

Abstract
Background
Profound edema or formation of a pseudocyst containing plasma proteins is a prominent characteristic of Nasal polyps (NP). However, the mechanisms underlying NP retention of plasma proteins in the submucosa remain unclear. Recently, we reported that impairment of fibrinolysis causes excessive fibrin deposition in NP and this might be involved in retention of plasma proteins. Although the coagulation cascade plays a critical role in fibrin clot formation at extravascular sites, the expression and role of coagulation factors in NP remain unclear.
Objective
The objective of this study was to investigate the expression of coagulation factors in patients with chronic rhinosinusitis (CRS).
Methods
Sinonasal tissues were collected from patients with CRS and control subjects. We assayed mRNA for factor XIII-A (FXIII-A) by using real-time PCR and measured FXIII-A protein by means of ELISA, immunohistochemistry and immunofluorescence.
Results
FXIII-A mRNA levels were significantly increased in NP from patients with CRS with nasal polyps (CRSwNP; P < .001) compared with uncinate tissue from patients with CRS or control subjects. Similarly, FXIII-A protein levels were increased in NP. Immunofluorescence analysis revealed FXIII-A expression in inflammatory cells, and FXIII-A+ cell numbers were significantly increased in NP. Most FXIII-A staining was observed within CD68+/CD163+ M2 macrophages in NP. Levels of FXIII-A correlated with markers of M2 macrophages, suggesting that M2 macrophages are major FXIIIA producing cells in NP.
Conclusion
Overproduction of FXIII-A by M2 macrophages might contribute to the excessive fibrin deposition in the submucosa of NP, which might contribute to the tissue remodeling and pathogenesis of CRSwNP.
Keywords: Chronic rhinosinusitis, nasal polyps, factor XIII-A (FXIII-A), M2 macrophages, fibrin, coagulation cascade
INTRODUCTION
Chronic rhinosinusitis (CRS) is a heterogeneous disease characterized by local inflammation of the upper airways and sinuses with symptoms lasting longer than 12 weeks despite medical management. CRS is one of the most common chronic diseases in adults in United States and affects up to 15% of the population.1–4 Primarily on the basis of physical examination, histology, and clinical course, CRS is typically classified into 2 types: CRS with nasal polyps (CRSwNP) and CRS without nasal polyps (CRSsNP). The etiology and pathogenesis of CRS remain controversial, however, allergy, bacterial and fungal infections, and structural abnormalities have all been theorized to play a role.5 In general, CRSwNP is associated more closely with clinical complaints of nasal obstruction and olfactory loss, and more frequently linked to comorbidities such as asthma and aspirin hypersensitivity. Sinonasal tissue from patients with CRSsNP displays a predominant infiltration of neutrophils and presence of Th1 cytokines, whereas CRSwNP tissue is characterized by more intense eosinophilic infiltration and a Th2-based cytokine profile.6
Nasal polyps (NP) usually present as edematous masses originating in and around the middle nasal meatus or paranasal sinuses. Histologically, NP are characterized by an infiltration by inflammatory cells, predominantly eosinophils, intense edematous stroma, and the formation of pseudocysts filled with plasma proteins, mainly albumin.7 Profound inflammation causes plasma exudation from capillaries, however, exuded plasma may not only induce edema, but may also pass through the airway epithelial layer.8 Antigen stimulation of the nasal cavity of patients with seasonal nasal allergy induces the influx of plasma proteins into the nasal lumen, as detected in nasal lavage.9 The mechanism responsible for retention of exuded plasma proteins in NP submucosa is not fully understood. Recently, we reported that excessive fibrin deposition and low levels of d-dimer, a fibrin degradation product, were observed in NP tissue from patients with CRSwNP. Our study suggested that downregulation of tissue plasminogen activator (t-PA) in NP may lead to insufficient fibrin degradation by plasmin, because t-PA converts plasminogen to plasmin and low levels of plasmin cause this excessive fibrin deposition, which would be expected to contribute to retention of plasma proteins in NP tissue.10
Activation of the coagulation cascade and deposition of fibrin as a consequence of inflammation is well known, and is thought to play a critical role in host defense and to be involved in containing microbial or toxic agents.11 However, dysregulation of the coagulation cascade may play an etiologic role in many diseases through excessive fibrin deposition, including rheumatoid arthritis, severe asthma, glomerulonephritis, delayed-type hypersensitivity, and Crohn’s diseases.12–16
In this study, we hypothesized that activation of coagulation factors is involved in excessive fibrin deposition in NP, leading to retention of plasma proteins and formation of intense edema and pseudocysts in NP. We sought to investigate the expression of coagulation factors in sinonasal tissue from CRS subjects. We found that factor XIII-A (FXIII-A) levels were upregulated in patients with CRSwNP and examined the cell types that produce this coagulation factor.
METHODS
Patients and biopsy specimens
Patients with CRS were recruited from the Allergy-Immunology and Otolaryngology Clinics of the Northwestern Medical Faculty Foundation (NMFF) and the Northwestern Sinus Center at NMFF. Sinonasal and NP tissues were obtained from routine Functional Endoscopic Sinus Surgery in patients with CRS. All patients met the criteria for CRS, as defined by American Academy of Otolaryngology-Head and Neck Surgery Chronic Rhinosinusitis Task Force.1, 17 Patients with an established immunodeficiency, pregnancy, coagulation disorder, diagnosis of classic allergic fungal sinusitis, Samter’s triad, Churg-Strauss syndrome, or cystic fibrosis did not participate in the study. Details of subjects’ characteristics are included in table I and in the Method sections of this article’s Online Repository. All subjects signed informed consent, and the protocol and consent forms governing procedures for study were approved by the Institutional Review Board of Northwestern University Feinberg School of Medicine.
