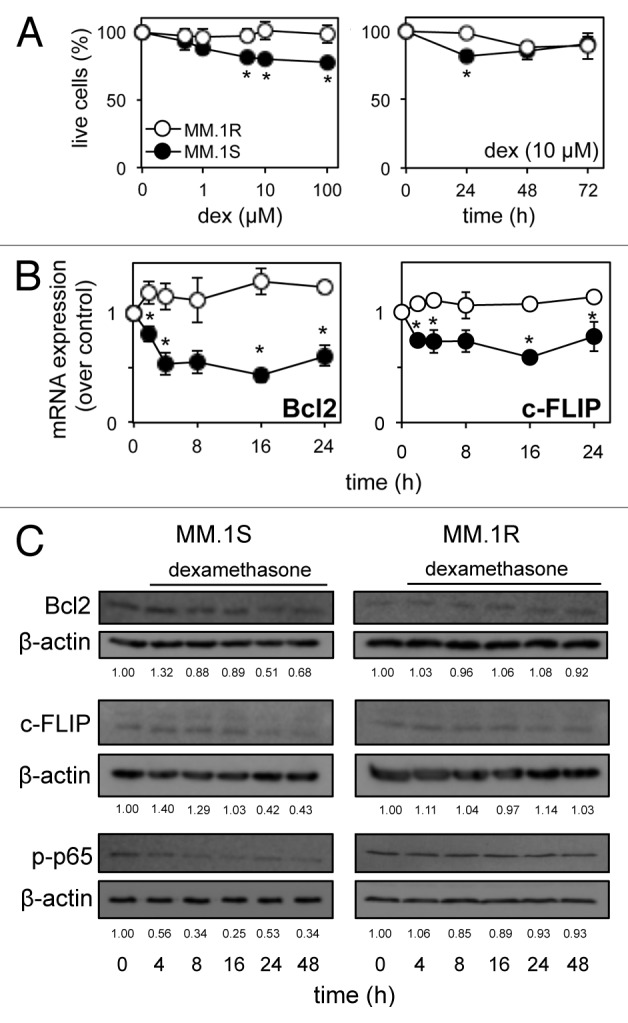
Figure 1. Dexamethasone fails to induce net cell death in multiple myeloma cells, despite repression of key anti-apoptotic genes. (A) Luminescent cell proliferation assays of dexamethasone-sensitive MM.1S and dexamethasone-resistant MM.1R cells with increased concentration and time of dexamethasone exposure. (B) qRT-PCR analysis of pro-survival Bcl2 and c-FLIP mRNA in response to dexamethasone (10 µM) over 24 h in MM.1S and MM.1R cells. Values indicate the mean ± SEM from three independent experiments (statistical significance calculated by Student’s t-test, with p ≤ 0.05 indicated,*). (C) Western blot analysis of total Bcl2 and c-FLIP protein and nuclear phosphorylated p65 (p-p65) in response to dexamethasone (10 μM) over 48 h in MM.1S and MM.1R cells. Membranes were re-probed for β-actin to confirm equal loading. Values indicate optical density of each band expressed as a fold change of untreated cells.
Dexamethasone induces miR-125b and miR-34a in human MM cells
To examine miRNA expression in MM.1S cells exposed to dexamethasone for 8 h, we utilized a commercial miRNA PCR array panel of 92 individual qRT-PCR primers for miRNA previously associated with cancer pathology and hematopoiesis. Despite a general trend for downregulation, there were high levels of expression and significant upregulation of oncomiR miR-125b (p ≤ 0.01) and tumor-suppressor miR-34a (p ≤ 0.001; Fig. 2A).
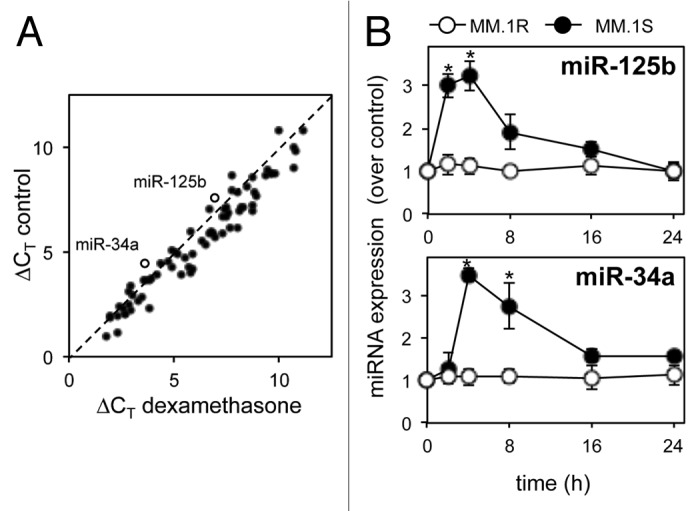
Figure 2. miRNA profiling of multiple myeloma cells in response to dexamethasone reveals regulation of miR-125b and miR-34a. (A) qRT-PCR analysis of 92 cancer-associated miRNAs in MM.1S cells in response to 8 h dexamethasone (10 µM). Values represent change in qRT-PCR cycle threshold normalized to an endogenous control (ΔCT). Dashed line indicates no change in expression. Open circles indicate miR-125b and miR-34a (labeled). (B) qRT-PCR analysis of miR-125b and miR-34a in response to dexamethasone (10 μM) over 24 h in MM.1S and MM.1R cells. Values indicate the mean ± SEM from three independent experiments (statistical significance calculated by Student’s t-test, * indicates p ≤ 0.05).
The functions and expression profile of miR-125b and miR-34a are well characterized, both in hematological malignancies26,27 and solid tumors.9,28 Both have been implicated in NFκB signaling29,30 and are emerging as important players in the pathogenesis of multiple myeloma.26 We therefore chose to further investigate the expression of miR-125b and miR-34a in MM.1S cells in response to dexamethasone over a time course of 24 h. qRT-PCR revealed peak expression of both miRs at 4 h post-dexamethasone, with basal expression levels returning by 24 h, compared with no change in expression in dexamethasone-resistant MM.1R cells (Fig. 2B).
Dexamethasone regulates the p53/miR-34a/SIRT1 signaling network in human MM cells
miR-34a is a key component of the p53/miR-34a/SIRT1 signaling network, through which it is directly trans-activated by p53 and targets the expression of the p53 deacetylase, SIRT1. Analysis of SIRT1 mRNA and protein expression in MM.1S and MM.1R cells reveals a decrease over time following dexamethasone treatment in MM.1S (Fig. 3A), coinciding with the induction of miR-34a expression (Fig. 2B). This suggested targeting of the SIRT1 3′ UTR by mature miR-34a, as shown previously.12 To confirm the interaction between miR-34a and the SIRT1 3′ UTR, we utilized a chemiluminescent reporter gene assay. MM.1S cells transfected with the pMIR-REPORT™, 3′UTR-SIRT1 plasmid (Fig. 3E) showed decreased luciferase reporter activity relative to β-galactosidase when co-transfected with synthetic miR-34a-mimics, compared with MM.1S transfected with a non-target control plasmid or scrambled (scrm)-miRNA control (Fig. 3B).

