

Abstract
The prokaryotic antiviral defense systems CRISPR (clustered regularly interspaced short palindromic repeats)/Cas (CRISPR-associated) employs short crRNAs (CRISPR RNAs) to target invading viral nucleic acids. A short spacer sequence of these crRNAs can be derived from a viral genome and recognizes a reoccurring attack of a virus via base complementarity. We analyzed the effect of spacer sequences on the maturation of crRNAs of the subtype I-B Methanococcus maripaludis C5 CRISPR cluster. The responsible endonuclease, termed Cas6b, bound non-hydrolyzable repeat RNA as a dimer and mature crRNA as a monomer. Comparative analysis of Cas6b processing of individual spacer-repeat-spacer RNA substrates and crRNA stability revealed the potential influence of spacer sequence and length on these parameters. Correlation of these observations with the variable abundance of crRNAs visualized by deep-sequencing analyses is discussed. Finally, insertion of spacer and repeat sequences with archaeal poly-T termination signals is suggested to be prevented in archaeal CRISPR/Cas systems.
Keywords: CRISPR, Cas6, endonuclease, crRNA, in-line probing, RNA binding, transcription termination
Introduction
The adaptive immune system CRISPR/Cas consists of an array of clustered, regularly interspaced, short palindrome repeats (CRISPR) and a set of CRISPR-associated (Cas) proteins. Spacer sequences that are present between the repeats can be derived from the genomes of mobile genetic elements (e.g., viruses) and are utilized to protect host organisms against recurring attacks from these elements. The activity of the CRISPR/Cas system within the cell is divided into three phases: (1) the adaptation of new spacers, (2) the maturation of small CRISPR RNAs (crRNAs) that contain a single spacer sequence and (3) the interference with foreign nucleic acids using crRNAs bound to a Cas protein interference complex.1-10
The diverse CRISPR/Cas systems are divided into three major types and at least 10 subtypes.11 In Clostridia, methanogens and halophiles, the subtype I-B is present, which is defined by the subtype-specific protein Cas8b. Methanococcus maripaludis C5 possesses a single subtype I-B CRISPR/Cas system with a minimal Cas protein set composed of the universal proteins Cas1, Cas2 and Cas4 (proposed to mediate spacer acquisition), as well as Cas3, Cas5, Cas7 and Cas8b, which are presumed to form the interference complex Cascade (Cas complex for antiviral defense). Cas6b completes the set of Cas proteins and was shown to process pre-crRNA in M. maripaludis.12 Cas6b belongs to a diversified family of Cas6 endonucleases that is responsible for crRNA processing. The first characterized Cas6 enzyme belongs to the subtype III-C system present in Pyrococcus furiosus (Pf Cas6). Pf Cas6 was described as a metal-independent endonuclease involved in the processing of pre-crRNAs into mature crRNAs.13-16 Different Cas6 enzymes were also characterized for subtype I-F (Cas6f, also known as Csy4 in Pseudomonas aerouginosa17-19) and subtype I-E (Cas6e or Cse3 in Escherichia coli and Thermus thermophilus20,21). For the subtype I-C that lacks a Cas6 protein, Cas5d appears to compensate for the functions of both Cas6e and Cas5e.22,23 Investigated Cas6 enzymes share a similar structure despite their low sequence similarities, feature a ferredoxin-like fold and operate in an analogous manner to create mature crRNA molecules. Nevertheless, significant differences in their catalytic site composition have been observed. In Pf Cas6, a catalytic triad formed by tyrosine, histidine and lysine residues can be found,13 while Cas6e utilizes a catalytic dyad of tyrosine and histidine residues.18 Two adjacent conserved histidine residues play a major role for crRNA processing by Cas6b.12 Despite these differences in their active sites, all investigated Cas6 proteins generate crRNAs with a 5′-terminal 8 nt repeat tag,12,14,17,20,21 whereas Cas5d processing yields an 11 nt 5′-terminal tag.22 The 3′ end of a crRNA contains the remaining repeat nucleotides, but is often subsequently trimmed by a currently unknown mechanism.12,24,25 It was shown that Cas6 enzymes bind their respective substrates as monomers13,15,17,19-21 and are proposed to deliver the mature crRNA to the Cascade complex.11,13 However, a non-cleaving homolog of Pf Cas6 was observed to form dimers upon binding of unspecific RNA substrates.16 Monomeric Cas6 activity is in agreement with a proposed wrap-around mechanism for Pf Cas6, in which Cas6 was suggested to be bound to the pre-crRNA in a bead chain-like manner using the spacer sequences as guide elements for repeat binding.15