TABLE 1.
Subjects’ characteristics
| Control | CRSsNP | CRSwNP | CRSwNP polyp | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total no. of subjects | n=35 (14M/21F) | n=56 (22M/34F) | n=95 (56M/39F) | — | ||||||
| Age (y), median (range) | 49 (16–72) | 36 (20–73) | 40 (23–72) | — | ||||||
| Y | N | U | Y | N | U | Y | N | U | — | |
| Atopy | 1 | 32 | 2 | 19 | 29 | 8 | 47 | 26 | 22 | — |
| Asthma | 0 | 35 | 0 | 5 | 48 | 3 | 45 | 47 | 3 | — |
| Methodology used | ||||||||||
| Tissue RNA | n=16 (7M/9F) | n=27 (8M/19F) | n=33 (21M/12F) | n=34 (22M/12F) | ||||||
| Age (y), median (range) | 45 (16–62) | 35 (20–59) | 38 (23–67) | 39 (23–67) | ||||||
| Tissue extract | n=14 (5M/9F) | n=20 (8M/12F) | n=19 (10M/9F) | n=24 (13M/11F) | ||||||
| Age (y), median (range) | 46 (35–72) | 30 (24–73) | 45 (26–68) | 44 (28–72) | ||||||
| Immunohistochemistry | n=10 (3M/7F) | n=11 (5M/6F) | n=10 (7M/3F) | n=12 (8M/4F) | ||||||
| Age (y), median (range) | 48 (27–64) | 44 (25–67) | 36 (28–55) | 42 (28–71) | ||||||
F, Female; M, male; N, no; U, unknown; Y, yes.
Microarray and real-time PCR
Total RNA from sinus tissue was extracted with QIAzol (Qiagen, Valencia, CA), and the quality of total RNA from sinus tissue was assessed with 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). A comprehensive microarray was performed as described previously, and gene expression was measured with GeneChip Human U133 Plus 2.0 probe arrays (Affymetrix, Santa Clara, CA).18, 19 Real-time RT-PCR was performed with a TaqMan method, as described previously.20 Detailed protocols are found in the Methods section of this article’s Online Repository. All microarray data have been deposited to gene expression omnibus: GSE36830.
ELISA
The concentration of FXIII-A in cell-free supernatant was determined using a specific ELISA kit (HYPHEN BioMed, Neuville-Sur-Oise, France). Details are in the Methods section of this article’s Online Repository.
Immunohistochemistry
Immunohistochemistry was performed as described previously.21 Briefly, blocked sections were incubated with anti-human FXIII-A antibody (CELL MARQUE, Rocklin, CA) at 4°C over night. After washing, sections were incubated in ABC reagent (Vector Laboratories, Burlingame, CA) for 1 hour. Sections were rinsed and incubated in DAB reagent (Invitrogen, Carlsbad, CA) and then counterstained with hematoxylin. Slides were blinded, and 10 photographic fields were randomly taken from each slide. The number of FXIII-A+ cells in the nasal mucosa was counted by a blinded observer. Details of the methods for immunofluorescence and immunohistochemistry are described in the Methods section of this article’s Online Repository.
Statistical analysis
All data are reported as mean ± SEM unless otherwise noted. Differences between groups were analyzed with the Kruskal-Wallis ANOVA with Dunnett post hoctesting. Correlations were assessedusing the Spearman rank correlation. A Pvalue of less than .05 was considered statistically significant.
RESULTS
Screen of the coagulation factors in sinonasal tissue
We analyzed data from a previously performed microarray analysis to compare coagulation factor gene expression in uncinate tissue (UT) from patients with CRSsNP, patients with CRSwNP, and control subjects, as well as in NP from patients with CRSwNP.19 We observed that mRNA levels of the FXIII-A were substantially increased in NP tissues from patients with CRSwNP in comparison with levels seen in UT from either patients with CRS or control subjects (Fig E1). Interestingly, we found no difference between patients with CRS and control subjects in UT mRNA levels of Factor XIII-B (FXIII-B), a subunit that forms a tight tetrameric complex with FXIII-A (FXIII-A2B2) in the plasma (Fig E1).
FXIII-A expression in patients with CRS
Sinonasal and polyp tissues were collected from 56 subjects with CRSsNP, 95 subjects with CRSwNP, and 35 control subjects to determine the presence of FXIII-A expression in patients with CRS. Subjects’ characteristics are shown in Table I.
We further assessed the expression of FXIII-A in UT from patients with CRSsNP, patients with CRSwNP, and control subjects, as well as in NP from patients with CRSwNP using real-time PCR. FXIII-A mRNA levels were significantly increased in NP tissues from patients with CRSwNP (p < .001) in comparison with levels seen in UT from either patients with CRS or control subjects (Fig 1, A). To confirm this observation at the protein level, we made detergent extracts from homogenates of UT and NP tissues and then measured the concentration of FXIII-A by ELISA. In agreement with the mRNA data, FXIII-A protein levels were significantly increased in NP from patients with CRSwNP (p < .05) in comparison with those seen in UT from either patients with CRS or control subjects (Fig 1, B).
Figure 1.