Figure 3. Dexamethasone-induced miR-125b and miR-34a expression are responsible for repression of SIRT1 and p53 target mRNA. (A) qRT-PCR shows time-dependant repression of SIRT1 and (C) p53 mRNA levels in response to dexamethasone. Dexamethasone-resistant MM.1R cells were used as a control. Insets show western blotting for SIRT1 and p53 protein at 24 h. (B) Chemiluminescent plasmid reporter gene assays confirm targeting of a fragment of the SIRT1 3′ UTR following transfection with synthetic miR-34a-mimics and (D) targeting of the p53 3′ UTR following transfection with synthetic miR-125b-mimics, compared with a non-target control 3′ UTR or scrambled-miR in MM.1S cells. (E) Schematic representation of the reporter constructs created and used. Values indicate the mean ± SEM from three independent experiments (statistical significance calculated by Student’s t-test; *, indicates p ≤ 0.05).
Unexpectedly, despite repression of its inhibitor SIRT1, tumor suppressor p53 also showed reduced mRNA and protein levels in response to dexamethasone over 24 h (Fig. 3C). This does, however, correspond with the early dexamethasone-driven induction of miR-125b, known to bind target sites in the p53 3′ UTR.6 Again, we utilized a chemiluminescent reporter gene assay to confirm the interaction between miR-125b and p53. MM.1S cells transfected with the pMIR-REPORT™-3′UTR-p53 (Fig. 3E) plasmid showed decreased luciferase reporter activity relative to β-galactosidase when co-transfected with synthetic miR-125b-mimics, compared with MM.1S transfected with a non-target control plasmid or scrm-miRNA scrambled control (Fig. 3D).
miR-125b modulates expression of the p53/miR-34a/SIRT1 signaling network
To determine if there is a relationship between novel regulation of miR-125b and the p53/miR-34a/SIRT1 signaling network, MM.1S and MM.1R cells were transfected with synthetic miR-34a-mimics or anti-miR-125b-transcripts. We hypothesized that repression of oncomiR miR-125b activity and enhancement of tumor suppressor miR-34a activity would have equivalent effects on MM cells through their regulation of p53 and SIRT1, respectively. In the presence of anti-miR-125b-transcripts, p53 mRNA and protein expression was increased, while SIRT1 mRNA and protein was downregulated (Fig. 4A and C). Similarly, the transfection of MM cells with synthetic miR-34a-mimics resulted in an induction of p53 and repression of SIRT1 mRNA and protein (Fig. 4A and C). Importantly, transfection of synthetic miR-125b-mimics or anti-miR-34a transcripts produced antagonistic effects, impairing expression of p53 mRNA while enhancing SIRT1 mRNA (Fig. 4B). Together with confirmation of 3′ UTR targeting (Fig. 3), these results show a novel relationship between miR-125b and the p53/miR-34a/SIRT1 signaling network.
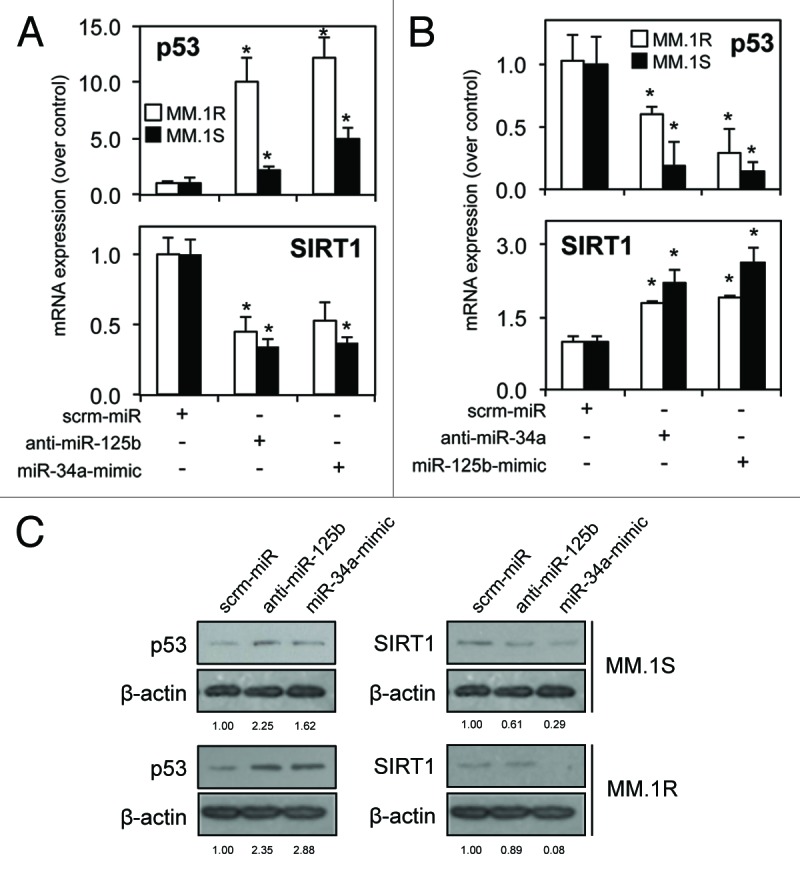
Figure 4. miR-125b and miR-34a have divergent effects on the p53/miR-34a/SIRT1 signaling network in multiple myeloma cells. (A) qRT-PCR analysis reveals enhanced p53 and repressed SIRT1 mRNA expression in MM.1S and MM.1R cells following transfection with anti-miR-125b-transcripts or synthetic miR-34a-mimics. (B) Transfection with anti-miR-34a-transcripts or synthetic miR-125b-mimics reveals an antagonistic effect. (C) Western blot analysis of total p53 and SIRT1 protein expression correlate with mRNA in response to anti-miR-125b or miR-34a-mimics in MM.1S and MM.1R cells. Values indicate the mean ± SEM from three independent experiments (statistical significance calculated by Student’s t-test; *, indicates p ≤ 0.05).
miR-125b attenuates MM cell death in response to dexamethasone
We hypothesized that dexamethasone-induced miR-125b expression led to targeting of tumor suppressor p53 and is therefore responsible for the net cell survival of MM.1S cells following dexamethasone treatment. To further address this, MM cells were again transfected with synthetic miR-34a-mimics or anti-miR-125b-transcripts and treated with dexamethasone for 24 h. While neither mimic nor transcript had any significant effect on cell death in untreated cells at this time point, the combination of dexamethasone with either anti-miR-125b-transcripts or miR-34a-mimics stimulated significantly (p ≤ 0.001) higher levels of cell death [approximately 60% compared with scrambled-miR and MM.1R controls (Fig. 5A)]. When the time course was extended to 72 h, the combination of dexamethasone and anti-miR-125b-transcripts enhanced further cell death [approximately 90% compared with scrambled-miR and MM.1R controls (Fig. 5B)]. Importantly, performing this experiment with anti-miR-34a-transcripts or miR-125b-mimics produced opposing results, with a significant (p ≤ 0.05) increase in live cells in response to dexamethasone (Fig. S1A). Further mRNA and protein analysis revealed the mechanism of anti-miR-125b-transcript enhancement of dexamethasone-induced cell death to be the alteration of p53, miR-34a and SIRT1 levels. MM cells transfected with anti-miR-125b-transcripts and then exposed to dexamethasone for 24 h, showed significant upregulation of p53 mRNA (p ≤ 0.05) and miR-34a (p ≤ 0.001), and downregulation of SIRT1 mRNA (p ≤ 0.001; Fig. 5C). This alteration by anti-miR125b-transcripts of the p53/miR-34a/SIRT1 signaling network was also confirmed at the protein expression level (Fig. 5D). Again, contrasting results were observed when mRNA analysis was performed following transfection with miR-125b-mimics and treatment with dexamethasone (Fig. S1B).
