

1. Introduction
There is an unmet clinical need for a non-invasive central biomarker for the metabolic degradation of endogenous cannabinoids (endocannabinoids). These retrograde lipid messengers of the cannabinoid system, with N-arachidonyl ethanolamide (anandamide; AEA) and 2-arachidonyl glycerol (2-AG) being the most abundant, regulate a variety of brain functions (e.g. cognition, emotions, motivations, motor control and pain) through the stimulation of cannabinoid receptors (CB1 and CB2) [1]. Both AEA and 2-AG are synthesized on demand and terminated by enzymatic hydrolysis via the serine hydrolases, fatty acid amide hydrolase (FAAH, EC3.5.1.99) and monoacyl glycerol lipase (MAGL, EC3.1.1.23), respectively [2]. Blockade of FAAH-mediated AEA degradation in animal models by genetic or pharmacological methods raised AEA levels up to fourteen-fold in the central nervous system (CNS) thereby demonstrating anti-inflammatory, analgesic, and anxiolytic results [3–5]. Importantly, these phenotypes were absent of the adverse side effects on motor control, appetite, memory and body temperature associated with direct CB1 agonists [6]. Dysregulation of FAAH has been associated with depression, neuropathic pain, addictions, and obesity in both animal models and humans [7–11]. As such, FAAH inhibitors are being actively pursued, not only as pharmacological tools, but also as potential therapeutics for CNS disorders [12].
Development of FAAH inhibitors has been extensively reviewed [13, 14] and the majority of those reported contain either a carbamate or a urea moiety. As suicide enzyme inhibitors, these compounds form an irreversible covalent bond with Ser241 of the FAAH catalytic triad (Ser241, Ser217, Lys142). The prototypical FAAH inhibitor, URB597 (3′-carbamoyl-[1,1′-biphenyl]-3-yl-cyclohexylcarbamate) is the most researched inhibitor to study FAAH in animal models [15]. Moreover, a small number of FAAH inhibitors have entered clinical trials with the most reported data on a urea-based inhibitor, Pfizer’s investigational drug PF-04457845 (N-(pyridazin-3-yl)-4-(3-((5-trifluoromethyl)pyridine-2-yl)oxy)benzylidene)piperidine-1-carboxamide) [16], which interacts with FAAH in an analogous method to carbamate-based inhibitors towards this enzyme [17]. From a Phase II crossover study as a treatment for pain associated with osteoarthritis, this compound was shown to modulate endocannabinoid levels in blood but did not induce an analgesic effect [18]. Two more Phase II trials investigating PF-04457845 are assessing the effects of FAAH inhibition on marijuana withdrawal and the role of endocannabinoids in extinction learning. Assessment of peripheral FAAH inhibition during such clinical trials can be quantitatively achieved by measuring enzyme activity in leukocytes via blood sampling, but quantifying local FAAH inhibition within the living brain requires a central biomarker. A non-invasive method to image and quantify FAAH expression in the CNS would improve the evaluation of potential treatments by directly observing changes in enzyme activity upon administration of FAAH inhibitors.
There are a limited number of reports outlining the preparation of positron emission tomography (PET) radiotracers targeting FAAH activity. [11C]1,1′-biphenyl-3-yl-(4-methoxyphenyl)carbamate, was prepared and evaluated in rodents; however it exhibited low brain uptake and no detectable specific binding, eliminating it as a potential PET radiotracer [19]. We have developed [11C]CURB ([11C-carbonyl]-6-hydroxy-[1,1′-biphenyl]-3-yl-cyclohexylcarbamate) [20], an analogue of URB597 possessing similar affinity and selectivity for FAAH to URB597 but exhibits greater brain penetration [21]. Ex vivo rodent studies of [11C]CURB demonstrated high brain uptake which was irreversible and highly selective for FAAH as shown by pharmacological blockade with a saturating intraperitoneal (ip) pre-treatment with FAAH inhibitors [20]. This radiotracer has recently been validated for PET imaging of FAAH in healthy human volunteers [22]. Recently we described the radiosynthesis and ex vivo properties (in rats) of a series of [11C-carbonyl]carbamates as potential FAAH radiotracers [23]. Most of these radiotracers had high brain uptake and specificity for FAAH but demonstrated variable binding kinetics, a property which is of important significance for irreversible ligands [24–26]. Skaddan et al. have recently reported a fluorine-18 labeled urea-based inhibitor [18F]PF-9811 (4-(3-((5-(2-[18F]fluoroethoxy)pyridine-2-yl)oxy)benzylidene)-N-(pyridazin-3-yl)piperidine-1-carboxamide) [27] which is an analogue of PF-04457845. [18F]PF-9811 demonstrated modest brain uptake (0.8 SUV in the cortex at 90 min) and specific to non-specific binding ratios (2.3 – 2.6) in rodents. A reversible radiotracer for FAAH, [11C]MK-3168 ((1S,2S)-2-(4-(5-((5-chloropyridin-2-yl)thio)-1-[11C]methyl-1H-imidazol-4-yl)phenyl)-N,N-dimethylcyclopropanecarboxamide), was recently reported in abstract form [28, 29].