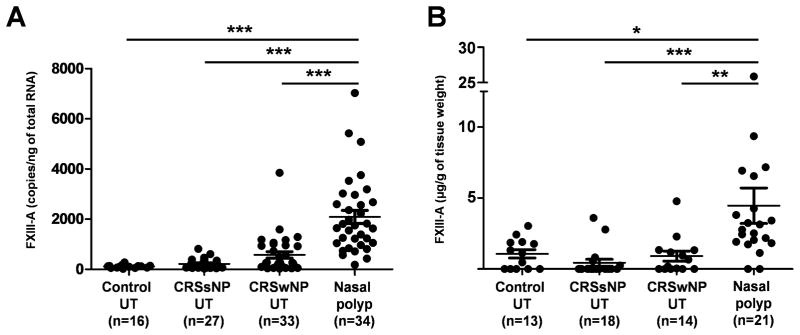
Increased expression of FXIII-A in NP tissue. A, Total RNA was extracted from UT and NP (Nasal polyp), and expression of FXIII-A was analyzed by real time PCR. B, Expression of FXIII-A protein in tissue homogenates of UT and NP from patients with CRSwNP was measured using ELISA. * p < .05, ** p < .01, *** p < .001.
Immunohistochemical analysis of FXIII-A in sinonasal tissue
To further characterize the expression of FXIII-A proteins in patients with CRS, we performed immunohistochemical analysis of surgical samples from control subjects and patients with CRS to determine whether FXIII-A expression could be detected. As shown in Fig 2, we detected FXIII-A staining mainly in submucosal inflammatory cells. We found that FXIII-A+ inflammatory cell numbers were highly elevated in NP (Fig 2, D). We counted the number of FXIII-A+ inflammatory cells using a semiquantitative method and confirmed that FXIII-A+ inflammatory cell numbers were significantly increased in NP from patients with CRSwNP compared to with those seen in UT from patients with either CRS or control subjects (p < .01; Fig 2, F).
Figure 2.

Immunohistochemistry of FXIII-A was performed with anti-human FXIII-A antibody. Representative immunostaining for FXIII-A in UT from control subject (A), a patient with CRSsNP (B), a patient with CRSwNP (C), and NP tissue (D). Negative control antibody staining in NP tissue from patient with CRSwNP (E) is shown. The number of FXIII-A+ cells in UT from control subjects (n = 10), patients with CRSsNP (n = 11), and patients with CRSwNP (n = 10) and NP (n = 12) was counted using a semiquantitative method (F). Magnification: ×400. ** p < .01, *** p < .001.
FXIII-A is expressed primarily in cells of bone marrow origin including platelets, megakaryocytes, and macrophages.22–25 We therefore focused on macrophages and performed dual-immunofluorescence analysis using anti-FXIII-A and antibody against markers of macrophages (CD68). We found a high degree of colocalization of FXIII-A with CD68+ macrophages in NP (Fig 3, A).
Figure 3.

Immunofluorescence of FXIII-A in NP tissue. Immunofluorescence assay was performed with anti FXIII-A (orange fluorescence) and anti-CD68 mAb (green fluorescence) for macrophages (A), and control IgG (B). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; blue fluorescence). The results are representative of 4 separate subjects.
Detection of FXIII-A in M2 macrophages
Macrophages are now widely recognized to be polarized by their microenvironment, especially by T helper cytokines and pathogens.26–30 Classically activated macrophage (also known as M1 macrophages) develop in response to pro-inflammatory stimuli, such as Th1 cytokines (IFN-γ) or bacterial products (LPS). In contrast, alternatively activated macrophages are induced by exposure to Th2 cytokines, including IL-4 and IL-13, and are therefore called M2 macrophages. Recent studies have suggested that increased expression of FXIII-A is present in M2 macrophages.25, 31 Therefore, we next examined whether M2 macrophages are major FXIII-A producing cells in NP. We first determined levels of M2 macrophage markers in NP using real-time PCR; the macrophage mannose receptor (MMR), CD163, and stabilin 1 (STAB1). Levels of mRNA for MMR, CD163, and STAB1 were significantly upregulated in NP (p < .01) in comparison with those seen in UT from either patients with CRS or control subjects (Fig 4, A). We also found that expression of FXIII-A significantly and positive correlated with expression of MMR (r = 0.8820, p < .0001), CD163 (r = 0.7797, p < .0001), and STAB1 (r = 0.5521, p = .0015; Fig 4, B). Levels of MMR also significantly correlated with levels of CD163 (r = 0.6434, p < .0001) and STAB1 (r = 0.4741, p = .0046).
Figure 4.

Correlation of FXIII-A with markers of M2 macrophages in NP tissue. A, Total RNA was extracted from UT from control subjects (n = 16), patients with CRSsNP (n = 27), and patients with CRSwNP (n = 33) and NP tissue (n = 34). The expression of FXIII-A and M2 macrophage markers MMR, CD163, and STAB1 was analyzed using real time PCR. B, the correlation in NP tissue was assessed using a Spearman rank correlation test. ** p < .01, *** p < .001.
To further investigate whether M2 macrophages were the FXIII-A producing cells in NP, we performed triple-immunofluorescence analysis using anti-FXIII-A and antibodies against a marker of M2 macrophages, CD163. We detected FXIII-A in CD68+ and CD163+ cells in NP (Fig 5). These results suggest that M2 macrophages are the sole or major FXIII-A producing cells in NP.
