

Abstract
The breakdown of L-arginine to ornithine and urea by host arginase supports Leishmania proliferation in macrophages. Studies using arginase-null mutants show that Leishmania-derived arginase plays an important role in disease pathogenesis. We investigated the role of parasite-derived arginase in secondary (memory) anti-Leishmania immunity in the resistant C57BL/6 mice. We found that C57BL/6 mice infected with arginase deficient (arg−) L. major failed to completely resolve their lesion and maintained chronic pathology after 16 weeks, a time when the lesion induced by wild type (WT) L. major is completely resolved. This chronic disease was associated with impaired antigen-specific proliferation and IFN-γ production, a concomitant increase in programmed cell death-1 (PD-1) expression on CD4+ T cells and failure to induce protection against secondary L. major challenge. Treatment with anti-PD-1 monoclonal antibody restored T cell proliferation and IFN-γ production in vitro and led to complete resolution of chronic lesion in arg− L. major-infected mice. These results show that infection with arg− L. major results in chronic disease due in part to PD-1-mediated clonal exhaustion of T cells, suggesting that parasite-derived arginase contributes to the overall quality of the host immune response and subsequent disease outcome in L. major-infected mice. They also indicate that persistent parasites alone do not regulate the quality of secondary anti-Leishmania immunity in mice and that the quality of the primary immune response may be playing a hitherto unrecognized dominant role in this process.
Keywords: Monocytes/macrophages, Th1/Th2 cells, parasitic-protozoan, cytokines, immunologic memory, T cell exhaustion
Introduction
Cutaneous leishmaniasis causes disfiguring lesions that often develop as a result of secondary bacterial infections. In majority of cases, primary infections are generally limited to localized cutaneous lesions that heal spontaneously after several months resulting in life-long immunity (1–3). This infection-induced immunity is dependent on a small number of persistent parasites that act as a source of continuous antigen stimulation to the host immune cells leading to maintenance of effector-like memory cells (1, 2, 4). Infection-induced immunity is lost if infected mice are manipulated to completely clear their parasites such as seen during an exclusive Th1 response to low dose infection or following anti-IL-10R antibody treatment (5, 6). In addition, recrudescence (relapse) of disease can occur when the host’s immune system becomes compromised (4, 7, 8). Thus, while the contribution of the host immune response to parasite persistence and maintenance of secondary immunity is known, little is known about the role played by parasite-derived factors in this process.
The outcome of infection with Leishmania major depends in part on the activation status of infected host macrophages (9–12). In infected animals, IFN-γ produced by CD4+ Th1 cells, classically activate macrophages leading to increased expression of inducible nitric oxide synthase (iNOS) (13–15). This enzyme acts on its substrate L-arginine to produce nitric oxide (NO) that is essential for parasite control (14). In contrast, CD4+ Th2 cell-derived cytokines, primarily IL-4 and IL-13, cause alternative activation of macrophages that favors parasite proliferation in infected cells. Alternative macrophage activation is accompanied by increased expression of arginase, which catalyzes the formation of ornithine from arginine leading to polyamine synthesis (10, 11, 14, 16, 17). iNOS and arginase are reciprocally regulated (13–15, 18), and the two enzymes compete directly for their common substrate, L-arginine, and indirectly because some of their intermediate products inhibit each other at several metabolic points (14, 16, 19, 20). Additionally, arginine catabolism may lead to metabolic stresses also leading to shifts in the immune response (21).