The proposed wrap-around mechanism correlates with the regularly interspaced arrangement of a pre-crRNA as determined by the CRISPR array. The repeats of a CRISPR are nearly uniform for one particular array and the spacer sequences are most often unique.1,5,6,8-10 The length of both spacers (26‒72 nt) and repeats (24‒47 nt) varies between different CRISPR systems8,26 and the arrays appear to follow restrictions regarding the preferred and allowed lengths of repeat and spacer sequences. Spacer acquisition studies in E. coli indicated a favored length of 32‒33 nt for newly integrated spacers, which is consistent with the length of previously existing spacers of the particular array.27 A Gaussian-like distribution of spacer length was found for the three CRISPR loci in Streptococcus thermophilus28 also showing a particular range of spacer lengths that is favored. In M. maripaludis, the spacer length varies between 34‒40 nt (Table S1). Apart from their length, repeat sequences can differ in proposed secondary structures, which have been shown to be important for processing of some repeat sequences.17,20-22 In other cases, the palindromic nature of repeats giving rise to small hairpin structures was shown to not be necessary for crRNA maturation.13-15 The position of the 5′ processing site within the repeat was shown to influence the length of mature crRNA, as a ruler mechanism is applied to ensure a consistent length.29 Based on their length, sequence and potential hairpin formation, 11 different repeat clusters were classified.30 Deep sequencing analyses identified highly variable crRNA abundance patterns for different organisms. A general observed trend, e.g., for both CRISPR loci of N. equitans31 and for the CRISPR locus of M. maripaludis (Table S1),12 is a gradual decline in abundance of crRNAs from the first (closest to the promoter) to the last spacer. Newly acquired spacers are inserted closest to the promoter and this abundance pattern infers that the most recent interactions with viruses cause a Cascade-targeting system that might be most effective against these recently spotted viruses. However, it is puzzling why some crRNAs do not follow this trend and are found to be severely underrepresented in vivo. The analysis of the abundance of crRNAs bound to CMR complexes in Sulfolobus solfataricus identified a variable distribution of crRNAs that are loaded into the interfering complex.32 Similar results were obtained by RNA sequencing of a total RNA isolation of S. solfataricus, where the crRNA abundances were highly variable.33,34
Both repeat and spacer sequences could affect crRNA abundance in the cell. To assess the variable crRNA abundance pattern of the single CRISPR system of M. maripaludis, we generated 26 spacer-repeat-spacer substrates and analyzed the influence of these spacer sequences on Cas6b cleavage activity in vitro. Possible influences of crRNA stability were monitored via in-line probing of crRNAs of M. maripaludis in the presence and absence of Cas6b. In addition, we observed dimerization of Cas6b upon binding of non-cleavable substrates while mature crRNA is bound by Cas6b monomers.
Results
Cas6b forms dimers upon binding to non-cleavable repeat substrates
Cas6b was previously identified to be the endonuclease responsible for the cleavage of repeat sequences of pre-crRNA molecules and for the generation of the crRNAs’ 5′-terminal 8 nt tag.12 In this work, we analyzed the binding of the enzyme to a native repeat substrate. To prevent endonucleolytic cleavage of this repeat sequence during the performed binding assays, a non-cleavable variant of the repeat served as a substrate. This variant harbors a 2'-deoxy modification at the position of cleavage (Fig. 1A), which was shown to abolish nuclease activity.12,17 Electrophoretic mobility shift assays (EMSA) were performed with varying concentrations of purified recombinant Cas6b. The EMSA assays revealed a band shift at low Cas6b concentrations that is fully converted to a distinct super shifted band with increasing Cas6b concentrations (Fig. 1B). This pattern of Cas6b repeat RNA binding is in accordance with the possible dimerization of the enzyme. Fractionation of a Cas6b-repeat complex via size-exclusion chromatography also supports Cas6b dimerization (Fig. S1). To prove that the observed Cas6b binding was specific, competition assays were performed. The addition of increasing concentrations of unlabeled 2'-deoxy-modified repeat RNA appears to preferentially diminish the occurrence of the super shift (Fig. 1C) and the total amount of unbound labeled RNA increased. The addition of up to 10 µg unlabeled yeast RNA did not influence the pattern of RNA shifts (Fig. 1C). Finally, we assayed the binding of a mature crRNA and observed a single band shift (Fig. 1D) that suggests the formation of a Cas6b monomer-crRNA complex after successful cleavage.

Figure 1. Cas6b-binding assays with non-cleavable native repeat RNA and mature crRNAs. (A) The employed RNA sequences are indicated. To prevent processing of the RNA repeat substrate, a deoxy substitution of the first base upstream of the 5′ tag (boxed) was used in the binding assays. (B) Binding assays using decreasing concentrations of Cas6b (50‒10 µM) and equal amounts of deoxy repeat RNA resulted in shift and super shift formation. The separation of samples was performed in native polyacrylamide gels (7%). Bands were visualized by phosphorimaging. (C) Competition assays with unchanged Cas6b concentration (20 µM) and increasing concentrations of unlabeled deoxy repeat RNA (0‒5 µM) and yeast RNA (10‒1 µg) show specificity of Cas6b binding. (D) Only a single shift is observed for Cas6b binding (30‒5 µM) with 5′-labeled crRNA substrate.