Figure 5.

Detection of FXIII-A in M2 macrophages in NP tissue. Immunofluorescence assay was performed with anti-FXIII-A (red fluorescence), anti-CD68 mAb (orange fluorescence) for macrophages, and anti-CD163 mAb (green fluorescence) for M2 macrophage (A), and control IgG (B). Nuclei were counterstained with DAPI (blue fluorescence). The results are representative of 4 separate subjects.
DISCUSSION
Previous studies have demonstrated that NP exhibit a high degree of tissue eosinophilia and mast cell infiltration as well as T cells cytokines demonstrating skewing toward a Th2 pattern.6, 32, 33 In NP tissue, activation of eosinophils and mast cells facilitates plasma exudation, intense edema or pseudocyst formation, which are major histopathological characteristics of NP.7, 34 We observed high levels of albumin, a major constituent of plasma, in NP compared to UT from patients with CRS and control subjects (Fig E2), in line with a previous report.7 However, the mechanism by which NP tissue retains plasma proteins in the stroma has not been explored. Most recently, we showed that excessive fibrin deposition is seen in NP tissue from patients with CRSwNP and is associated with a reduction of t-PA, which is involved in fibrinolysis by converting plasminogen to plasmin.10 We hypothesized that profound fibrin deposition is responsible for retention of exuded plasma proteins, forming of intense edema and pseudocysts in NP tissue.10 To further test this hypothesis in the current study, we evaluated the components of the coagulation cascade in control and nasal polyp tissues. We demonstrated that FXIII-A levels are increased in NP tissue from patients with CRSwNP (Fig 1). Coagulation factor XIII is a transglutaminase that participates in the final stage of the coagulation cascade. There are two forms of FXIII. Plasma FXIII consists of two enzymatically active A subunits (FXIII-A) and two inhibitory/carrier B subunits (FXIII-B), whereas cellular FXIII is a dimer of FXIII-A, present in platelets, monocytes and macrophages.35, 36 During the process of plasma FXIII activation, thrombin cleaves off an activation peptide from FXIII-A, then FXIII-B dissociates in the presence of Ca2+ and FXIII-A becomes transformed into an active transglutaminase. In contrast, cellular FXIII, which lacks the inhibitory B subunit, does not require the cleavage of the activation peptide for its activation, and typical levels of Ca2+ present in tissue are sufficient to convert this protransglutaminase into an active transglutaminase. Activated-FXIII catalyzes the formation of covalent cross-links between γ-glutamyl and ε-lysyl residues on adjacent fibrin chains in polymerized fibrin to yield the mature clot, and also cross-links α2-plasmin inhibitor (α2PI) with fibrin. The cross-linking of fibrin enhances its stiffness and rigidity, which allows it to retain plasma proteins. Cross-linking of α2PI to fibrin in the matrix has the predominant role of protecting newly formed fibrin from degradation by the fibrinolytic enzyme, plasmin.25, 35 We also observed a significant positive correlation between the protein levels of albumin and FXIII-A in nasal tissues (r = 0.441, p < .0001, Fig E3). Taken together, these data support a hypothesis that the upregulation of FXIII-A plays a critical role in forming excessive fibrin deposition, which is involved in retention of exuded plasma proteins in NP tissue.
Growing evidence indicates that FXIII-A is a multifunctional protein that plays an important role in a wide variety of physiological and pathological process.25 Using FXIII-A deficient mice, an essential role of FXIII-A in the wound healing process was clearly demonstrated, and it was shown that angiogenesis plays a critical role in this process.37, 38 It has been reported that activated-FXIII regulates the key steps of angiogenesis by increasing endothelial cell migration, proliferation, and survival, which are found to be dependent on transglutaminase activity of FXIII-A. These effects of FXIII-A on endothelial cells are accompanied by downregulation of Thrombospondin-1 (THBS1), one of the best characterized antiangiogenic factors.39 Activated FXIII binding to αvβ3 integrin elicited an intracellular signaling cascade leading to c-Jun upregulation, which in turn downregulated THBS1.40 High levels of angiogenesis are one of the reported characteristics of NP tissue.41, 42 Taken together, upregulation of FXIII-A in NP may induce fibrin deposition and plasma protein retention as well as angiogenesis, which might also be involved in tissue remodeling in NP tissue. A previous report demonstrated that the induction of fibrin- fibronectin cross-linking by FXIII also plays a critical role in migration, proliferation, and survival of fibroblasts.43 Since activation of fibroblasts is considered to be an important event leading to NP development,32 the role of FXIII-A in these processes is worthy of further investigation.