Pursuant to our efforts to develop FAAH radiotracers for PET in vivo imaging studies, we identified PF-04457845 as a potential candidate due to its favorable pharmacokinetic properties (high bioavailability and brain penetration), high selectivity, and known safety in humans [30, 31]. To circumvent modifications to the structure of PF-04457845, we elected to prepare the carbon-11 isotopologue by radiolabeling at the urea carbonyl via [11C]CO2 fixation. Herein we report the radiosynthesis of [11C-carbonyl]PF-04457845 ([11C]PF-04457845, Scheme 1) along with the ex vivo brain biodistribution and blocking studies in conscious rodents.
Scheme 1.
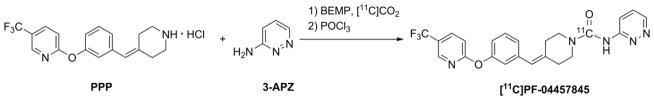
One pot radiosynthesis of [11C]PF-04457845. With 2.63 ± 0.58 GBq in the final bottle, the isolated radiochemical yield was 4.5 ± 1.3% based upon starting [11C]CO2 (uncorrected for decay) with a radiochemical purity of 98.4 ± 1.3%, and specific activity of 73.5 ± 8.2 GBq/μmol at the end of synthesis (25 ± 2 min; n = 4).
2. Methods
2.1 General materials and methods
All reagents were used as purchased from Sigma Aldrich. Solvents used were either reagent or HPLC grade and purchased from Caledon Laboratories or Sigma Aldrich. A Scanditronix MC 17 cyclotron was used for radionuclide production. [11C]CO2, produced by the 14N(p,α)11C nuclear reaction, was concentrated from the gas target in a stainless steel coil cooled to −178 °C. Upon warming, the [11C]CO2 in a stream of N2 gas was passed through a NOx trapping column and a drying column of P2O5 prior to use [32]. Purifications and analyses of radioactive mixtures were performed by high performance liquid chromatography (HPLC) with an in-line UV detector (254 nm) in series with a NaI crystal radioactivity detector. Isolated radiochemical yields were determined with a dose calibrator (Capintec CRC-712M). Automated radiosyntheses were controlled by Labview™ software. Unless otherwise stated, all radioactivity measurements were corrected for radioactive decay. POCl3 was distilled under reduced pressure prior to use. All animal experiments were carried out under humane conditions, with approval from the Animal Care Committee at the Centre for Addiction and Mental Health and in accordance with the guidelines set forth by the Canadian Council on Animal Care. Rats (male, Sprague Dawley) were kept on a reversed 12 h light/12 h dark cycle and allowed food and water ad libitum. The radiolabeling precursor, 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride (PPP) and the authenticated standard (PF-04457845) were prepared via a literature procedure [16]. [11C]CURB was prepared via the literature procedure [20].
2.2 Radiosynthesis of [11C]PF-04457845
Carbon-11 labeled CO2 was dispensed in a stream of nitrogen (7 mL/min) into a 1 mL conical vial charged with phosphazene base BEMP (2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine; 6.92 μmol) and PPP (2.70 μmol) in anhydrous CH3CN (100 μL) as well as 3–aminopyridazine (3-APZ, 52.6 μmol) in anhydrous 1:1 CH3CN:DMSO (100 μL). One min after radioactivity peaked, a 0.2% (v/v) solution of POCl3 (2.15 μmol) in CH3CN (100 μL) was added followed 1 min later by a 1:1 mixture of mobile phase in water (800 μL). The quenched reaction mixture was puri ed by reverse-phase HPLC with a Phenomenx Luna C18 10 μm, 10 x 250 mm column and a mobile phase of 55/45 v/v acetonitrile/water containing 1% formic acid (pH = 3) at 7 mL min−1. The desired product was collected, evaporated, and formulated in 5% v/v Tween80® in saline (10 mL) and passed through a Millipore 0.22 μm filter into a sterile vial containing 1N sodium bicarbonate solution (0.5 mL). The identity and radiochemical purity of the radiotracer were verified by co-injection with PF-04457845 using several reverse-phase HPLC conditions (solvents, pH, wavelength). Lipophilicity was determine as log P7.4 by a literature shake flask procedure (n = 16) [33].
2.3 Ex vivo biodistribution studies
Concious male Sprague-Dawley rats (330 – 370 g), held in a restraining box, received 33 – 44 MBq of high specific activity [11C]PF-04457845 (0.88 – 1.1 μg/kg; 1.9 – 2.4 nmol/kg) in 0.3 mL of 5% v/v Tween80® buffered saline via the tail-vein which had been vasodilated in a warm water bath. Rats were sacrificed by decapitation at various time points after radiotracer administration and blood was collected from the trunk. The brain was quickly removed, stored on ice and dissected. Brain regions (whole cortex, cerebellum, hypothalamus and remainder of brain) were excised, blotted, and weighed. Radioactivity in the tissues was assayed in an automated gamma counter, back corrected to the time of injection using diluted aliquots of the initial injected dose as standards. Tails were counted in a dose calibrator and the injected dose was corrected for residual activity. Experiments were conducted in groups of four rats per time point with results expressed as standard uptake values (SUV; mean ± standard deviation) defined as % injected dose/g of tissue per 100 g of rat body weight.