Leishmania also express an arginase enzyme (22, 23), related to the mammalian arginases 1 and 2 (23). Importantly, parasite-derived arginase is not stage-specific as expression has been detected in both amastigotes and promastigotes at similar levels (23). It has been proposed that parasite-derived arginase is a virulence factor, which may act to deprive iNOS of L-arginine availability thereby limiting host NO production (24). Indeed, the proliferation and survival of Leishmania arginase null mutants (arg−) is completely dependent on exogenous polyamine supplementation in vitro (22). In the susceptible BALB/c mice infected with arg− L. major, lesion pathology and parasite burden were significantly reduced compared to those infected with WT parasites. Interestingly, there was no significant difference in the production of NO by macrophages infected with arg–and WT L. major (25), suggesting that parasite-derived arginase does not limit host NO production but enhances the establishment of a favorable environment for parasite survival and proliferation through increased polyamine synthesis. Previously, the contribution of arginase in the pathogenesis of cutaneous leishmaniasis has been investigated by using pharmacologic inhibitors (9–11). Since mammalian and Leishmania arginase show considerable homology (23), such approach does not permit the understanding of the specific role of parasite-derived arginase in disease pathogenesis. The availability of arginase mutant parasites provides an excellent resource for evaluating the relative contributions of parasite-derived arginase in a way not feasible by pharmacological means.
In addition to directly inhibiting NO production thereby enhancing parasite proliferation, excessive host arginase activity contributes to non-healing disease by causing suppression of T cell proliferation and effector cytokine response (26). This is consistent with the observation showing that host arginase 1 impairs T cell responses by depleting the bioavailability of L-arginine, a key amino acid critical for optimal cell division (21, 27). Indeed, deprivation of L-arginine has been associated with impaired T cell response observed in many pathological conditions, including asthma (28), psoriasis (29) and tuberculosis (30). However, a recent report found that in vivo inhibition of arginase activity has no effect on skin allograft rejection or systemic T cell proliferation (31).
Recent reports suggest that T cell exhaustion, which is characterized by the presence of antigen-specific T cells exhibiting poor effector functions including proliferation and cytokine responses (32), is a hallmark of many protozoan diseases including malaria (33), toxoplasmosis (34, 35) and leishmaniasis (36, 37). In murine model of visceral leishmaniasis, CD8+ T cell exhaustion due to high PD-1 expression was shown to be responsible for severe disease outcome (37). Similarly, decreased CD8 T cell response and loss of effector cytokine production (including IFN-γ, TNF-α, and IL-2) was associated with the development of diffuse cutaneous leishmaniasis in patients infected with L. mexicana (38). Although T-cell exhaustion has been mostly described for CD8+ T cells in leishmaniasis, no report has demonstrated CD4+ T cell exhaustion in this disease although dysfunctional CD4+ T-cell compartment has been also observed in other chronic infections (32).
In this report, we investigated the influence of parasite-derived arginase on host T cell responses in vivo. Surprisingly, we found that infection with arg− L. major causes chronic infection in C57BL/6 mice and fails to protect infected mice against virulent challenge. This inability of arg− L. major to induce protection was related to clonal exhaustion of CD4+ T cells resulting in impaired antigen-specific proliferation and IFN-γ production by T cells from arg− -infected mice. Thus, contrary to the effects of host arginase, our findings show that parasite-derived arginase plays a critical role in enhancing the host immune response to the parasite and its absence results in impaired CD4+ T cell response leading to chronic disease.
Materials and Methods
Reagents
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich, St. Louis MO, and all antibodies are from eBiosciences, San Diego, CA. Media, penicillin/streptomycin, FBS, and glutamine were purchased from Invitrogen, Carlsbad, CA.
Mice
Six to eight week old female C57BL/6 mice were purchased from Charles River Laboratories (Montreal, Canada) or Central Animal Care Services, University of Manitoba. Female Thy1.1 C57BL/6 mice (6 to 8 weeks old) were purchased from The Jackson Laboratories, Bar Harbor ME. All mouse experiments were approved by the University of Manitoba Animal Care Committee in accordance with the regulation of the Canadian Council on Animal Care.