Recently, Krysko et al44 and Peterson et al45 showed that numbers of MMR+ M2 macrophages, but not M1 macrophages, were increased in NP. In the current study, we also confirmed that the M2 macrophage markers MMR, CD163, and STAB1 were significantly upregulated in NP (Fig 4, A). It is possible that the increased number of M2 macrophages could be explained by either the Th2 milieu of CRSwNP or by the presence of mast cell and type 2 innate lymphoid cells, as demonstrated in recent reports.28, 46, 47 However they are recruited to NP, the role of macrophages in the pathogenesis of CRS remains unclear. Importantly, it has been reported that M2 macrophages express FXIII-A.48–50 We found that FXIII-A was detected in CD163+ macrophages but not in CD163− macrophages in NP (Fig 5 and data not shown). We also showed that levels of M2 macrophage markers correlated well with levels of FXIII-A (Fig 4, B). These results indicate that M2 macrophages are the sole or major FXIII-A producing cell type in NP. Although there is a general agreement on the cytoplasmic localization of FXIII-A in macrophages, FXIII-A lacks an identifiable endoplasmic reticulum signal sequence and it is not clear how it can be released from macrophages to the extracellular milieu.35 Recent reports demonstrate the appearance of FXIII-A in bronchoalveolar lavage fluid and in the culture medium of macrophages, and there is evidence that FXIII-A is released by an alternative secretory pathway in human macrophages.35, 49, 51 In fact, extravascular fibrin deposition is frequently observed within and around tumor matrix, and tumor associated macrophages containing profound levels of FXIII-A have been reported.52, 53 It is reasonable to speculate that infiltrating M2 macrophages might contribute to excessive fibrin deposition by secreting FXIII-A in NP tissue, but this will required further experiments to test. In spite of presenting prominent inflammation, NP tissue shows low levels of fibrosis.4, 10 A previous report suggested that downregulation of transforming growth factorbeta (TGF-β) may partially explain the low levels of collagen detected in NP tissue.4 It has also been reported that M2 macrophages are involved in suppression of tissue fibrosis by production of IL-10, resistin-like molecule alpha and Arginase-1.28 On the other hand, M1 macrophages, which have been identified as key regulators in demyelinating diseases of the central nervous system, produce significant amounts of TGF-β.54 Thus, predominant infiltration of M2 macrophages might prevent or diminish fibrosis in NP tissue. Phagocytosis is one of the most important functions of macrophages, in which the rearrangement of cell cytoskeleton is deeply involved, and FXIII-A is implicated in phagocytic activities by catalyzing alterations in certain cytoskeletal components, including actin, myosin, vinculin, small heat shock protein HSP27 and thymosin β4.24, 50 Macrophages from FXIII-A deficient patients showed an impaired capacity of Fcγ, complement, and lectin-like receptor mediated phagocytosis.55 FXIII-A plays a critical intracellular role in receptor-mediated phagocytosis of macrophages. However, a recent study suggested that the phagocytic capacity of M2 macrophages is impaired in NP tissue and facilitated the increased presence of S. aureus in CRSwNP.44 Further studies are required to determine how and whether FXIII-A participates in alteration of phagocytosis in M2 macrophages associated with nasal polyposis.
We found that M2 macrophages were major FXIII-A-expressing cells in NP. However the regulation of macrophage recruitment in NP is poorly understood. We recently demonstrated that CCL23, known as a chemokine for macrophage, is elevated in NP tissue and CCL23 might play critical role in macrophage infiltration.56 It is possible that FXIII-A may play some role. Activated-FXIII enhances the proliferation of peripheral blood monocytes, accelerates their migration, and inhibits monocyte apoptosis through the downregulation of THBS1 and due to the upregulation of c-Jun and Egr-1.25, 43 Previous reports demonstrated that activated-FXIII is involved in the generation of the complement C5-derived monocyte chemotactic factor.57 Recently, it has also been reported that ribosomal protein S19 forms a dimer with activated FXIII and converts it into a monocyte-selective chemoattractive factor.58 Since it is well known that formation of a fibrin clot facilitates infiltration and activation of monocytes and macrophages,16, 59, 60 it is possible that a feed-forward mechanism exists in NP in which M2 macrophages induce activation of FXIII-A and the actions of activated FXIII-A lead to further recruitment of macrophages into the NP tissue.
Most recently, we reported that stimulation of Th2 cytokines downregulates the expression of t-PA in cultured nasal epithelial cells.10 Since t-PA converts plasminogen to plasmin and induces fibrinolysis, downregulation of t-PA may therefore be involved in formation of the excessive fibrin deposition that we observed. Based on the current observation and our recent report, it is reasonable to speculate that the Th2 milieu facilitates excessive fibrin deposition by both acceleration of the coagulation cascade and reduction of the process of fibrinolysis in NP tissue (summarized in Figure 6). It is well known that infection with many parasitic helminths induces activation of Th2 immunity in mucosal tissue and M2 macrophages are mobilized in the development of this Th2 polarization.61 It seems possible that Th2 immunity, and subsequent production of extravascular fibrin deposition, take part in mucosal innate immunity by forming a fibrin mesh and impeding the motility or feeding of parasitic helminths. This process may promote fibrogenesis via the activation of FXIII-A and the suppression of the fibrinolytic protein t-PA. In this scenario, the excessive activation of type 2 cytokines that occurs in CRSwNP and may lead to the formation of NP can be viewed as a localized sterile anti-parasite response.
Figure 6.

Hypothetical model in which the Th2 milieu facilitates excessive fibrin deposition in NP tissue. Th2 inflammation leads to recruitment of M2 macrophages and subsequent production of FXIII-A that induces excessive fibrin deposition by cross-linking of fibrin and via the anti-fibrinolytic effect through binding α2PI to fibrin. In the presence of Th2 cytokines, t-PA levels are reduced, causing impaired plasmin generation, which in turn decreases fibrinolysis.
We report here that tissue levels of FXIII-A were profoundly increased in NP tissue and that M2 macrophages are the sole or major FXIII-A producing cell in nasal polyps. Overproduction of FXIII-A may lead to acceleration of the coagulation cascade, resulting in excessive fibrin deposition, which in turn retains exuded plasma proteins and participates in tissue remodeling, intense edema or pseudocyst formation in submucosa of NP tissue. Our results imply that targeting the local production of FXIII-A from M2 macrophage might be therefore be of therapeutic value for treating patients with CRSwNP.