2.4 Pharmacological blocking of [11C]PF-04457845 or [11C]CURB uptake in rat brains
Groups of male Sprague-Dawley rats (300 – 350 g, n = 4) received an ip injection of vehicle (5% v/v Tween80® buffered saline), PF-04457845 (0.1 or 1.0 mg/kg) or URB597 (2 mg/kg) 1 h before radiotracer administration. Rats received either [11C]PF-04457845 or [11C]CURB in 0.3 mL of 5% v/v Tween80® buffered saline via the tail-vein. Temporal and regional distribution of the radiotracers were determined in a similar manner as described above and reported as SUV (mean ± standard deviation). Student’s t-tests were performed to compare regional uptake of [11C]PF-04457845 or [11C]CURB in groups receiving ip pre-treatments to the group receiving vehicle and p-values less than 0.05 were considered significant.
2.5 HPLC analysis of plasma metabolites
The blood from the trunk of decapitated male Sprague-Dawley rats was collected into heparinized tubes and centrifuged to separate the plasma. Aliquots of plasma were treated with 20% (v/v) of 50% aqueous acetic acid to disrupt plasma protein binding and injected directly onto an HPLC loop (Valco, Texas, USA). Analyses of plasma were performed with minor modifications of the method described by Hilton et al. [34]. The HPLC loop was flushed onto a small capture column (4.6 x 20 mm) packed in house with OASIS HLB 30 μm (Waters, New Jersey, USA). The capture column was eluted with 1% acetonitrile (2 mL/min) for 4 min then backflushed (60% acetonitrile/40% water + 0.1 N ammonium formate, pH = 4, 2 mL/min) onto a Phenomenex Luna C18 10μm, 250 x 4.6 mm column. Both column effluents were monitored through a flow detector (Bioscan Flow-Count) operated in coincidence mode. To monitor for highly lipophilic metabolites, the HPLC eluent was switched to 100% ethanol after the parent radiotracer eluted. All radioactivity data were corrected for physical decay and integrated.
2.6 Irreversible binding of [11C]PF-04457845 to FAAH in the rat brain
Following tail-vein injection of [11C]PF-04457845 groups of 3 conscious male Sprague-Dawley rats were sacrificed and the whole brain was surgically removed from the skull, washed in saline, and kept on ice. To measure specific binding, rats in one group were pretreated with URB597 (2 mg/kg in saline with 5% Tween80®, ip) 1 h prior to radiotracer injection. Brains were then homogenized (Polytron, setting 7) in 5 mL of cold 80% acetonitrile/20% aqueous hydrochloric acid (0.01%) and centrifuged (17000 rpm, 10 min). Following careful decantation of the supernatants, the pellets were resuspended in extraction solvent (5 mL) and centrifuged again. After repeating the extraction procedure once more, an aliquot from the combined supernatants from each rat was removed, weighed and counted for radioactivity. Pellets were also counted for radioactivity.
3. Results
3.1 Blocking [11C]CURB with PF-04457845
We synthesized the known FAAH inhibitor PF-04457845 as previously reported by Johnson et al [16]. To verify its ability to cross the blood-brain barrier and block FAAH, conscious male Sprague-Dawley rats were pretreated with PF-04457845 (ip) at two different doses (0.1 or 1.0 mg/kg) then injected with [11C]CURB via the tail-vein and sacrificed 40 min post injection. Depending upon the region, uptake of radioactivity in rat brain regions decreased 53 – 83% for both ip doses of PF-04457845 (Fig. 1, p < 0.05).
Figure 1.

Effect of 1 h pre-dosing with PF-04457845 (ip) on brain biodistribution of [11C]CURB (SUV with standard deviation bars) in rats, 40 min post radiotracer injection (n = 4/group). *p < 0.05 compared to the vehicle group.
3.2 Radiochemistry
To radiolabel PF-04457845, we employed a [11C]CO2 fixation method used previously to prepare [11C]carbamates [35–37], [11C]ureas [37, 38] and [11C]oxazolidinones [39]. All experiments were carried out by bubbling [11C]CO2 into a conical vial containing a fixating base (BEMP) and 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride (PPP) in acetonitrile. Following HPLC purification and formulation, [11C]PF-04457845 was prepared in 4.5 ± 1.3% radiochemical yield, based on starting [11C]CO2 (uncorrected for decay) and a radiochemical purity of 98.4 ± 1.3 % with a total synthesis time of 25 ± 2 min (n = 4, Scheme 1). The reaction was carried out using an automated synthesis module which required no heating/cooling or manual manipulations, as previously described [20, 37–39]. Clinically useful amounts (2.63 ± 0.58 GBq) of [11C]PF-04457845, with a specific activity of 73.5 ± 8.2 GBq/μmol at end of synthesis, were obtained as a final formulated solution, suitable for animal studies.