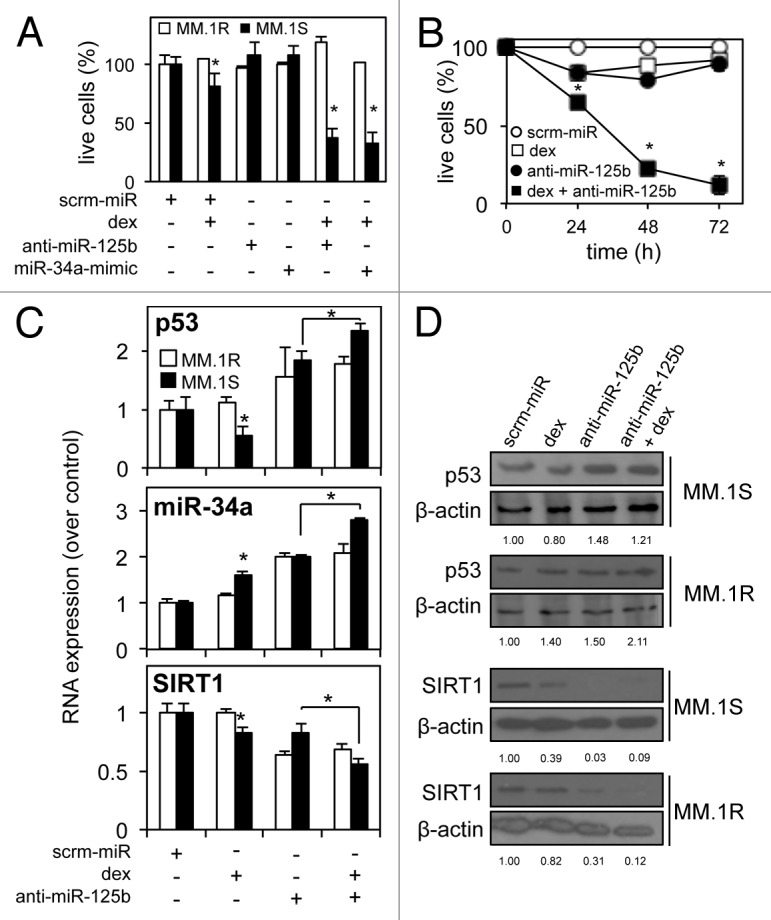
Figure 5. Repression of miR-125b allows dexamethasone-induced cell death by enhancing tumor suppressor p53 expression in multiple myeloma cells. (A) Luminescent cell death analysis shows significantly enhanced levels of dexamethasone-induced cell death in MM.1S cells transfected with anti-miR-125b-transcripts or synthetic miR-34a-mimics after 24 h against a scrambled-miR control and compared with dexamethasone-resistant MM.1R cells. (B) Dexamethasone-induced cell death is further enhanced with anti-miR-125b-transcript up to 72 h after dexamethasone exposure in MM.1S cells. (C) qRT-PCR reveals regulation of the p53/miR-34a/SIRT1 signaling network in MM.1S cells transfected with anti-miR-125b and exposed to dexamethasone: p53 mRNA and miR-34a levels are enhanced, while SIRT1 mRNA is repressed. (D) Western blot analysis of total p53 and SIRT1 protein confirms divergent regulation, in response to anti-miR-125b and dexamethasone. Dexamethasone-resistant MM.1R cells were used as a control. Values indicate the mean ± SEM from 3 independent experiments (statistical significance calculated by Student’s t-test; *, indicates p ≤ 0.05).
Ultimately the importance of our findings could only be confirmed by analysis of miR-125b and the p53/miR-34a/SIRT1 axis in primary human bone marrow derived from MM patients. Markedly enhanced dexamethasone-induced cytotoxicity, increased miR-34a and decreased SIRT1 mRNA expression was observed in primary bone marrow cells from 6 MM patients transfected with anti-miR125b-transcripts (Fig. 6A and B). Interestingly we were unable to detect p53 expression by qRT-PCR in 4 of the 6 samples analyzed (data not shown). We therefore repeated the analysis of anti-miR-125b-transcript enhanced dexamethasone-induced cytotoxicity in the p53WT myeloma-derived B-lymphocyte cell line H929, and the p53MUT myeloma-derived B-lymphocyte cell lines U266 and LP-1, as classified by the IARC TP53 database.31 While H929 cells responded to the combination of dexamethasone and anti-miR-125b-transcripts in the same way as the p53WT MM.1S cells, with enhanced cell death, increased miR-34a expression and repression of SIRT1 mRNA, the p53MUT U266 and LP-1 cells showed a trend toward increased cell proliferation but no significant change in miR-34a or SIRT1 expression (Fig. 6C and D).