Clinical implications.
Overexpression of FXIII-A may have a pathogenic role in CRSwNP and strategies to reduce the activity of the coagulation cascade might have therapeutic value in the treatment of CRSwNP.
Acknowledgments
Supported byNIH grants R37HL068546-27, R01HL078860, RO1AI072570 and the Ernest S. Bazley Trust
Abbreviations
- CRS
Chronic rhinosinusitis
- CRSwNP
CRS with nasal polyps
- CRSsNP
CRS without nasal polyps
- NP
Nasal polyps
- t-PA
Tissue plasminogen activator
- FXIII-A
Factor XIII-A
- FXIII-B
Factor XIII-B
- UT
Uncinate tissue
- α2PI
α2-plasmin inhibitor
- MMR
Macrophage mannose receptor
- STAB1
Stabilin 1
- THBS1
Thrombospondin-1
- TGF-β
transforming growth factor beta
- DAPI
4′,6-diamidino-2-phenylindole
Footnotes
Disclosure of potential conflict of interest:None – the work has no commercial value.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Meltzer EO, Hamilos DL, Hadley JA, Lanza DC, Marple BF, Nicklas RA, et al. Rhinosinusitis: Establishing definitions for clinical research and patient care. Otolaryngol Head Neck Surg. 2004;131:S1–S62. doi: 10.1016/j.otohns.2004.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kern RC, Conley DB, Walsh W, Chandra R, Kato A, Tripathi-Peters A, et al. Perspectives on the etiology of chronic rhinosinusitis: an immune barrier hypothesis. Am J Rhinol. 2008;22:549–59. doi: 10.2500/ajr.2008.22.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schleimer RP, Kato A, Peters A, Conley D, Kim J, Liu MC, et al. Epithelium, inflammation, and immunity in the upper airways of humans: studies in chronic rhinosinusitis. Proc Am Thorac Soc. 2009;6:288–94. doi: 10.1513/pats.200808-088RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Bruaene N, Derycke L, Perez-Novo CA, Gevaert P, Holtappels G, De Ruyck N, et al. TGF-beta signaling and collagen deposition in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124:253–9. doi: 10.1016/j.jaci.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Bachert C, Gevaert P, van Cauwenberge P. Staphylococcus aureus superantigens and airway disease. Curr Allergy Asthma Rep. 2002;2:252–8. doi: 10.1007/s11882-002-0027-9. [DOI] [PubMed] [Google Scholar]
- 6.Van Zele T, Claeys S, Gevaert P, Van Maele G, Holtappels G, Van Cauwenberge P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. 2006;61:1280–9. doi: 10.1111/j.1398-9995.2006.01225.x. [DOI] [PubMed] [Google Scholar]
- 7.Bachert C, Gevaert P, Holtappels G, Cuvelier C, van Cauwenberge P. Nasal polyposis: from cytokines to growth. Am J Rhinol. 2000;14:279–90. doi: 10.2500/105065800781329573. [DOI] [PubMed] [Google Scholar]
- 8.Persson CG. Plasma exudation in the airways: mechanisms and function. Eur Respir J. 1991;4:1268–74. [PubMed] [Google Scholar]
- 9.Hanf G, Noga O, O’Connor A, Kunkel G. Omalizumab inhibits allergen challenge- induced nasal response. Eur Respir J. 2004;23:414–8. doi: 10.1183/09031936.04.00024504. [DOI] [PubMed] [Google Scholar]
- 10.Takabayashi T, Kato A, Peters AT, Hulse KE, Suh LA, Carter R, et al. Excessive fibrin deposition in nasal polyps caused by fibrinolytic impairment through reduction of t-PA expression. Am J Respir Crit Care Med. 2012;187:49–57. doi: 10.1164/rccm.201207-1292OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jennewein C, Tran N, Paulus P, Ellinghaus P, Eble JA, Zacharowski K. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med. 2011;17:568–73. doi: 10.2119/molmed.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabazza EC, Osamu T, Yamakami T, Ibata H, Sato T, Sato Y, et al. Correlation between clotting and collagen metabolism markers in rheumatoid arthritis. Thromb Haemost. 1994;71:199–202. [PubMed] [Google Scholar]
- 13.de Boer JD, Majoor CJ, van ‘t Veer C, Bel EHD, van der Poll T. Asthma and coagulation. Blood. 2012;119:3236–44. doi: 10.1182/blood-2011-11-391532. [DOI] [PubMed] [Google Scholar]
- 14.Neale TJ, Tipping PG, Carson SD, Holdsworth SR. Participation of cell-mediated immunity in deposition of fibrin in glomerulonephritis. Lancet. 1988;2:421–4. doi: 10.1016/s0140-6736(88)90413-8. [DOI] [PubMed] [Google Scholar]
- 15.Hudson M, Hutton RA, Wakefield AJ, Sawyerr AM, Pounder RE. Evidence for activation of coagulation in Crohn’s disease. Blood Coagul Fibrinolysis. 1992;3:773–8. doi: 10.1097/00001721-199212000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Szaba FM, Smiley ST. Roles for thrombin and fibrin(ogen) in cytokine/chemokine production and macrophage adhesion in vivo. Blood. 2002;99:1053–9. doi: 10.1182/blood.v99.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearlman AN, Conley DB. Review of current guidelines related to the diagnosis and treatment of rhinosinusitis. Curr Opin Otolaryngol Head and Neck Surg. 2008;16:226–30. doi: 10.1097/MOO.0b013e3282fdcc9a. [DOI] [PubMed] [Google Scholar]