Establishment of an experimental approach to assess crRNA processing in M. maripaludis
To assess the previously reported highly variable abundance pattern of crRNAs isolated from M. maripaludis,12 we specifically designed an experimental setup to individually assay the influence of the two neighboring spacer sequences on Cas6b repeat RNA processing and crRNA stability in vitro (Fig. 2). Individual primer pairs were designed to amplify spacer-repeat-spacer sequences from M. maripaludis genomic DNA. One primer contained the T7 RNA polymerase promoter sequence. PCR products were generated for each spacer(n)-repeat-spacer(n+1) combination that were subsequently used as DNA templates for T7 RNA polymerase mediated in vitro run-off transcription. This approach allowed us to produce all individual RNA substrates. The addition of (α-32P)-ATP in the transcription mixture yielded radioactively labeled RNA substrates that were used for Cas6b endonuclease assays. In an alternative approach, we generated unlabeled RNA molecules, processed these with an excess amount of Cas6b to obtain mature crRNA, which then were radioactively labeled at their 5′-termini and used in in-line probing assays35 for structural analyses (Fig. 4). Even though the cleavage of spacer-repeat-spacer substrates yields two fragments, the T4 polynucleotide kinase labeling reaction strongly favored the crRNA product, which demonstrates the differences in the 5′-terminal phosphorylation state (5′-ppp vs. 5′-OH) of the two fragments.
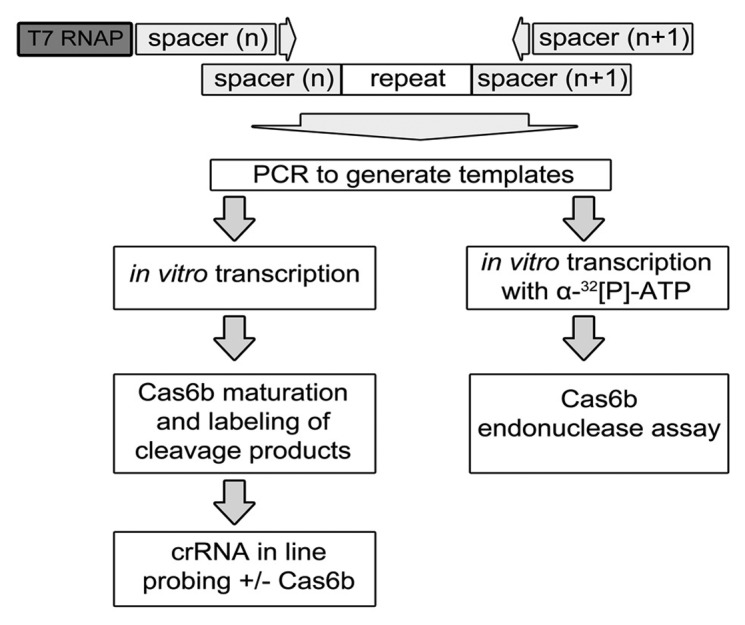
Figure 2. Experimental setup to assess the influence of adjacent spacers on repeat RNA processing by Cas6b. Individual primers for each spacer(n)-repeat-spacer(n+1) combination were designed and each forward primer contained a T7 RNA polymerase promoter sequence. The primers were used in PCR reactions with genomic DNA to generate individual spacer-repeat-spacer templates for in vitro run-off transcription. Adding (α-32P)-ATP to the reaction yields radioactively labeled RNAs that were used for Cas6b endonuclease assays, whereas unlabeled RNAs were treated with Cas6b to generate mature crRNAs, which subsequently were labeled at the 5′ tag and used for in-line probing reactions to evaluate crRNA stability.

Figure 4. In-line crRNA probing assays. (A) Individual spacer(n)-repeat-spacer(n+1) RNAs were generated by in vitro run-off transcription, processed by Cas6b to yield mature crRNAs. The crRNAs were 5′ labeled with (γ-32P)-ATP and incubated for 16h at room temperature using a mild alkaline buffer to aim for partial digestion (lane “I”). Cas6b influence in the probing reaction was determined by addition of 20 µM Cas6b (lane “C”), untreated crRNA served as control (“-“). The probing reactions were separated by denaturing polyacrylamide gel-electrophoresis (20%) and visualized by autoradiography. (B) A time-course experiment employing crRNA26 was performed taking samples at the indicated time spots. (C) Exemplary RNase T1 (Ambion) digests of the indicated crRNAs using 1 U of the enzyme were performed to identify specific cleavage of guanine bases and to determine the sizes of the RNA fragments. Two enzyme concentrations were tested for crRNA11 (1 U, 0.1 U).