Parasites and Infection
All parasites were derived from the wild-type Leishmania major line LV39cl5 (RHO/SU/59/P). The generation and biochemical characteristics of homozygous null mutants lacking arginase Δarg::HYG/Δarg::PAC, referred to as arg−, and its complemented line Δarg::HYG/−arg::PAC/+pXG-ARG (referred to arg− /+ARG) are described elsewhere (39). Parasites were grown at 25°C in M199 medium (Hyclone, Logan, UT) supplemented with 10% heat-inactivated FBS, 2μg/ml biopterin, 100 U/ml penicillin, 100 μg/ml streptomycin, 5 μg/ml hemin, 1 μg/ml biotin, 0.1mM adenine, 40 mM HEPES (pH 7.4), 20 mM L-glutamine and 50 mM putrescine (for arg−-L. major). For infection, stationary phase promastigotes were washed three times in PBS and 2 million parasites (suspended in 50μl PBS) were injected into the right hind footpad. After infection, the development and progression of footpad lesion was monitored weekly by measuring the diameter of infected footpad with calipers. Uninfected contra-lateral footpads served as controls. In some experiments, mice were challenged in the contra-lateral footpad with 2 million parasites 14 weeks after the primary infection and footpad thickness was measured in the challenged footpad three days later to quantify delayed type hypersensitivity (DTH) response.
Generation and infection of bone marrow-derived dendritic cells (BMDCs)
Bone marrow-derived dendritic cells were differentiated from the femur and tibia of 6–8 weeks old C57BL/6 mice. Briefly, marrow cells were prepared, depleted of erythrocytes with ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, pH 7.2–7.4), and seeded in 100 ×15 mm Petri dishes (BD Falcon, cat #351029) at 2 ×105/ml and incubated at 37°C in CO2 incubator. The cells were differentiated into immature DCs with recombinant murine GM-CSF (20 ng/ml, Peprotech, Indianapolis, IN). The culture media were changed twice on day 3 and on day 6, and on day 7, the non-adherent cells (dendritic cells) were collected for in vitro experiments. Immature DCs expressed CD11c (>98%), and high levels of CD40, CD80, CD86 and MHC class II as assessed by flow cytometry. For infection, BMDCs were incubated with WT parasites for 5 hours at a BMDC to parasite ratio of 1:10. The free parasites were washed away (3 times washing with complete media) and infected cells were cultured at 37 °C for 24 hours.
In vitro co-culture experiments with BMDCs and T cells from infected mice
T cells were purified from the spleens or dLNs of infected or naive mice by positive selection using CD90.2 coated microbeads and autoMACS separator system according to manufacturer’s suggested protocols (Miltenyi, Auburn, CA). Purified (> 98%) T cells were labeled with CFSE dye and co-cultured with L. major-infected BMDCs for 5 days and cell proliferation and intracellular IFN-γ production were assessed by flow cytometry.
Direct ex vivo and in vitro recall responses and intracellular cytokine staining
At various times after infection, spleens and dLNs were harvested and made into single cell suspensions. The cells were directly cells stimulated with PMA (50 ng/ml), ionomycin (500 ng/ml) and BFA (10 μg/ml) for 4–6 hr (all from Sigma) for 4–6 hours and stained intracellularly for IFN-γ (as below). The remaining cells were resuspended at 4 ×106 cells/ml in complete medium (DMEM supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin), plated at 1 ml/well in 24-well tissue culture plates (Falcon) and stimulated with soluble leishmanial antigen (SLA, 50 μg/ml). After 72 hr, the culture supernatants were collected and stored at –20 °C until assayed for cytokines by ELISA. Some cells were used for intracellular cytokine staining as previously described (40). Briefly, cells were stimulated PMA, ionomycin and Brefeldin A (all from Sigma), for 4–6 hours, washed and fixed with 2% paraformaldehyde for 10 mins, then permeablized with 0.1% saponin (Sigma) in staining buffer for another 10 mins. The cell were then stained with specific fluorochrome-conjugated mAbs against IFN-γ (XMG1.2) and TNF (Biolegend, San Diego, CA). Samples were acquired on a FACSCanto II flow cytometer (BD Bioscience, Mississauga, ON, Canada) and analyzed with FlowJo software (TreeStar, Ashland, OR).
In vitro proliferation assays
To measure proliferation, single cell suspension of spleen or lymph node cells from WT or arg- L. major-infected mice were labeled with 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE, Molecular Probes, Eugene, OR) as previously described (41). Labeled cells were resuspended at 106/ml, plated onto 96-well round bottom plates (200 μl/well) and stimulated with or without SLA (50 mg/ml), anti-PD1 mAb (10 μg/ml) or anti-CD3 and anti-CD28 (1 μg/ml). In some experiments, CFSE labeled cells were co-cultured with infected BMDCs at 100:1 (cells to infected BMDC) ratio. After 5 days, proliferation was analyzed by flow cytometry.