3.3 Lipophilicity as measured by Log P7.4
The partition coefficient, between 1-octanol and 0.02 M phosphate buffer at pH 7.4, of [11C]PF-04457845 was measured via a shake-flask method [33] to be 3.48 ± 0.08 (n = 16).
3.4 Regional and temporal distribution of [11C]PF-04457845 in rat brain
Following tail-vein injections of [11C]PF-04457845 into conscious rats, brain uptake was high with SUV ranging from 1.2 to 4.4, reaching a plateau 40 min post injection (Table 1). Radioactivity was significantly lower in the plasma than the brain with cortex-to-plasma ratios increasing from 2:1 to 34:1 between 2 and 40 min post injection. A heterogeneous uptake of radioactivity was observed with highest levels in the cortex, intermediate amounts in the cerebellum and lowest uptake in the hypothalamus. This distribution of radioactivity in various brain regions is similar to [11C]CURB and in accordance with the known expression of FAAH in the rat brain (Fig. 2) [40–42].
Table 1.
Ex vivo biodistribution (SUV ± standard deviation) of [11C]PF-04457845 in conscious rat brain at 2, 15, 40, and 60 min post iv injection (n = 4 per time-point).
| Tissue | 2 min | 15 min | 40 min | 60 min |
|---|---|---|---|---|
| Hypothalamus | 1.29 ± 0.05 | 1.96 ± 0.10 | 2.18 ± 0.20 | 2.10 ±0.31 |
| Cortex | 2.22 ± 0.06 | 3.64 ± 0.24 | 4.11 ± 0.30 | 3.92 ± 0.37 |
| Cerebellum | 1.87 ± 0.05 | 3.17 ± 0.10 | 3.56 ± 0.10 | 3.42 ± 0.34 |
| Rest of Brain | 1.51 ± 0.08 | 2.44 ± 0.07 | 2.77 ± 0.24 | 2.59 ± 0.19 |
| Plasma | 1.03 ± 0.19 | 0.33 ± 0.04 | 0.12 ± 0.01 | 0.09 ± 0.03 |
Figure 2.

Correlation between regional brain uptake of FAAH radiotracers and regional FAAH enzyme activity in rat brain. Data taken from this work ([11C]PF-004457845 uptake; --●-- ; y = 1.85x + 0.59, R2 = 0.91), Wilson et al. [20] ([11C]CURB uptake; –■–; y = 1.22x + 0.23, R2 = 0.93), and Thomas et al. [40] (FAAH enzyme activity). Error bars represent standard deviation from the mean.
3.5 Specificity of binding of [11C]PF-04457845
To demonstrate that binding of [11C]PF-04457845 is saturable, rats were pretreated (ip) with two doses of PF-04457845 (0.05 or 0.5 mg/kg; 0.11 or 1.1 μmol/kg) 1h prior to injection with the radiotracer (Fig. 3). At both of the doses used, uptake of radioactivity was reduced by 67 – 85%, depending on the region. Binding specificity of [11C]PF-04457845 was further accessed by pretreating rats (ip; 1h prior) with the selective FAAH inhibitor URB597 at a dose (2 mg/kg; 5.9 μmol/kg) known to inactivate > 90% of the enzyme in rodent brains [21]. Brain uptake was lowered by 71 – 81%, depending upon the region. Similar low and homogenous regional distribution was observed after treatment with either URB597 or PF-04457845. Comparing the uptake of the control group to that of the group pretreated with URB597, the specific to non-specific binding ratio in the cortex, cerebellum, and hypothalamus were 4.2, 3.4 and 2.5, respectively. In the plasma, levels of radioactivity increased with all pre-treatment protocols compared to controls (Fig. 3, p < 0.05). Control and blocking groups both were sacrificed 40 min after iv injection of [11C]PF-04457845.
Figure 3.
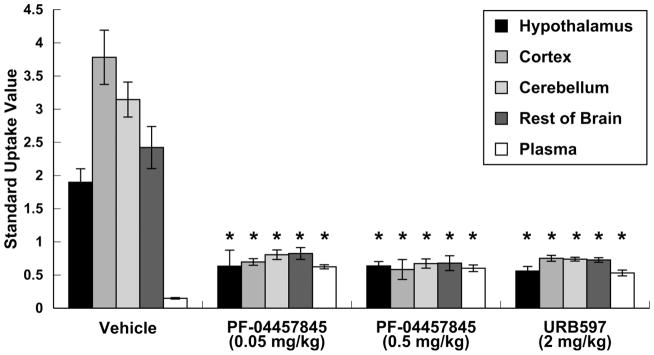
Pharmacological blockade of radiotracer uptake (SUV with standard deviation bars) of radioactivity following iv injection of [11C]PF-04457845. Intraperitoneal pre-treatment with vehicle, PF-04457845 or URB597 occurred 1 h pre-radiotracer injection and rats were sacrificed 40 min post-radiotracer injection (n = 4/group). *p < 0.05 compared to vehicle group.