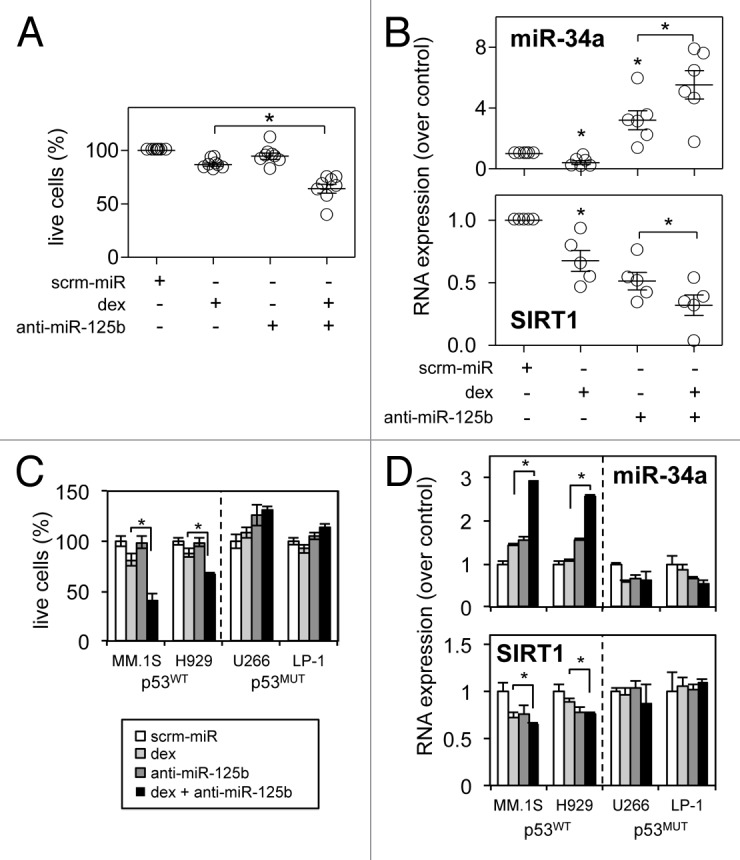
Figure 6. miR-125b modulates expression of the p53/miR-34a/SIRT1 signaling network and dexamethasone-induced cell death in primary MM samples and is dependent on p53WT expression. (A) Luminescent cell death analysis reveals enhanced dexamethasone-induced cell death with anti-miR-125b-transcript in six primary bone marrow MM patient samples. (B) qRT-PCR reveals regulation of the p53/miR-34a/SIRT1 signaling network in the same primary MM patient samples: miR-34a levels are enhanced, while SIRT1 mRNA is repressed. (C) Luminescent cell death analysis reveals enhanced dexamethasone-induced cell death with anti-miR-125b-transcript in p53WT MM cell lines while p53MUT MM cells are unable to respond. (D) A similar lack of response to anti-miR-125b-transcripts and dexamethasone exposure is seen in the regulation of miR-34a expression and SIRT1 mRNA levels in p53MUT compared with p53WT cell lines. Values indicate the mean ± SEM from three independent experiments (statistical significance calculated by Student’s t-test; *, indicates p ≤ 0.05).
Nutlins are pharmacological agents that enhance p53 function by disrupting its interaction with the ubiquitin ligase MDM2. Here we observed that 16 h pre-treatment with 2.5 μM nutlin3 was capable of enhancing p53 mRNA and miR-34a levels and repressing SIRT1 mRNA levels in MM.1S cells (Fig. 7A). Manipulation of the p53/miR-34a/SIRT1 axis alone, however, is not sufficient to enhance dexamethasone-induced MM cell death (Fig. 7B), proving that the key to enable greater dexamethasone sensitivity in MM cells lies with miR-125b, which is not regulated by nutlin3 (Fig. 7A). Indeed, the recent successful MM chemotherapeutic bortezomib (Vecade) also acts to significantly (p ≤ 0.01) inhibit miR-125b expression in MM.1S and MM1.R cells over a range of concentrations (Fig. 7C), an effect that can be significantly enhanced by the addition of dexamethasone in dexamethasone-sensitive MM1.S cells (p ≤ 0.001). Other recent potential MM chemotherapeutics, lenalidomide and ibrutinib,32 showed mixed effects on miR-125b levels (data not shown). This suggests a potential mechanism by which a synergism between bortezomib and dexamethasone is established and supports the notion that pharmacological targeting of miR-125b in vivo is a legitimate strategy in the battle to eradicate MM.
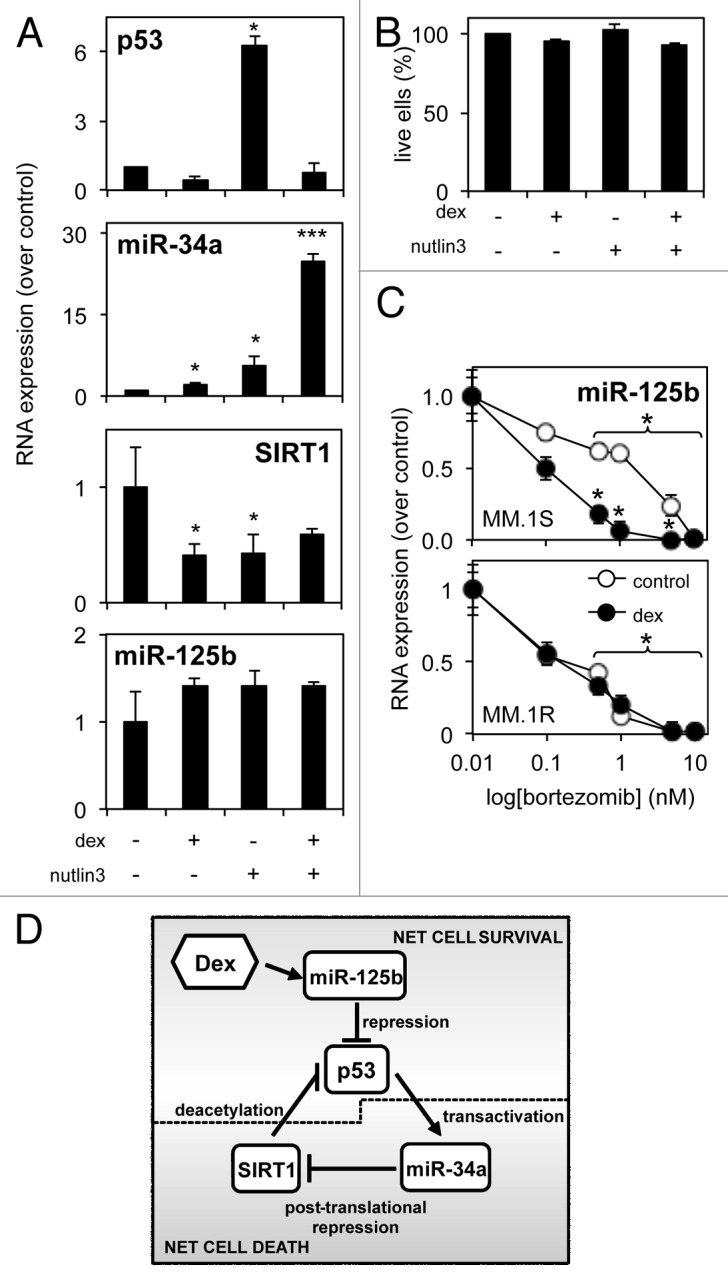
Figure 7. A potential role for miR-125b in the p53/miR-34a/SIRT1 signaling network in MM. (A) qRT-PCR reveals regulation of the p53/miR-34a/SIRT1 signaling network following direct p53 stabilization and activation by 16 h nutlin3 pre-treatment before a further 24 h exposure to dexamethasone. (B) Luminescent cell death analysis reveals nutlin3 pre-treatment to have no effect of on dexamethasone-induced cytotoxic responses. (C) qRT-PCR reveals the effect of bortezomib treatment on miR-125b levels in MM.1S cells is dose-dependent, and can be significantly enhanced by addition of dexamethasone, compared with MM.1R cells. (D) Proposed scheme of how the p53/miR-34a/SIRT1 signaling network is regulated by dexamethasone in MM and intervention of miR-125b. Several solid tumors and hematological malignancies, display a balance between net cell survival and net cell death.13,14 Tumor suppressor p53 transcription factor transactivates pro-apoptotic miR-34a, that in turn targets numerous 3′ UTR sites of pro-survival histone deacetylase SIRT1. Active SIRT1 deacetylates p53’s C terminus to prevent its transcriptional activity. Dexamethasone represses numerous pro-survival proteins; however, MM cells exposed to dexamethasone demonstrate only low levels of cell death. Dexamethasone induced expression of oncogenic miR-125b triggers net cell survival in MM cells by targeting p53. We show here that repression of miR-125b activity with synthetic anti-miRs enhances p53 expression, shifting the balance back toward net cell death.