Spacer sequences influence crRNA maturation in vitro
Three independent Cas6b endonuclease assays were performed with 26 consecutive spacer(n)-repeat-spacer(n+1) RNA substrates of the M. maripaludis CRISPR locus (spanning spacer 1‒26) to screen for the influences of flanking spacer pairs on the efficiency of repeat RNA processing. Each RNA species was incubated with three different concentrations of Cas6b (10 µM, 1 µM and 0.1 µM) to evaluate and compare the cleavage efficiency for each substrate. All substrates displayed complete cleavage with 10 µM Cas6b. Some differences were observed for the other two concentrations that were quantified in triplicate (Fig. 3B). Cleavage assays with 1 µM Cas6b revealed that the two substrates containing spacer 8 (sp8) (60% and 70% substrate conversion, respectively) and the sp18-repeat-sp19 (79% substrate conversion), as well as the sp25-repeat-sp26 pre-crRNAs (72% substrate conversion), were less efficiently processed (Fig. 3). The addition of 0.1 µM Cas6b showed further differences with reduced crRNA maturation for some of the substrates (e.g., sp3-repeat-sp4, sp18-repeat-sp19, sp25-repeat-sp26) while other substrates displayed a comparatively increased product formation (e.g., sp2-repeat-sp3 or sp9-repeat-sp10).

Figure 3. The influence of spacer sequences on crRNA maturation. (A) In three independent endonucleolytic cleavage assays, each internally labeled spacer(n)-repeat-spacer(n+1) substrate was processed using 10 µM, 1 µM and 0.1 µM Cas6b (“C” indicates controls). The RNAs were separated in denaturing polyacrylamide gels (12%) running at 12 W and visualized by autoradiography. (B) Quantification of product formation. In three independent cleavage assays, the intensities of formed products in relation to the total intensity of observed bands was determined.
Minor differences of crRNA stability were observed in vitro
To analyze other possible factors that might influence the abundance of crRNA in M. maripaludis in vivo, the stability of the individual crRNAs was compared in in-line probing assays (Fig. 4). In these assays, the natural instability of crRNA is observed over time (16 h) to achieve partial digestion of the RNA, which can be slowed down or prevented by the formation of RNA secondary structures.35 In a second assay, Cas6b was added to the reaction in order to investigate potential stabilizing or destabilizing effects of the endonuclease binding to mature crRNAs. The probing assays for most of the crRNAs of the M. maripaludis CRISPR locus showed no obvious differences in crRNA stability since all reactions resulted in uniform laddered degradation that is expected for unstructured RNAs (Fig. 4A, lane “I”). A potential small hairpin formed by inverted 5′-CUUG-3′ sequences in the repeat was not apparent. Interestingly, the addition of Cas6b (Fig. 4A, lane “C”) to the probing assay causes, for most crRNAs, a change of the degradation pattern with the accumulation of smaller RNA fragments (most prominently e.g., for crRNAs containing spacer 10 or 23). Parallel assays with equal amounts of Cas6b storage buffer alone did not cause this destabilizing effect on the crRNAs. The crRNA24 was not degraded and the crRNA26 exhibits degradation that differs from the degradation pattern of the other crRNAs. In the probing reaction of this substrate with Cas6b, the accumulation of smaller RNA degradation products was not observed. However, the formation of a distinct larger RNA fragment can be seen. A time course experiment using this particular crRNA confirmed this observation (Fig. 4B). With longer incubation times, the amount of the large crRNA fragment increased while the substrate amount decreased. The degradation patterns for the probing reaction without Cas6b remained nearly identical during the investigated time period. RNase T1 digests were utilized to identify guanine bases in the RNA in order to pinpoint the sites of increased differential RNA stability found for probing with Cas6b (Fig. 4C). The RNase T1 digestion revealed that most of the accumulated 5′-labeled small RNA fragments have a length of 8‒11 bases indicating the repeat tag of the crRNA. The accumulated crRNA26 fragment is cleaved between bases 22‒23 of the crRNA. This is a very G-rich region of this spacer with the highest GC-content of all M. maripaludis spacers.