- 18.Kato A, Chustz RT, Ogasawara T, Kulka M, Saito H, Schleimer RP, et al. Dexamethasone and FK506 inhibit expression of distinct subsets of chemokines in human mast cells. J Immunol. 2009;182:7233–43. doi: 10.4049/jimmunol.0801375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seshadri S, Lin DC, Rosati M, Carter RG, Norton JE, Suh L, et al. Reduced expression of antimicrobial PLUNC proteins in nasal polyp tissues of patients with chronic rhinosinusitis. Allergy. 2012;67:920–8. doi: 10.1111/j.1398-9995.2012.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato A, Truong-Tran AQ, Scott AL, Matsumoto K, Schleimer RP. Airway epithelial cells produce B cell-activating factor of TNF family by an IFN-beta-dependent mechanism. J Immunol. 2006;177:7164–72. doi: 10.4049/jimmunol.177.10.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato A, Peters A, Suh L, Carter R, Harris KE, Chandra R, et al. Evidence of a role for B cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2008;121:1385–92. doi: 10.1016/j.jaci.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katona EE, Ajzner E, Toth K, Karpati L, Muszbek L. Enzyme-linked immunosorbent assay for the determination of blood coagulation factor XIII A-subunit in plasma and in cell lysates. J Immunol Methods. 2001;258:127–35. doi: 10.1016/s0022-1759(01)00479-3. [DOI] [PubMed] [Google Scholar]
- 23.Malara A, Gruppi C, Rebuzzini P, Visai L, Perotti C, Moratti R, et al. Megakaryocyte-matrix interaction within bone marrow: new roles for fibronectin and factor XIII-A. Blood. 2011;117:2476–83. doi: 10.1182/blood-2010-06-288795. [DOI] [PubMed] [Google Scholar]
- 24.Adany R, Bardos H. Factor XIII subunit A as an intracellular transglutaminase. Cell Mol Life Sci. 2003;60:1049–60. doi: 10.1007/s00018-003-2178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 2011;91:931–72. doi: 10.1152/physrev.00016.2010. [DOI] [PubMed] [Google Scholar]
- 26.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 28.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fairweather D, Cihakova D. Alternatively activated macrophages in infection and autoimmunity. J Autoimmun. 2009;33:222–30. doi: 10.1016/j.jaut.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 31.Torocsik D, Szeles L, Paragh G, Jr, Rakosy Z, Bardos H, Nagy L, et al. Factor XIII-A is involved in the regulation of gene expression in alternatively activated human macrophages. Thromb Haemost. 2010;104:709–17. doi: 10.1160/TH09-11-0805. [DOI] [PubMed] [Google Scholar]
- 32.Pawliczak R, Lewandowska-Polak A, Kowalski ML. Pathogenesis of nasal polyps: an update. Curr Allergy Asthma Rep. 2005;5:463–71. doi: 10.1007/s11882-005-0027-7. [DOI] [PubMed] [Google Scholar]
- 33.Takabayashi T, Kato A, Peters AT, Suh LA, Carter R, Norton J, et al. Glandular mast cells with distinct phenotype are highly elevated in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2012;130:410–20.e5. doi: 10.1016/j.jaci.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10:440–52. doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bagoly Z, Katona E, Muszbek L. Factor XIII and inflammatory cells. Thromb Res. 2012;129 (Suppl 2):S77–81. doi: 10.1016/j.thromres.2012.02.040. [DOI] [PubMed] [Google Scholar]
- 36.Derrick EK, Barker JN, Khan A, Price ML, Macdonald DM. The tissue distribution of factor XIIIa positive cells. Histopathology. 1993;22:157–62. doi: 10.1111/j.1365-2559.1993.tb00095.x. [DOI] [PubMed] [Google Scholar]
- 37.Lauer P, Metzner HJ, Zettlmeissl G, Li M, Smith AG, Lathe R, et al. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88:967–74. [PubMed] [Google Scholar]
- 38.Kondo T, Ishida Y. Molecular pathology of wound healing. Forensic Sci Int. 2010;203:93–8. doi: 10.1016/j.forsciint.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Dardik R, Solomon A, Loscalzo J, Eskaraev R, Bialik A, Goldberg I, et al. Novel proangiogenic effect of factor XIII associated with suppression of thrombospondin 1 expression. Arterioscler Thromb Vasc Biol. 2003;23:1472–7. doi: 10.1161/01.ATV.0000081636.25235.C6. [DOI] [PubMed] [Google Scholar]
- 40.Dardik R, Loscalzo J, Eskaraev R, Inbal A. Molecular mechanisms underlying the proangiogenic effect of factor XIII. Arterioscler Thromb Vasc Biol. 2005;25:526–32. doi: 10.1161/01.ATV.0000154137.21230.80. [DOI] [PubMed] [Google Scholar]
- 41.Hirshoren N, Neuman T, Gross M, Eliashar R. Angiogenesis in chronic rhinosinusitis with nasal polyps and in antrochoanal polyps. Inflamm Res. 2011;60:321–7. doi: 10.1007/s00011-010-0271-8. [DOI] [PubMed] [Google Scholar]