Discussion
Dexamethasone is routinely used as a front-line adjuvant therapy in combination with classic MM chemotherapies, but shows little cytotoxicity to MM cells on its own. In this study we have uncovered novel dexamethasone-induced p53/miR-34a/SIRT1 activity in the MM/B-lymphoblast cell line MM.1S and established important signaling mechanisms behind the net cell survival of MM cells when exposed to dexamethasone alone. Most importantly we have shown for the first time that the oncomiR miR-125b can feed into this signaling network, targeting p53 tumor suppressor function and effecting MM cell death response through SIRT1 expression. Crucially, the action of miR-125b can be repressed by transient transfection of anti-miR-125b-transcripts, which enhance p53 mRNA and protein levels and increase cell death in response to dexamethasone in both MM cell lines, and in primary MM patient samples. We propose that miR-125b is a key player in the delicate balance between net cell survival and net cell death in MM in response to dexamethasone and should therefore be the focus of future studies (Fig. 7).
Array analysis of miRNA expression in neoplastic tissues, cell lines and in response to various chemotherapies is frequently used to elucidate potential disease biomarkers, and some of the earliest miRNAs to be classified as such were investigated in B-cell malignancies.33 Evidence has since emerged of a deregulation of miR-125b in cancerous B-cells, specifically during differentiation34,35 and pre-leukemic oncogenesis.27,30 Significantly, transgenic mice overexpressing miR-125b develop lethal B-cell malignancies, produce apoptosis-resistant B-cells and display repression of a stress-induced p53 target gene, Trp35inp1.36 In this study, we have shown a novel dexamethasone-induced upregulation of oncogenic miR-125b in a miRNA array of MM.1S cells. This further substantiates a role for miR-125b in B-cell malignancies, suggesting a function in the response to drug therapy, as well as the pathogenesis of MM.
The enhanced expression of dexamethasone-induced oncogenic miR-125b we have observed in MM cells initially appeared inconsistent with the similarly enhanced levels of tumor suppressor miR-34a. Likewise, the net cell survival achieved following exposure to dexamethasone is paradoxical to the downregulation of pro-survival NFκB subunit p65 and the repression of anti-apoptotic Bcl2 and c-FLIP, but reflects levels of apoptotic (sub-G1) MM1.S cells observed in previous dexamethasone studies using highly accurate flow cytometric analysis.37 This also highlights the sensitivity of the luminescent cell death assay utilized, which quantifies metabolic activity of live cells as opposed to transient caspase activity of apoptotic cells, traditionally measured in such studies. When presented together with the miR-125b-driven repression of tumor suppressor p53, however, our results appear to suggest a level of dexamethasone resistance coordinated through changes in miRNA expression.
Both miR-125b and miR-34a have been previously implicated in drug resistance in solid tumors and leukemia.28,38 miR-34a, for example, has been shown to enhance the chemo-sensitivity of paclitaxel-resistant prostate cancer cells through decreased levels of SIRT1 and Bcl2,39 while upregulation of miR-125b confers paclitaxel-resistance to breast cancer cells through targeting of a pro-apoptotic Bcl2-family members.7 This demonstrates a clear potential for activity of miR-125b and the p53/miR-34a/SIRT1 signaling network, and its downstream targets, in drug response and resistance. Similarly, activation of GR-α, the glucocorticoid receptor to which dexamethasone binds, has been found to have protective effects on cancer cells, albeit in solid tumors.40 Interestingly, this may provide some explanation for the enhanced p53 response observed in GR-α-null MM1.R cells (Fig. 4A) in addition to the loss of negative crosstalk between p53 and GR-α in response to stress-induced endogenous glucocorticoid release.41 Despite this, it is well established that the use of dexamethasone as an adjuvant therapy alongside primary chemotherapeutics in vivo is of benefit in multiple myeloma.18 Our work suggests, however, that dexamethasone will be of even greater use in combination with miR-125b inhibition.
The ability to reverse miRNA-driven drug resistance and induce apoptosis with the use of an inhibitor against miR-125b has been demonstrated previously in vitro, in glioblastoma.42 Similarly, we have shown significantly enhanced dexamethasone-induced apoptosis in MM cells through anti-miR-driven repression of miR-125b or enhanced expression of miR-34a through the use of synthetic mimics, an effect that can be countered with the use of anti-miR-34a transcripts or miR-125b-mimics. This shift toward net cell death is time-dependant and correlates with significantly enhanced expression of p53 and downregulation of SIRT1 mRNA and protein. The ability to target disease-associated miRNAs in vivo has been proven, although remains a relatively novel field. For example, studies have shown reduced tumor burden in vivo in chemotherapy-resistant cancers treated with an “antagomiR” against the oncogenic miRNA 17-5p-92 polycistron.43 Similarly, antagomiRs against the pro-metastatic miR-10b have displayed a significant reduction in metastasis and tumor size in vivo and, perhaps most promisingly, showed minimal toxic effects in animals treated.44 Contrary to our findings, recent studies have shown overexpression of miR-125b in vivo to have a protective effect, repressing cell growth and angiogenesis.45 The ability of miR-125b to function as both a tumor suppressor and oncomiR demonstrates an apparent inconsistency in miRNA function; however, this is to be expected due to the low-specificity binding potential of mammalian miRNAs and the numerous different tissues targeted. For example, it is possible that the role of miR-125b varies considerably between solid tumors and hematological malignancies due to the differing challenges of each microenvironment. Similarly, it is likely that cancers lacking p53 expression, or expressing a mutant variant, display differing miR-125b profiles due to the loss of a major miRNA target. As p53 mutations are relatively rare in MM at the point of initial diagnosis,46 and we were consistently able to detect p53 mRNA and protein expression in our p53WT MM cell lines, we are confident that miR-125b provides a viable oncogenic target for potential future in vivo experimentation in MM. Importantly, by investigating dexamethasone-induced cell death and the expression of the p53/miR-34a/SIRT1 signaling network in both p53WT and p53MUT cell lines, we have revealed a potential requirement for p53 expression for optimum response to dexamethasone. It is of note that there was no difference in response to combined dexamethasone and anti-miR-125b treatment in the six primary MM patient samples tested, despite an inability to detect p53 expression via qRT-PCR in 4 out of the 6 samples. This, combined with the relative rarity of p53 mutations in early stage MM, allows us to hypothesize that these samples were expressing p53, but at levels too low detect via mRNA analysis.