3.6 Metabolite analysis
Following tail-vein injection of [11C]PF-04457845 and decapitation at various time points, trunk blood was collected and total radioactivity in the plasma was analyzed by radioHPLC [34]. At 2 min post injection, 82% of the parent radiotracer remained which slowly decreased to 82%, 73% and 66% at 15, 40 and 60 min post injection, respectively. A small amount of a lipophilic metabolite representing 3 – 3.5% of the total radioactivity present in plasma was detected at later time-points.
3.7 Determination of irreversible binding
Excised rat brains were homogenized and exhaustively extracted with 0.01% aqueous HCl in acetonitrile (20/80 v/v) following tail-vein injection with [11C]PF-04457845 [20, 24, 25]. Measuring the amount of radioactivity in the extract and fixed to the residual pellet provided a ratio of radiotracer irreversibly bound to brain parenchyma at the various time points. After 2 min, 84% of the radioactivity was irreversibly bound to brain tissue and this value increased to 98% after 40 min (Fig. 4a). The specificity of this binding for FAAH was determined by pretreating one group of rats with URB597 (ip), resulting in a decrease in radiotracer binding to brain tissue from 2.5 ± 0.4 SUV 40 min post injection for the control group to 0.028 ± 0.009 (Fig. 4b).
Figure 4.
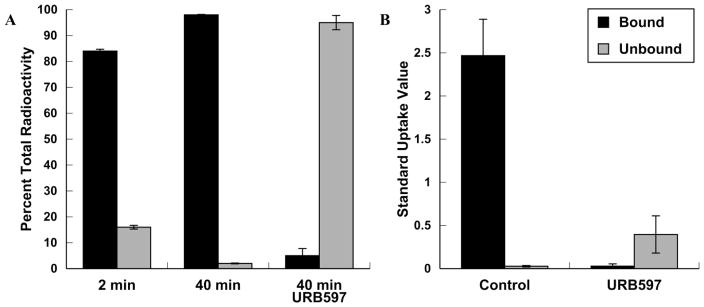
Amount of radioactivity bound to rat brain tissue at 2 and 40 min post iv injection. A) Percentage with standard deviation bars of radioactivity bound to rat brain tissue at 2 and 40 min post iv injection or after pre-treatment with URB597 (ip 2 mg/kg; 5.9 μmol/kg; n = 3/group). B) Absolute amounts of radioactivity bound to rat brain tissue 40 min post injection (SUV with standard deviation bars) in control and after pre-treatment with URB597 (ip 2 mg/kg; 5.9 μmol/kg; n = 3/group).
4. Discussion
Recent work in our laboratory led to the discovery of a radiolabeled irreversible FAAH inhibitor, [11C]CURB [20], which has been validated in healthy human volunteers [22]. Our continuing efforts towards the development of a PET radiotracer targeting FAAH includes seven other [11C]carbamates (described elsewhere [23]) and a [11C]urea, [11C]PF-04457845, described herein. As PF-04457845 has undergone clinical evaluation in human subjects for safety and efficacy, a positron emitting isotopologue has a high probability of rapid translation to clinical use at multiple PET centers for non-invasive visualization of FAAH in humans. To prepare [11C]PF-04457845, we adapted the [11C]CO2 fixation method used to radiolabel other [11C-carbonyl]ureas [37, 38]. The mechanism of inhibition of FAAH by ureas such as PF-04457845 involves covalent attachment of Ser241 to the carbamoyl carbon with expulsion of the N-aryl residue [17]. Thus the enzymes can be covalently labeled with carbon-11 if the radiotracer is radiolabeled at the carbonyl position. Non-nucleophilic aromatic amines such as 3-APZ are problematic in the radiosynthesis of [11C-carbonyl]ureas by [11C]CO2 fixation [37], but their inherent lack of reactivity can be overcome by using them in large excess (compared with aliphatic amine PPP) [38].
A plausible mechanism for the radiosynthesis of [11C]PF-04457845 is depicted in Scheme 2. Cyclotron produced [11C]CO2 is captured in solution by BEMP, forming a BEMP-[11C]CO2 adduct which rapidly exchanges with PPP forming a [11C]carbamic salt that is then dehydrated to a mixed [11C]anhydride (Scheme 2) [43]. The aromatic amine 3-APZ, present in 20-fold excess compared to PPP, then reacts with the anhydride to form the [11C]urea bond. In the current work, both PPP and 3-APZ were present in the conical vial receiving [11C]CO2 (Scheme 1), prior to the formation of the mixed [11C]anhydride without detriment to radiochemical yield, purity or specific activity. This method allowed for a simple, one-pot automated reaction requiring only one reagent addition and no heating or cooling, yielding sufficient quantities of [11C]PF-04457845 to complete animal or human studies.
Scheme 2.