Discussion
Different oligomerization states are reported for members of the Cas6 family of crRNA processing endonucleases. While most Cas6 proteins are reported to act as monomeric proteins, a non-catalytic Cas6 homolog from P. horikoshii was shown to feature RNA sequence-dependent dimerization.16 The active site of Cas6 can feature a catalytic triad that is also commonly found in tRNA splicing endonucleases.14 These splicing endonucleases always act as multimers with different families that form either heterotetrameric,36-39 homotetrameric40 or dimeric41,42 assemblies. Our binding assays with M. maripaludis Cas6b and a non-hydrolysable repeat RNA clearly show two distinct shifts and, together with gel-filtration analysis, suggest dimerization of Cas6b upon substrate binding. However, binding assays with mature crRNA revealed only a single shift, which suggests monomeric crRNA binding of Cas6b after processing. Cas6b dimerization is observed in a state of stalled substrate processing, similar to the results obtained for P. horikoshii. Here, the non-catalytic active site of Cas6 hinders processing, while in the case of Cas6b, the 2'-deoxy modification of the repeat RNA prevents endonucleolytic cleavage. Pull-down assays with either Cas7 or Cas5 from S. solfataricus did not lead to co-purification of Cas6 proteins, suggesting that Cas6 does not interact (or interacts only transiently) with these Cascade proteins.43 In conclusion, we support a model that requires Cas6b dimerization during repeat RNA cleavage. A long CRISPR RNA precursor could potentially facilitate the binding of several Cas6b dimers to the various repeat sequences. Finally, Cas6 enzymes might act as a transporter to deliver the crRNA to the Cascade complex. Thus, commonly observed monomeric Cas6-crRNA complexes might act as a scaffold for Cascade formation around the mature crRNA.
The processing of pre-crRNAs by Cas6 enzymes is highly dependent on the repeat sequence, but it is not fully understood which role spacer sequences play in influencing efficient crRNA processing and the total abundance of different crRNAs in the cell. Different factors determine which spacer is integrated into a CRISPR array. A crucial element is the protospacer adjacent motif (PAM), which is essential for recognition of protospacer sequences in the DNA of mobile genetic elements and, therefore, for the adaptation of new spacers into the growing CRISPR array.11,44 PAM sequences were shown to be very small, only 2 nt45 or 3 nt46 in length, and are also needed for the interference step to distinguish between self and non-self nucleic acids.45-47 In addition, it was shown that the length of the spacer is significant. Spacer acquisition assays in E. coli revealed that the majority of newly acquired spacers had a length between 32‒33 nt with only few exceptions of longer or shorter spacers.27 A similar spacer distribution was identified for the three CRISPR loci of S. thermophilus.28 Spacer length in M. maripaludis shows an analogous pattern with most of the spacers being 36‒38 nt in length (Table S1). The longest spacer is spacer 8, which is the only 40 nt spacer in the investigated CRISPR. Interestingly, both spacer-repeat-spacer RNA substrates that contained spacer 8 showed a reduced Cas6b processing efficiency, which suggests that spacer sequences or spacer length play a role in Cas6b activity. Our comparative analysis of all potential spacer-repeat-spacer combinations revealed additional reproducible differences in the efficiency of Cas6b processing of the enclosed repeat. However, these differences are often minor and, most importantly, do not clearly match with the highly variable crRNA abundance pattern observed in vivo (Table S1).12 The crRNAs that are underrepresented in deep-sequencing data showed no reduced 5′-terminal maturation by Cas6b. However, potential sequencing biases in RNA-Seq studies should always be considered. Variable stability of crRNAs might also influence the crRNA abundance in vivo. However, our in-line probing experiments indicated a rather uniform pattern of crRNA degradation without observing stable secondary structure elements within the crRNAs. It was surprising to see that Cas6b destabilizes crRNAs, which could e.g., be explained by bending of the phosphate backbone upon Cas6b binding. Most retaining RNA degradation products show that the 5′-terminal repeat tag is protected from further degradation. In our experiment, the influence of spacer sequences on the stability of the RNA is obvious as the single spacer 26 with an unusually high GC-content forms a distinct RNA fragment that is protected from further degradation. However, crRNA26 appears not to be overrepresented in the cell (Table S1). In conclusion, these results show that while spacer sequences do have an effect on Cas6b processing and Cas6b-crRNA stability, these effects do not fully explain the in vivo crRNA abundance patterns. These patterns are suggested to be determined by several factors that include the ones we measured with the presented methodology. Additional parameters that could not be addressed with the current experimental setup are (1) the establishment of the interaction of crRNAs with Cas protein interference complexes in the cell, (2) differential turnover of crRNAs depending on their utilization in the cell and (3) different intrinsic stabilities of crRNAs after Cas protein interference complex binding. It could be shown for S. solfataricus that the abundance of crRNAs bound to the interference Cas protein complex CMR was also very diverse.32 Therefore, the distance of the crRNA’s spacer to the promoter and the success rate of the loading of crRNAs into a CMR or Cascade complex might have the strongest influences on crRNA abundance in the cell.