Perhaps unsurprisingly, the primary MM chemotherapies used in conjunction with dexamethasone, lenalidomide and bortezomib have also been found to influence the expression of the p53/miR-34a/SIRT1 signaling network. For example, lenalidomide promotes degradation of p53 protein by stabilizing its endogenous inhibitor MDM2,47 while bortezomib stabilizes p53 itself and enhances DNA binding.48 Interestingly, expression profiles of p53 have proven to be predictive of poor response to lenalidomide and dexamethasone treatment in relapsed/refractory MM, further confirming a pivotal role for p53 in response to MM therapies.49 A recent study aiming to stabilize miR-34a/p53 expression in lymphoma B-cells overexpressing oncogenic Myc revealed unexpected repression of steady-state p53 in response to miR-34a-mimics, while the converse inhibition of miR-34a lead to enhanced sensitivity to bortezomib.50 This hints at a level of complexity not yet understood and suggests that miRNA function is highly dependent on cell and tissue type, disease state and previous exposure to chemotherapies.
As our understanding of the complexities of miRNA function and our ability to successfully target miRNAs in vivo is still in its infancy, it is important to investigate novel chemotherapies that may provide the desired effect on miR-125b expression and the p53/miR-34a/SIRT1 signaling network without the need for intensive clinical studies. For example, a recent study has shown repression of SIRT1 and induction of miR-34a resulting in synergistic induction of apoptosis in CLL cells in response to safe and inexpensive nicotinamide therapy alongside front-line chemotherapies.14 Similarly, the classic topoisomerase I inhibitor camptothecin has been shown to downregulate miR-125b expression during apoptosis of CML cells, while ectopic expression of miR-125b was able to reverse the apoptotic response.51 We believe promising results may be found through the screening of novel, but clinically available, MM drugs for their effect on the expression of key miRNAs, in conjunction with dexamethasone and other front-line therapeutics. Likewise, future work should focus on the expression profile of miR-125b and the p53/miR-34a/SIRT1 axis in in vivo models of MM to better understand the discrepancy between oncogenic and tumor suppressor functions of these genes.
In summary, we have highlighted the importance of the p53/miR-34a/SIRT1 signaling network in MM, in response to front-line therapy dexamethasone. We have shown that miR-125b functions as an oncomiR in the MM/B-lymphoblast cell line, MM.1S and in MM patient cells, and revealed enhanced cell death when dexamethasone is used in combination with anti-miR-125b-transcripts. We propose that miR-125b feeds into the p53/miR-34a/SIRT1 axis by targeting the 3′UTR of p53 controlling a delicate balance between net cell survival and net cell death that needs to be exploited to develop better MM chemotherapies in the future.
Materials and Methods
Materials
All antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz). Ambion® Anti-miR-125b inhibitor and mirVana™ miR-34a mimic sequences were purchased from Invitrogen Life Technologies. All other reagents were obtained from Sigma-Aldrich, unless stated otherwise. All MM/B-lymphoblast cell lines were obtained from the European Collection of Cell Cultures, with cell identity authenticated by cytogenetic analysis. Cells were cultured in the laboratory for a maximum of 6 mo post-revival from liquid nitrogen storage to ensure low passage number and retention of their true characteristics, testing regularly for Mycoplasma infection.
Cell culture
Primary MM cells were obtained under local ethical approval (LREC ref. 07/H0310/146) and processed as described.32 MM.1S and MM.1R MM cell lines were cultured in a humidified atmosphere at 37°C with 5% CO2 in RPMI-1640 containing L-Glutamine from Gibco Life Technologies supplemented with 10% fetal bovine serum (FBS).52
Transient transfection of miRNA mimic and anti-miR transcripts
MM.1S and MM.1R cells were transfected with 30 nM Ambion® anti-miR-125b or anti-miR-34a inhibitor, or mirVana™ miR-34a or miR-125b mimic transcripts by nucleofection using the Amaxa™ Nucleofector™ according to manufacturer’s instructions (Lonza Biologics). Cells were maintained for 24 h post-transfection before further treatment and RNA or protein extraction.
RNA extraction and quantitative real time-PCR
Total RNA was extracted from 1 × 106 MM cells using Total RNA Lysis Solution (Applied Biosystems) and the mirVana™ miRNA Isolation Kit (Ambion® Life Technologies), according to the manufacturer’s instructions. Reverse transcription of total RNA (containing miRNA) to cDNA was performed using the miScript II RT Kit (Qiagen). Relative quantitative real-time PCR (qRT-PCR) using LightCycler® 480 SYBR Green I Master (Roche) was performed on cDNA produced via the reverse transcription reaction using real-time PCR primers purchased from Invitrogen. Primer sequences are as follows: GAPDH, forward 5′-ACCAGCCTCAAGATCATCAGC-3′ and reverse 5′-TGCTAAGCAGTTGGTGGTGC-3′; SIRT1, forward 5′-GCGATTGGGTACCGAGATAA-3′ and reverse 5′-TTGCATGTGAGGCTCTATCC-3′; Bcl2, forward 5′-CACCTGTGGTCCACCTGAC-3′ and reverse 5′-AGCTGGCTGGACATCTCG-3′; p53, forward 5′-CCCCAGCCAAAGAAGAAAC-3′ and reverse 5′-GCCCACGGATAAT-3′. qRT-PCR of miRNA was performed as above with miScript Universal Primer (Qiagen) and the OncomiR collection Cancer miRNA qPCR Array (SBI). After pre-amplification (95°C for 2 min) the cDNA was amplified for 45 cycles (95°C for 15 sec, 60°C for 10 sec, 72°C for 10 sec) on the Roche LightCycler® 480 Real-Time PCR system as described.53 Gene expression was analyzed using the comparative cycle threshold algorithm (ΔΔCT); mRNA expression was standardized against GAPDH expression, while miRNA expression was standardized against U6 snRNA expression.
Luminescent cell death analysis
1 × 105 MM.1S and MM.1R cells were treated with dexamethasone as described above. Cell viability was measured using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega) as per the manufacturer’s instructions. Luminescence was measured with the FLUOstar Omega Microplate Reader (BMGLabtech).
Chemiluminescent reporter gene assay
A 750 nucleotide fragment of the SIRT1 3′ UTR with predicted miR-34a interaction regions, or the entire p53 3′ UTR, was amplified from human genomic DNA, using the following PCR primers: SIRT1; forward 5′- AAAGAGCTCTCAGGGAGAATAATTCTGATAGTGC -3′ and reverse 5′- AATAAGCTTTACTCATAATCCTGTCATTTGTCTTCG -3′; p53; forward 5′- AAAGAGCTCCATTCTCCACTTCTTGTTCCC -3′ and reverse 5′- AATAAGCTTCACCCCTCAGACACACAGG -3′. The 3′ UTR fragments were then subcloned into the SacI/HindIII site in the expression vector pMIR-REPORT™-Luciferase (pMIR-REPORT™, 3′UTR-SIRT1/p53; Applied Biosystems). 2 × 106 MM.1S cells were transfected with a total of 500 ng DNA (250 ng pMIR-REPORT™, Luciferase or pMIR-REPORT™-3′UTR-SIRT1/p53 and 250 ng pMIR-REPORT™, β-galactosidase control plasmid) with 30–50 nM miR-34a or miR-125b mimic (Ambion® Life Technologies). Luciferase and β-galactosidase activities were measured 48 h after transfection using the Dual-Light® Chemiluminescent Reporter Gene Assay System Kit (Promega) with the EnVision 2103 Multilabel Plate Reader (Perkin Elmer)
Western immunoblotting
Protein fractions were extracted from MM.1S and MM.1R cells using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific) according to the manufacturer’s instructions. SDS/PAGE and western blot analyses were performed as described previously.54-56 ECL detection using Pierce ECL Western Blotting Substrate (Thermo Scientific) and subsequent quantification were performed with the Chemi-Stage CC-16 mini imaging system (Fujifilm).