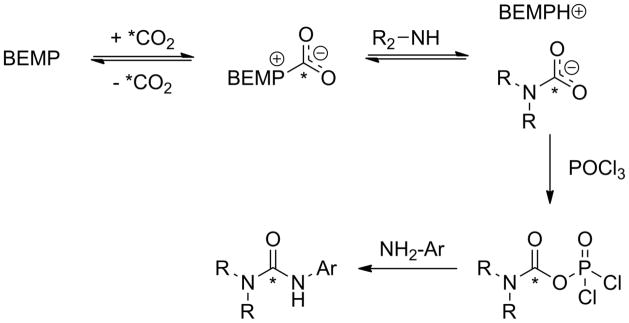
Representation of the proposed mechanism of [11C]CO2 fixation to form asymmetric arylalkyl [11C]ureas. The * indicates the position of the radiolabel.
The high uptake of [11C]PF-04457845 and heterogeneous distribution reflective of known FAAH expression in the rat brain (Fig. 2) [40–42] suggests it has good potential as a FAAH targeted PET radiotracer (Table 1). High blood-brain barrier penetration was anticipated based upon the potency of PF-04457845 to block FAAH binding of [11C]CURB (Fig. 1). The selectivity of [11C]PF-04457845 binding to FAAH was established as uptake of the radiotracer within the rat CNS was effectively blocked and the distribution of radioactivity became homogeneous following ip pre-treatment with a low and high dose of PF-04457845 or a dose of URB597 known to inhibit 90% of FAAH activity (Fig. 3) [21]. This provided strong support that the uptake of [11C]PF-04457845 into the rodent brain is mediated by FAAH. The irreversibility of binding was demonstrated by comparing the level of bound and unbound radiotracer within the rat brain following an exhaustive extraction process. (Fig. 4a). In the same study, it was shown that an ip pre-treatment with URB597 reduced the amount of [11C]PF-04457845 bound to brain parenchyma from 98% to 5% (Fig. 4a). This decrease in irreversible binding is even more drastic when comparing the absolute amount of bound radioactivity between the 40 min control group and the group receiving ip pre-treatment of URB597 (2.5 to 0.028 SUV, respectively; Fig. 4b). As URB597 is highly selective for FAAH in the brain [15, 17, 44, 45], this provides further evidence that [11C]PF-04457845 brain uptake is mediated by FAAH.
No true reference region can be applied to calculate the specific to non-specific binding ratio (SBR) of [11C]PF-04457845 as there is no brain tissue devoid of FAAH. A commonly used method under such circumstances is to designate the regional brain uptake in the control animals to represent the specific plus the non-specific binding and the regional brain uptake in animals receiving a blocking dose of drug to represent the non-specific binding. Thus, we can estimate that at 40 min post injection of [11C]PF-04457845, the SBR in the cortex, cerebellum and hypothalamus were 4.2, 3.4 and 2.5, respectively. However, these values are likely a gross underestimation of the SBR as the amount of [11C]PF-04457845 in the plasma compartment increased substantially during the challenge studies (Fig. 3), which would imply a dramatic increase in brain input. Our extraction studies are more revealing and showed that (i) 98% of radioactivity was irreversibly bound to brain tissue at 40 min post injection of [11C]PF-04457845 (Fig. 4a) and (ii) essentially all (98.9%) of the irreversible binding could be eliminated by pre-treatment of animals with URB597 (Fig. 4b). Clearly >95% of the radioactivity in whole brain is FAAH-bound and thus the SBR of [11C]PF-04457845 to FAAH is >18 ([95/5]-1). This level of specific binding is higher than that previously reported for [11C]CURB (80%) [20].
Chromatographic analysis of rat plasma revealed the presence of modest levels of metabolites within the plasma with 73% of the parent radiotracer remaining at 40 min post injection. All of the metabolites were more polar than [11C]PF-04457845 and demonstrated a lack of brain penetration when the unbound fraction from the brain extraction studies was analyzed. With extractable brain radioactivity of only 2% (Fig. 4a), the transfer of polar metabolites across the blood-brain barrier and the potential involvement of [11C]PF-04457845 in other metabolic pathways apart from FAAH in the CNS are both nominal. A similar metabolic analysis was observed for [11C]CURB [20]. When the results from metabolic analysis are combined with the observed high specificity and selectivity, the likelihood is very high that the observed radioactivity uptake into the rat brain is principally attributable to binding of [11C]PF-04457845 to FAAH.
5. Conclusion
The breadth of clinical data for PF-04457845 made it an attractive candidate as a PET radiotracer and led us to pursue the radiosynthesis of [11C]PF-04457845. Results from ex vivo rat studies demonstrated high brain uptake of this radiotracer which binds selectively, specifically, and irreversibly to FAAH without troublesome brain-penetrant metabolites. The differentiation between regions of high and low FAAH expression and the subsequent reduction in uptake across all regions upon pharmacological blockade both suggest the kinetics are favorable for in vivo imaging. The facile radiosynthesis of [11C]PF-04457845, encouraging preclinical results and known safety information of PF-04457845 warrant further evaluation of this radiotracer in higher species.