Finally, the sequences of potential spacers have a huge impact on the global CRISPR transcription if they contain elements that might either serve as promoters or terminators. Recent studies revealed internal promotion of CRISPR array transcription12,33 suggesting that spacer sequences with promoter elements could be selected for during evolution if the increased transcription of crRNAs with older spacers was beneficial. A problematic scenario is the possible integration of spacer sequences with transcription termination sequences into a CRISPR. Many questions remain regarding the termination signals in archaeal genomes. However, one commonly observed feature are poly-T stretches at termination sites.48,49 We observed that M. maripaludis and many archaea avoid the adaptation of spacer sequences with long poly-T stretches. In addition, several long poly-A stretches are found in the newly acquired spacers of the M. maripaludis CRISPR. These poly-A stretches specify poly-T stretches in the reverse direction and might provide beneficial termination signals for the prevention of anti-crRNA production. In addition, repeat sequences appear to have evolved to lack poly-T (more than 3 T bases in a row) motifs in Archaea while these are commonly found in bacterial repeats.30 Careful manual inspection of archaeal repeats identified only two sequences with four consecutive T residues while the majority of them (over 300) contained poly-A sequences. Future research on the loading efficiency of Cascade complexes, internal promotion and termination signal will be required to fully assess the diverse influences that spacer sequences can have on crRNA abundance and their effectiveness against viral attacks.
Material and Methods
Production of Cas6b
Production and purification of recombinant Cas6b via Ni-NTA chromatography was performed as described earlier.12 Oligomeric states of Cas6b were determined by size-exclusion chromatography. Ni-NTA purified Cas6b in lysis buffer [10 mM TRIS-HCl (pH 8.0); 300 mM NaCl; 10% glycerol; 0.5 mM DTT] or Cas6b incubated with repeat RNA harboring a 2'-deoxy modification (see below) was applied to an analytical Superdex column (200 10/300 GL, GE Healthcare) and fractionated using a FPLC Äkta-Purification system (GE Healthcare). Proteins of collected fractions were precipitated by addition of one quarter volume 100% TCA (trichloracetic acid) and 15 min incubation on ice. The precipitated proteins were washed twice with 200 µl acetone (100%, ice-cold) and dried at 95°C for 5 min before resuspension in SDS loading buffer. Molecular weight of the fractions was determined by comparison to a calibration of the column with molecular weight markers, as described in the manufacturer manual (Kit for Molecular Weights, Sigma-Aldrich).
Generation of RNA substrates
PCR reactions with genomic DNA, isolated from Methanococcus maripaludis C5 and containing forward primers encoding the T7 RNA polymerase (RNAP) promoter, yielded spacer(n)-repeat-spacer(n+1) templates for in vitro run-off transcription (Fig. 2). Transcriptions for unlabeled RNAs were performed for 1 h at 37°C in a final volume of 20 µl [40 mM HEPES-KOH (pH 8.0); 22 mM MgCl2; 5 mM DTT; 1 mM spermidine; 4 mM UTP, CTP, GTP, ATP; 20 U RNase Inhibitor, 1 µg T7 RNAP, 1 µg PCR product]. Radiolabeled spacer-repeat-spacer substrates were generated by in vitro run-off transcription reducing the amount of ATP to 2 mM and adding 25 µCi (α-32P) adenosine triphosphate (ATP) (5,000 Ci/mmol, Hartman Analytic). For the production of crRNA substrates, unlabeled spacer-repeat-spacer RNAs were processed with 15 µM Cas6b and purified by phenol-chloroform extraction. The obtained crRNAs were 5′-end labeled using (γ-32P) ATP (5,000 Ci/mmol) and T4 polynucleotide kinase (PNK) in a volume of 20 µl [15 µl of purified RNA; 2 µl PNK buffer (New England Biolabs) and 25 U T4 PNK (Ambion)] for 1 h at 37°C. Synthesis of repeat RNA with a deoxy substitution of the first unprocessed nucleotide was done by Eurofins MWG Operon. 5′-end labeling of the deoxy-modified repeat RNA was achieved as mentioned above. Labeled RNAs were separated by denaturing PAGE (8 M urea; 1 × TBE; 10% polyacrylamide) and visualized by autoradiography. Identified bands were excised from the gels and RNA species were eluted by overnight incubation on ice using 500 µl RNA elution buffer [250 mM NaOAc; 20 mM TRIS-HCl (pH 7.5); 1.5 mM ehtylenediaminetetraacetic acid (EDTA) (pH 8.0); 0.25% SDS]. After addition of two volumes, EtOH (100%, ice cold) and 1/100 glycogen (Roche) the RNAs were precipitated for 1 h at -20°C. Subsequent washing with 70% EtOH was followed by pelleting of the RNA.