Supplementary Material
Acknowledgments
We are grateful to Professor Richard Ball, Norfolk and Norwich University Hospitals Human Tissue Bank for sample storage. This work was supported by the Flexibility and Sustainability Funding from the National Institutes of Health Research (NIHR), Association for International Cancer Research, The Big C and Leukaemia Lymphoma Research.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authorship Contributions
M.Y.M., S.A.R., L.Z. and D.J.M. designed the research and analyzed data. M.Y.M., S.A.R., K.M.B. and D.J.M. wrote the paper. K.M.B. contributed vital new reagents. M.Y.M., S.A.R. and L.Z. performed the research.
Footnotes
This work was funded by the National Institutes of Health Research, Big C Appeal, Leukaemia and Lymphoma Research, and the Association for International Cancer Research. The authors declare no competing financial interests.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25251
References
- 1.Marcucci G, Mrózek K, Radmacher MD, Garzon R, Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011;117:1121–9. doi: 10.1182/blood-2010-09-191312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 3.Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C, et al. Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA. 2008;105:3945–50. doi: 10.1073/pnas.0800135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melino G, Knight RA. MicroRNAs meet cell death. Cell Death Differ. 2010;17:189–90. doi: 10.1038/cdd.2009.122. [DOI] [PubMed] [Google Scholar]
- 5.Shaham L, Binder V, Gefen N, Borkhardt A, Izraeli S. miR-125 in normal and malignant hematopoiesis. Leukemia. 2012;26:2011–8. doi: 10.1038/leu.2012.90. [DOI] [PubMed] [Google Scholar]
- 6.Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–76. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem. 2010;285:21496–507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schotte D, De Menezes RX, Akbari Moqadam F, Khankahdani LM, Lange-Turenhout E, Chen C, et al. MicroRNA characterize genetic diversity and drug resistance in pediatric acute lymphoblastic leukemia. Haematologica. 2011;96:703–11. doi: 10.3324/haematol.2010.026138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan K, Gao J, Yang T, Ma Q, Qiu X, Fan Q, et al. MicroRNA-34a inhibits the proliferation and metastasis of osteosarcoma cells both in vitro and in vivo. PLoS One. 2012;7:e33778. doi: 10.1371/journal.pone.0033778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–22. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 11.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–6. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peurala H, Greco D, Heikkinen T, Kaur S, Bartkova J, Jamshidi M, et al. miR-34a expression has an effect for lower risk of metastasis and associates with expression patterns predicting clinical outcome in breast cancer. PLoS One. 2011;6:e26122. doi: 10.1371/journal.pone.0026122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Audrito V, Vaisitti T, Rossi D, Gottardi D, D’Arena G, Laurenti L, et al. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res. 2011;71:4473–83. doi: 10.1158/0008-5472.CAN-10-4452. [DOI] [PubMed] [Google Scholar]
- 15.Vinall RL, Ripoll AZ, Wang S, Pan CX, deVere White RW. miR-34a chemosensitizes bladder cancer cells to cisplatin treatment regardless of p53-Rb pathway status. Int J Cancer. 2012;130:2526–38. doi: 10.1002/ijc.26256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weeraratne SD, Amani V, Neiss A, Teider N, Scott DK, Pomeroy SL, et al. miR-34a confers chemosensitivity through modulation of MAGE-A and p53 in medulloblastoma. Neuro Oncol. 2011;13:165–75. doi: 10.1093/neuonc/noq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 18.Bird JM, Owen RG, D’Sa S, Snowden JA, Pratt G, Ashcroft J, et al. Haemato-oncology Task Force of British Committee for Standards in Haematology (BCSH) and UK Myeloma Forum Guidelines for the diagnosis and management of multiple myeloma 2011. Br J Haematol. 2011;154:32–75. doi: 10.1111/j.1365-2141.2011.08573.x. [DOI] [PubMed] [Google Scholar]
- 19.Rowland TL, McHugh SM, Deighton J, Ewan PW, Dearman RJ, Kimber I. Differential effect of thalidomide and dexamethasone on the transcription factor NFkappa B. Int Immunopharmacol. 2001;1:49–61. doi: 10.1016/S0162-3109(00)00265-4. [DOI] [PubMed] [Google Scholar]
- 20.Murphy SH, Suzuki K, Downes M, Welch GL, De Jesus P, Miraglia LJ, et al. Tumor suppressor protein (p)53, is a regulator of NFkappaB repression by the glucocorticoid receptor. Proc Natl Acad Sci USA. 2011;108:17117–22. doi: 10.1073/pnas.1114420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langenau DM, Jette C, Berghmans S, Palomero T, Kanki JP, Kutok JL, et al. Suppression of apoptosis by bcl-2 overexpression in lymphoid cells of transgenic zebrafish. Blood. 2005;105:3278–85. doi: 10.1182/blood-2004-08-3073. [DOI] [PubMed] [Google Scholar]
- 22.Norman M, Hearing SD. Glucocorticoid resistance - what is known? Curr Opin Pharmacol. 2002;2:723–9. doi: 10.1016/S1471-4892(02)00232-1. [DOI] [PubMed] [Google Scholar]
- 23.Kotani A, Ha D, Hsieh J, Rao PK, Schotte D, den Boer ML, et al. miR-128b is a potent glucocorticoid sensitizer in MLL-AF4 acute lymphocytic leukemia cells and exerts cooperative effects with miR-221. Blood. 2009;114:4169–78. doi: 10.1182/blood-2008-12-191619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chauhan D, Auclair D, Robinson EK, Hideshima T, Li G, Podar K, et al. Identification of genes regulated by dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene. 2002;21:1346–58. doi: 10.1038/sj.onc.1205205. [DOI] [PubMed] [Google Scholar]
- 25.Rushworth SA, Zaitseva L, Langa S, Bowles KM, MacEwan DJ. FLIP regulation of HO-1 and TNF signalling in human acute myeloid leukemia provides a unique secondary anti-apoptotic mechanism. Oncotarget. 2010;1:359–66. doi: 10.18632/oncotarget.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chim CS, Wong KY, Qi Y, Loong F, Lam WL, Wong LG, et al. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis. 2010;31:745–50. doi: 10.1093/carcin/bgq033. [DOI] [PubMed] [Google Scholar]