Acknowledgments
This work was supported by NIH Grant # 1R21MH094424-01 to AAW, an Ontario Ministry of Research and Development Early Researcher award to NV, and a University of Toronto Institute of Medical Science Open Fellowship award to JWH. We would like to thank Armando Garcia, Winston Stableford, Min Wong, Virginia S. Wilson, Patrick McCormick, and Alvina Ng for their assistance with the radiochemistry and animal dissection experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–58. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luchicchi A, Pistis M. Anandamide and 2-arachidonoylglycerol: pharmacological properties, functional features, and emerging specificities of the two major endocannabinoids. Mol Neurobiol. 2012;46:374–92. doi: 10.1007/s12035-012-8299-0. [DOI] [PubMed] [Google Scholar]
- 3.Murphy N, Cowley TR, Blau CW, Dempsey CN, Noonan J, Gowran A, et al. The fatty acid amide hydrolase inhibitor URB597 exerts anti-inflammatory effects in hippocampus of aged rats and restores an age-related deficit in long-term potentiation. J Neuroinflamm. 2012;9:79–90. doi: 10.1186/1742-2094-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–13. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 6.Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonist as medicines. Br J Pharmacol. 2009;156:397–411. doi: 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, et al. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007;62:1103–10. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Polklis JL, Boger DL, et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–10. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sipe JC, Chiang K, Gerber AL, Beutler E, Cravatt BF. A missense mutation in human fatty acid amide hydrolase associated with problem drug use. PNAS. 2002;99:8394–9. doi: 10.1073/pnas.082235799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwasaki S, Ishiguro H, Higuchi S, Onaivi ES, Arinami T. Association between alcoholism and endocannabinoid metabolic enzyme genes encoding fatty acid amide hydrolase and monoglyceride lipase in Japanese population. Psychiatr Genet. 2007;17:215–20. doi: 10.1097/YPG.0b013e32809913d8. [DOI] [PubMed] [Google Scholar]
- 11.Sipe JC, Waalen J, Gerber A, Beutler E. Overweight and obesity associated with a missense polymorphism in fatty acid amide hydrolase (FAAH) Int J Obes. 2005;29:755–9. doi: 10.1038/sj.ijo.0802954. [DOI] [PubMed] [Google Scholar]
- 12.Ramos JA, González S, Sagredo O, Gómez-Ruiz M, Fernández-Ruiz J. Therapeutic potential of the endocannabinoid system in the brain. Mini-Rev Med Chem. 2005;5:609– 17. doi: 10.2174/1389557054368817. [DOI] [PubMed] [Google Scholar]
- 13.Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51:7327–43. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 14.Khanna IK, Alexander CW. Fatty acid amide hydrolase inhibitors - progress and potential. CNS Neurol Disord Drug Targets. 2011;10:545–58. doi: 10.2174/187152711796234989. [DOI] [PubMed] [Google Scholar]
- 15.Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, et al. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597) CNS Drug Reviews. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, et al. Discovery of PF-04457845: A highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med Chem Lett. 2011;2:91–6. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, et al. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–30. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 18.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-044577845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–46. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 19.wyffels L, Muccioli GG, Kapanda CN, Labar G, De Bruyne S, De Vos F, et al. PET imaging of fatty acid amide hydrolase in the brain: synthesis and biological evaluation of an 11C-labelled URB597 analogue. Nucl Med Biol. 2010;37:665–75. doi: 10.1016/j.nucmedbio.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Wilson AA, Garcia A, Parkes J, Houle S, Tong J, Vasdev N. [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl Med Biol. 2011;38:247–53. doi: 10.1016/j.nucmedbio.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, et al. A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem. 2009;4:1505–13. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rusjan P, Wilson AA, Mizrahi R, Boileau I, Chavez S, Lobaugh N, et al. Initial evaluation of [11C]CURB in human brain using positron emission tomography. J Cereb Blood Flow Metab. 2012 doi: 10.1038/jcbfm.2012.180. [DOI] [Google Scholar]
- 23.Wilson AA, Hicks JW, Sadovski O, Parkes J, Tong J, Houle S, et al. Radiosynthesis and evaluation of [11C-carbonyl]-labeled carbamates as fatty acid amide hydrolase radiotracers for positron emission tomography. J Med Chem. 2013;56:201–9. doi: 10.1021/jm301492y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacGregor RR, Halldin C, Fowler JS, Wolf AP, Arnett CD, Langstrom B, et al. Selective, irreversible in vivo binding of [11C]L-deprenyl in mice: potential for measurement of functional monoamine oxidase activity in brain using positron emission tomography. Biochem Pharmacol. 1985;34:3207–10. doi: 10.1016/0006-2952(85)90173-x. [DOI] [PubMed] [Google Scholar]