Endonuclease assay
In endonucleolytic cleavage assays the indicated Cas6b concentrations were incubated for 10 min at 37°C together with the RNA substrates in cleavage buffer [250 mM KCl; 1.875 mM MgCl2; 1 mM DTT; 20 mM HEPES-KOH (pH 8.0)]. The reaction was quenched by adding 2 × formamide buffer [95% formamide; 5 mM EDTA (pH 8.0); 2.5 mg bromophenol blue; 2.5 mg xylene cyanol] and incubation at 95°C for 5 min. Separation of RNA species was achieved by a denaturing 12‒20% polyacrylamide gel running at 12 W. Visualization of RNA was done by autoradiography. In order to obtain full processing of precursor RNA to generate mature crRNAs suitable for 5′ labeling with (γ-32P) ATP, the reaction time was increased to 1 h with 15 µM Cas6b.
Electrophoretic mobility shift assay (EMSA)
In EMSA reactions, indicated Cas6b concentrations were mixed with radiolabeled RNA substrates in binding buffer [10 mM TRIS-HCl (pH 8.0); 200 mM KCl; 5% Glycerol; 0.5 mM DTT; 0.5 mM EDTA (pH 8.0); 1 µg BSA]. The reaction mix was incubated for 1 h at 37°C and immediately mixed with 6 × DNA loading dye (4 g sucrose; 25 mg bromophenol blue; 25 mg xylene cyanol in 10 ml H2O). Separation of the reaction was done with a 7% native polyacrylamide gel running in 1 × TBE at 8 W for 2.5 h. Visualization was achieved by autoradiography.
In-line probing
Structural analysis of crRNAs was analyzed via in-line probing35 incubating crRNA in probing buffer [5 mM TRIS-HCl (pH 8.5), 0.5 µg yeast RNA (Ambion), 100 mM KCl, 20 mM MgCl2] for 16 h at room temperature. To test Cas6b influence on crRNA stability, 20 µM Cas6b was added to the probing reaction. The reaction was stopped by addition of 2 × formamide buffer and incubation at 95°C for 5 min. Visualization was achieved by phosphorimaging after separation of RNA using 20% denaturing polyacrylamide gels.
Supplementary Material
Acknowledgments
We thank André Plagens for advice and discussions. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG grants FOR1680 RA 2169/1-1 to L.R. and BA 2168/5-1 to R.B.) and the Max-Planck Society.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/23715
References
- 1.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Barrangou R, Horvath P. CRISPR: new horizons in phage resistance and strain identification. Annu Rev Food Sci Technol. 2012;3:143–62. doi: 10.1146/annurev-food-022811-101134. [DOI] [PubMed] [Google Scholar]
- 3.Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–61. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 4.Cui Y, Li Y, Gorgé O, Platonov ME, Yan Y, Guo Z, et al. Insight into microevolution of Yersinia pestis by clustered regularly interspaced short palindromic repeats. PLoS One. 2008;3:e2652. doi: 10.1371/journal.pone.0002652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–70. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 6.Koonin EV, Makarova KS. CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol Rep. 2009;1:95. doi: 10.3410/B1-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plagens A, Tjaden B, Hagemann A, Randau L, Hensel R. Characterization of the CRISPR/Cas subtype I-A system of the hyperthermophilic crenarchaeon Thermoproteus tenax. J Bacteriol. 2012;194:2491–500. doi: 10.1128/JB.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorek R, Kunin V, Hugenholtz P. CRISPR--a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6:181–6. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 9.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321–7. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–7. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–77. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res. 2012;40:9887–96. doi: 10.1093/nar/gks737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carte J, Pfister NT, Compton MM, Terns RM, Terns MP. Binding and cleavage of CRISPR RNA by Cas6. RNA. 2010;16:2181–8. doi: 10.1261/rna.2230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–96. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R, Preamplume G, Terns MP, Terns RM, Li H. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011;19:257–64. doi: 10.1016/j.str.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R, Zheng H, Preamplume G, Shao Y, Li H. The impact of CRISPR repeat sequence on structures of a Cas6 protein-RNA complex. Protein Sci. 2012;21:405–17. doi: 10.1002/pro.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–8. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haurwitz RE, Sternberg SH, Doudna JA. Csy4 relies on an unusual catalytic dyad to position and cleave CRISPR RNA. EMBO J. 2012;31:2824–32. doi: 10.1038/emboj.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sternberg SH, Haurwitz RE, Doudna JA. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA. 2012;18:661–72. doi: 10.1261/rna.030882.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gesner EM, Schellenberg MJ, Garside EL, George MM, Macmillan AM. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat Struct Mol Biol. 2011;18:688–92. doi: 10.1038/nsmb.2042. [DOI] [PubMed] [Google Scholar]
- 21.Sashital DG, Jinek M, Doudna JA. An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol. 2011;18:680–7. doi: 10.1038/nsmb.2043. [DOI] [PubMed] [Google Scholar]