- 42.Lee HS, Myers A, Kim J. Vascular endothelial growth factor drives autocrine epithelial cell proliferation and survival in chronic rhinosinusitis with nasal polyposis. Am J Respir Crit Care Med. 2009;180:1056–67. doi: 10.1164/rccm.200905-0740OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dardik R, Krapp T, Rosenthal E, Loscalzo J, Inbal A. Effect of FXIII on monocyte and fibroblast function. Cell Physiol Biochem. 2007;19:113–20. doi: 10.1159/000099199. [DOI] [PubMed] [Google Scholar]
- 44.Krysko O, Holtappels G, Zhang N, Kubica M, Deswarte K, Derycke L, et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy. 2011;66:396–403. doi: 10.1111/j.1398-9995.2010.02498.x. [DOI] [PubMed] [Google Scholar]
- 45.Peterson S, Poposki JA, Nagarkar DR, Chustz RT, Peters AT, Suh LA, et al. Increased expression of CC chemokine ligand 18 in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2012;129:119–27. e1–9. doi: 10.1016/j.jaci.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–62. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 47.Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The Transcription Factor GATA3 Is Essential for the Function of Human Type 2 Innate Lymphoid Cells. Immunity. 2012;37:649–59. doi: 10.1016/j.immuni.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 48.Ichinose A. Factor XIII is a key molecule at the intersection of coagulation and fibrinolysis as well as inflammation and infection control. Int J Hematol. 2012;95:362–70. doi: 10.1007/s12185-012-1064-3. [DOI] [PubMed] [Google Scholar]
- 49.Katona E, Nagy B, Kappelmayer J, Baktai G, Kovacs L, Marialigeti T, et al. Factor XIII in bronchoalveolar lavage fluid from children with chronic bronchoalveolar inflammation. Journal of thrombosis and haemostasis: JTH. 2005;3:1407–13. doi: 10.1111/j.1538-7836.2005.01353.x. [DOI] [PubMed] [Google Scholar]
- 50.Torocsik D, Bardos H, Nagy L, Adany R. Identification of factor XIII-A as a marker of alternative macrophage activation. Cell Mol Life Sci. 2005;62:2132–9. doi: 10.1007/s00018-005-5242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cordell PA, Kile BT, Standeven KF, Josefsson EC, Pease RJ, Grant PJ. Association of coagulation factor XIII-A with Golgi proteins within monocyte-macrophages: implications for subcellular trafficking and secretion. Blood. 2010;115:2674–81. doi: 10.1182/blood-2009-08-231316. [DOI] [PubMed] [Google Scholar]
- 52.Hao N-B, Lu M-H, Fan Y-H, Cao Y-L, Zhang Z-R, Yang S-M. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098. doi: 10.1155/2012/948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quatresooz P, Paquet P, Hermanns-Le T, Pierard GE. Molecular mapping of Factor XIIIa-enriched dendrocytes in the skin (Review) Int J Mol Med. 2008;22:403–9. [PubMed] [Google Scholar]
- 54.Kiefer R, Kieseier BC, Stoll G, Hartung HP. The role of macrophages in immune-mediated damage to the peripheral nervous system. Prog Neurobiol. 2001;64:109–27. doi: 10.1016/s0301-0082(00)00060-5. [DOI] [PubMed] [Google Scholar]
- 55.Sarvary A, Szucs S, Balogh I, Becsky A, Bardos H, Kavai M, et al. Possible role of factor XIII subunit A in Fcgamma and complement receptor-mediated phagocytosis. Cell Immunol. 2004;228:81–90. doi: 10.1016/j.cellimm.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 56.Poposki JA, Uzzaman A, Nagarkar DR, Chustz RT, Peters AT, Suh LA, et al. Increased expression of the chemokine CCL23 in eosinophilic chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128:73–81.e4. doi: 10.1016/j.jaci.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okamoto M, Yamamoto T, Matsubara S, Kukita I, Takeya M, Miyauchi Y, et al. Factor XIII-dependent generation of 5th complement component(C5)-derived monocyte chemotactic factor coinciding with plasma clotting. Biochim Biophys Acta. 1992;1138:53–61. doi: 10.1016/0925-4439(92)90151-c. [DOI] [PubMed] [Google Scholar]
- 58.Semba U, Chen J, Ota Y, Jia N, Arima H, Nishiura H, et al. A plasma protein indistinguishable from ribosomal protein S19: conversion to a monocyte chemotactic factor by a factor XIIIa-catalyzed reaction on activated platelet membrane phosphatidylserine in association with blood coagulation. Am J Pathol. 2010;176:1542–51. doi: 10.2353/ajpath.2010.090720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akassoglou K, Adams RA, Bauer J, Mercado P, Tseveleki V, Lassmann H, et al. Fibrin depletion decreases inflammation and delays the onset of demyelination in a tumor necrosis factor transgenic mouse model for multiple sclerosis. Proc Natl Acad Sci U S A. 2004;101:6698–703. doi: 10.1073/pnas.0303859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Del Rosso M, Fibbi G, Pucci M, Margheri F, Serrati S. The plasminogen activation system in inflammation. Front Biosci. 2008;13:4667–86. doi: 10.2741/3032. [DOI] [PubMed] [Google Scholar]
- 61.Taylor MD, van der Werf N, Maizels RM. T cells in helminth infection: the regulators and the regulated. Trends Immunol. 2012;33:181–9. doi: 10.1016/j.it.2012.01.001. [DOI] [PubMed] [Google Scholar]