- 27.Bousquet M, Harris MH, Zhou B, Lodish HF. MicroRNA miR-125b causes leukemia. Proc Natl Acad Sci USA. 2010;107:21558–63. doi: 10.1073/pnas.1016611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Tan G, Dong L, Cheng L, Li K, Wang Z, et al. Circulating miR-125b as a marker predicting chemoresistance in breast cancer. PLoS One. 2012;7:e34210. doi: 10.1371/journal.pone.0034210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Wang K, Chen X, Meng H, Song M, Wang Y, et al. Transcriptional activation of microRNA-34a by NFkappa B in human esophageal cancer cells. BMC Mol Biol. 2012;13:4. doi: 10.1186/1471-2199-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC. MicroRNAs miR-125a and miR-125b constitutively activate the NFκB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20) Proc Natl Acad Sci USA. 2012;109:7865–70. doi: 10.1073/pnas.1200081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 32.Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NFκB. Cell Signal. 2013;25:106–12. doi: 10.1016/j.cellsig.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gururajan M, Haga CL, Das S, Leu CM, Hodson D, Josson S, et al. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int Immunol. 2010;22:583–92. doi: 10.1093/intimm/dxq042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaudhuri AA, So AY, Mehta A, Minisandram A, Sinha N, Jonsson VD, et al. Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc Natl Acad Sci USA. 2012;109:4233–8. doi: 10.1073/pnas.1200677109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Enomoto Y, Kitaura J, Hatakeyama K, Watanuki J, Akasaka T, Kato N, et al. Eμ/miR-125b transgenic mice develop lethal B-cell malignancies. Leukemia. 2011;25:1849–56. doi: 10.1038/leu.2011.166. [DOI] [PubMed] [Google Scholar]
- 37.Cai B, Wang S, Huang J, Lee CK, Gao C, Liu B. Cladribine and bendamustine exhibit inhibitory activity in dexamethasone-sensitive and -resistant multiple myeloma cells. Am J Transl Res. 2013;5:36–46. [PMC free article] [PubMed] [Google Scholar]
- 38.Kastl L, Brown I, Schofield AC. miRNA-34a is associated with docetaxel resistance in human breast cancer cells. Breast Cancer Res Treat. 2012;131:445–54. doi: 10.1007/s10549-011-1424-3. [DOI] [PubMed] [Google Scholar]
- 39.Kojima K, Fujita Y, Nozawa Y, Deguchi T, Ito M. miR-34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC3 cells through direct and indirect mechanisms. Prostate. 2010;70:1501–12. doi: 10.1002/pros.21185. [DOI] [PubMed] [Google Scholar]
- 40.Moran TJ, Gray S, Mikosz CA, Conzen SD. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 2000;60:867–72. [PubMed] [Google Scholar]
- 41.Sengupta S, Vonesch JL, Waltzinger C, Zheng H, Wasylyk B. Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. EMBO J. 2000;19:6051–64. doi: 10.1093/emboj/19.22.6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi L, Zhang S, Feng K, Wu F, Wan Y, Wang Z, et al. MicroRNA-125b-2 confers human glioblastoma stem cells resistance to temozolomide through the mitochondrial pathway of apoptosis. Int J Oncol. 2012;40:119–29. doi: 10.3892/ijo.2011.1179. [DOI] [PubMed] [Google Scholar]
- 43.Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One. 2008;3:e2236. doi: 10.1371/journal.pone.0002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–7. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muramatsu F, Kidoya H, Naito H, Sakimoto S, Takakura N. microRNA-125b inhibits tube formation of blood vessels through translational suppression of VE-cadherin. Oncogene. 2013;32:414–21. doi: 10.1038/onc.2012.68. [DOI] [PubMed] [Google Scholar]
- 46.Stühmer T, Chatterjee M, Hildebrandt M, Herrmann P, Gollasch H, Gerecke C, et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood. 2005;106:3609–17. doi: 10.1182/blood-2005-04-1489. [DOI] [PubMed] [Google Scholar]
- 47.Wei S, Chen X, McGraw K, Zhang L, Komrokji R, Clark J, et al. Lenalidomide promotes p53 degradation by inhibiting MDM2 auto-ubiquitination in myelodysplastic syndrome with chromosome 5q deletion. Oncogene. 2013;32:1110–20. doi: 10.1038/onc.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cross B, Chen L, Cheng Q, Li B, Yuan Z-M, Chen J. Inhibition of p53 DNA binding function by the MDM2 protein acidic domain. J Biol Chem. 2011;286:16018–29. doi: 10.1074/jbc.M111.228981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen MH, Qi CX, Saha MN, Chang H. p53 nuclear expression correlates with hemizygous TP53 deletion and predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with lenalidomide. Am J Clin Pathol. 2012;137:208–12. doi: 10.1309/AJCPHC85DGAXZDBE. [DOI] [PubMed] [Google Scholar]
- 50.Sotillo E, Laver T, Mellert H, Schelter JM, Cleary MA, McMahon S, et al. Myc overexpression brings out unexpected antiapoptotic effects of miR-34a. Oncogene. 2011;30:2587–94. doi: 10.1038/onc.2010.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng CW, Zhang XJ, Lin KY, Ye H, Feng SY, Zhang H, et al. Camptothecin induces apoptosis in cancer cells via microRNA-125b-mediated mitochondrial pathways. Mol Pharmacol. 2012;81:578–86. doi: 10.1124/mol.111.076794. [DOI] [PubMed] [Google Scholar]
- 52.Rae C, MacEwan DJ. Granulocyte macrophage-colony stimulating factor and interleukin-3 increase expression of type II tumor necrosis factor receptor, increasing susceptibility to tumor necrosis factor-induced apoptosis. Control of leukaemia cell life/death switching. Cell Death Differ. 2004;11(Suppl 2):S162–71. doi: 10.1038/sj.cdd.4401494. [DOI] [PubMed] [Google Scholar]
- 53.Rushworth SA, Bowles KM, Raninga P, MacEwan DJ. NFkappaB-inhibited acute myeloid leukemia cells are rescued from apoptosis by heme oxygenase-1 induction. Cancer Res. 2010;70:2973–83. doi: 10.1158/0008-5472.CAN-09-3407. [DOI] [PubMed] [Google Scholar]
- 54.Rushworth SA, Bowles KM, MacEwan DJ. High basal nuclear levels of Nrf2 in acute myeloid leukemia reduces sensitivity to proteasome inhibitors. Cancer Res. 2011;71:1999–2009. doi: 10.1158/0008-5472.CAN-10-3018. [DOI] [PubMed] [Google Scholar]
- 55.Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NFκB and underlies its chemo-resistance. Blood. 2012;120:5188–98. doi: 10.1182/blood-2012-04-422121. [DOI] [PubMed] [Google Scholar]
- 56.Barrera LN, Rushworth SA, Bowles KM, MacEwan DJ. Bortezomib induces heme oxygenase-1 expression in multiple myeloma. Cell Cycle. 2012;11:2248–52. doi: 10.4161/cc.20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.