- 22.Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, et al. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure. 2012;20:1574–84. doi: 10.1016/j.str.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garside EL, Schellenberg MJ, Gesner EM, Bonanno JB, Sauder JM, Burley SK, et al. Cas5d processes pre-crRNA and is a member of a larger family of CRISPR RNA endonucleases. RNA. 2012;18:2020–8. doi: 10.1261/rna.033100.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–56. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hale C, Kleppe K, Terns RM, Terns MP. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA. 2008;14:2572–9. doi: 10.1261/rna.1246808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–76. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horvath P, Romero DA, Coûté-Monvoisin AC, Richards M, Deveau H, Moineau S, et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190:1401–12. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatoum-Aslan A, Maniv I, Marraffini LA. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci USA. 2011;108:21218–22. doi: 10.1073/pnas.1112832108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunin V, Sorek R, Hugenholtz P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007;8:R61. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Randau L. RNA processing in the minimal organism Nanoarchaeum equitans. Genome Biol. 2012;13:R63. doi: 10.1186/gb-2012-13-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Rouillon C, Kerou M, Reeks J, Brugger K, Graham S, et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol Cell. 2012;45:303–13. doi: 10.1016/j.molcel.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng L, Kenchappa CS, Peng X, She Q, Garrett RA. Modulation of CRISPR locus transcription by the repeat-binding protein Cbp1 in Sulfolobus. Nucleic Acids Res. 2012;40:2470–80. doi: 10.1093/nar/gkr1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons BA, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–41. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soukup GA, Breaker RR. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999;5:1308–25. doi: 10.1017/S1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshinari S, Fujita S, Masui R, Kuramitsu S, Yokobori S, Kita K, et al. Functional reconstitution of a crenarchaeal splicing endonuclease in vitro. Biochem Biophys Res Commun. 2005;334:1254–9. doi: 10.1016/j.bbrc.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 37.Calvin K, Hall MD, Xu F, Xue S, Li H. Structural characterization of the catalytic subunit of a novel RNA splicing endonuclease. J Mol Biol. 2005;353:952–60. doi: 10.1016/j.jmb.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 38.Tocchini-Valentini GD, Fruscoloni P, Tocchini-Valentini GP. Structure, function, and evolution of the tRNA endonucleases of Archaea: an example of subfunctionalization. Proc Natl Acad Sci USA. 2005;102:8933–8. doi: 10.1073/pnas.0502350102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Randau L, Calvin K, Hall M, Yuan J, Podar M, Li H, et al. The heteromeric Nanoarchaeum equitans splicing endonuclease cleaves noncanonical bulge-helix-bulge motifs of joined tRNA halves. Proc Natl Acad Sci USA. 2005;102:17934–9. doi: 10.1073/pnas.0509197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Trotta CR, Abelson J. Crystal structure and evolution of a transfer RNA splicing enzyme. Science. 1998;280:279–84. doi: 10.1126/science.280.5361.279. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Abelson J. Crystal structure of a dimeric archaeal splicing endonuclease. J Mol Biol. 2000;302:639–48. doi: 10.1006/jmbi.2000.3941. [DOI] [PubMed] [Google Scholar]
- 42.Fujishima K, Sugahara J, Miller CS, Baker BJ, Di Giulio M, Takesue K, et al. A novel three-unit tRNA splicing endonuclease found in ultrasmall Archaea possesses broad substrate specificity. Nucleic Acids Res. 2011;39:9695–704. doi: 10.1093/nar/gkr692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lintner NG, Kerou M, Brumfield SK, Graham S, Liu H, Naismith JH, et al. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE) J Biol Chem. 2011;286:21643–56. doi: 10.1074/jbc.M111.238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–7. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gudbergsdottir S, Deng L, Chen Z, Jensen JV, Jensen LR, She Q, et al. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol Microbiol. 2011;79:35–49. doi: 10.1111/j.1365-2958.2010.07452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischer S, Maier LK, Stoll B, Brendel J, Fischer E, Pfeiffer F, et al. An archaeal immune system can detect multiple protospacer adjacent motifs (PAMs) to target invader DNA. J Biol Chem. 2012;287:33351–63. doi: 10.1074/jbc.M112.377002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci USA. 2011;108:10098–103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santangelo TJ, Reeve JN. Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J Mol Biol. 2006;355:196–210. doi: 10.1016/j.jmb.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 49.Thomm M, Hauser W, Hethke C. Transcription Factors and Termination of Transcription in Methanococcus. Syst Appl Microbiol. 1993;16:648–55. doi: 10.1016/S0723-2020(11)80336-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.