- 25.Fowler JS, Wolf AP, MacGregor RR, Dewey SL, Logan J, Schlyer DJ, et al. Mechanistic positron emission tomography studies: demonstration of a deuterium isotope effect in the monoamine oxidase-catalyzed binding of [11C]L-deprenyl in living baboon brain. J Neurochem. 1988;51:1524–34. doi: 10.1111/j.1471-4159.1988.tb01121.x. [DOI] [PubMed] [Google Scholar]
- 26.Koeppe RA, Frey KA, Synder SE, Meyer P, Kilbourn MR, Kuhl DE. Kinetic modeling of N-[11C]methylpiperdin-4-yl propionate: alternatives for analysis of an irreversible positron emission tomography trace for measurement of acetylcholinesterase activity in human brain. J Cereb Blood Flow Metab. 1999;19:1150–63. doi: 10.1097/00004647-199910000-00012. [DOI] [PubMed] [Google Scholar]
- 27.Skaddan MB, Zhang L, Johnson DS, Zhu A, Zasadny KR, Coelho RV, et al. The synthesis and in vivo evaluation of [18F]PF-9811: a novel PET ligand for imaging brain fatty acid amide hydrolase (FAAH) Nucl Med Biol. 2012;39:1058–67. doi: 10.1016/j.nucmedbio.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Sanabria-Bohórquez S, Joshi A, Cook J, Holahan M, Posavec D, et al. The discovery and characterization of [11C]MK-3168, a novel PET tracer for imaging fatty acid amide hydrolase (FAAH) J Labelled Compd Radiopharm. 2011;53:S38. [Google Scholar]
- 29.Joshi A, Li W, Sanabria S, Holahan M, Purcell M, Declercq R, et al. Translational studies with [11C]MK-3168, a PET tracer for fatty acid amide hydrolase (FAAH) J Nucl Med. 2012;53 (Supplemental 1):397. [Google Scholar]
- 30.Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338:114–24. doi: 10.1124/jpet.111.180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li GL, Winter H, Arends R, Jay GW, Le V, Young T, et al. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharmacol. 2012;73:706–16. doi: 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tewson TJ, Banks W, Franceschini M, Hoffpauir J. A trap for the removal of nitrogen oxides from carbon-11 carbon dioxide. Appl Radiat Isot Int J Radiat Appl Instrum Part A. 1989;40:765–8. [Google Scholar]
- 33.Wilson AA, Jin L, Garcia A, DaSilva JN, Houle S. An admonition when measuring the lipophilicity of radiotracers using counting techniques. Appl Radiat Isot. 2001;54:203–8. doi: 10.1016/s0969-8043(00)00269-4. [DOI] [PubMed] [Google Scholar]
- 34.Hilton J, Yokoi F, Dannals RF, Ravert HT, Szabo Z, Wong DF. Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl Med Biol. 2000;27:627–30. doi: 10.1016/s0969-8051(00)00125-6. [DOI] [PubMed] [Google Scholar]
- 35.Hooker JM, Reibel AT, Hill SM, Schueller MJ, Fowler JS. One-pot direct incorporation of [11C]CO2 into carbamates. Angew Chem Int Ed. 2009;48:3482–5. doi: 10.1002/anie.200900112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson AA, Garcia A, Houle S, Vasdev N. Direct fixation of [11C]-CO2 by amines: formation of [11C-carbonyl]-methylcarbamates. Org Biomol Chem. 2010;8:428–32. doi: 10.1039/b916419g. [DOI] [PubMed] [Google Scholar]
- 37.Wilson AA, Garcia A, Houle S, Sadovski O, Vasdev N. Synthesis and application of isocyanates radiolabeled with carbon-11. Chem-Eur J. 2011;17:259–64. doi: 10.1002/chem.201002345. [DOI] [PubMed] [Google Scholar]
- 38.Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N. Towards the preparation of radiolabeled 1-aryl-3-benzyl ureas: Radiosynthesis of [11C-carbonyl] AR-A014418 by [11C]CO2 fixation. Bioorg Med Chem Lett. 2012;22:2099–101. doi: 10.1016/j.bmcl.2011.12.139. [DOI] [PubMed] [Google Scholar]
- 39.Vasdev N, Sadovski O, Garcia A, Dolle F, Meyer JH, Houle S, et al. Radiosynthesis of [11C]SL25. 1188 via [11C]CO2 fixation for imaging monoamine oxidase B. J Labelled Compd Radiopharm. 2011;54:678–80. [Google Scholar]
- 40.Thomas EA, Cravatt BF, Danielson PE, Gilula NB, Sutcliffe JG. Fatty acid amide hydrolase, the degradative enzyme for anandamide and oleamide, has selective distribution with the rat central nervous system. J Neurosci Res. 1997;50:1047–52. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1047::AID-JNR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 41.Egertová M, Giang DK, Cravatt BF, Elphick MR. A new prespective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci. 1998;265:2081–5. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueda N, Puffenbarger RA, Yamamoto S, Deutsch DG. The fatty acid amide hydrolase (FAAH) Chem Phys Lipids. 2000;108:107–21. doi: 10.1016/s0009-3084(00)00190-0. [DOI] [PubMed] [Google Scholar]
- 43.Paz J, Pérez-Balado C, Iglesias B, Muñoz L. Carbon dioxide as a carbonylating agent in the synthesis of 2-oxazolidinones, 2-oxazinones, and cyclic ureas: scope and limitation. J Org Chem. 2010;75:3037–46. doi: 10.1021/jo100268n. [DOI] [PubMed] [Google Scholar]
- 44.Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, et al. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52:1095–105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 45.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, et al. Discovery and characterization of highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–20. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]