Adoptive Transfer Studies
Infected CD90.2+ mice (Thy1.2) were sacrificed at least 14 weeks after primary infection and single cell suspensions were made of the spleens and lymph nodes draining the infection site. Forty million cells in 100 μl of PBS were injected into the tail vein of naïve CD90.1+ (Thy1.1) recipient mice. Cells from naive age-matched mice were used controls. In some experiments, CD90.2+ T cells were purified by positive selection using kits and reagents from Miltenyi according to manufacturers suggested protocol. CD90.2 enriched cells were greater than 98% CD3+ as assessed by flow cytometry (data not shown). To monitor proliferation in vivo, donor cells were sometimes labeled with CFSE dye prior to transfer. Mice were infected as described above 24 hours after cell transfer.
Treatment with anti-PD-1 monoclonal antibody (mAb) in vivo
To investigate the role of PD1 in lesion chronicity, arg- L. major-infected mice PD-1 were injected weekly (starting at week 14 post-infection) with rat anti-mouse PD-1 mAb (1 mg/mouse intraperitoneally, BioXCell, West Lebanon, NH). Lesion size was monitored weekly for additional 8 weeks.
Estimation of Parasite Burden
At various times after infection or challenge, mice were sacrificed and parasite burden in the footpads of infected mice was quantified by limiting dilution analysis as previously described (42, 43). If the footpad was from mice infected with arg−-L. major, the media was also supplemented with 50 mM putrescine.
Cytokine ELISAs
The levels of IL-4, IL-10 and IFN-γ n dLN or spleen cell culture supernatant fluids were determined by sandwich ELISA using antibody (Ab) pairs (BD Biosciences San Jose, CA) according to the manufacturer’s suggested protocols. The sensitivities of the ELISA were 31.25 pg/ml (IL-4), and 15.65 pg/ml for both IL-10 and IFN-γ.
Arginase Activity Assay
Arginase activity in vitro was quantified as previously described by Corraliza et al. (44). Briefly, homogenate of infected feet was first centrifuged at low speed (500 rpm for 5 min) to remove large tissue debris and the supernatant was then centrifuged again at high speed (3000 rpm for 15 min) to pellet the cells and amastigotes. Both pellets were incubated for 30 min in 2 ml lysis buffer (0.1 M Tris-HCl, 300 μM NaCl, 1 μM phenylmethanesulfonyl fluoride, 1% TritonX) and the lysate was assayed for arginase as previously described (44).
Statistical Analysis
A two-tailed Student’s t-test or analysis of variance (ANOVA) was used to compare means of lesion sizes, parasites burden, and cytokine production from different groups of mice. Significance was considered if p < 0.05.
Results
Infection of C57BL/6 mice with arg− L. major results in chronic infection
We previously showed that arg− L. major are impaired in their macrophage infectivity in vitro and their growth could be rescued by L-ornithine, agmatine, putrescine or spermidine supplementation (39). In addition, their in vivo infectivity is also highly compromised since they fail to induce progressive disease in the susceptible BALB/c mice (25). Here, we characterized the outcome of primary infection of C57BL/6 mice with arg− L. major. We used C57BL/6 mice because this strain heals wild-type (WT) L. major infection and generate life-long immunity, akin to most infected humans. C57BL/6 mice infected with WT L. major developed progressive lesion starting from 2 weeks post-infection, which was associated with increased parasite burden (Fig. 1A and B). Lesions in arg− L. major-infected animals were slow to develop but surprisingly while never reaching the severity seen in WT infection, remained stable until the termination of the experiment (> 16 weeks post-infection, Fig. 1 A). Consistent with lesion sizes, parasite burden was significantly (p < 0.05) lower in arg− L. major-infected than WT infected mice at 2, 4 and 8 weeks post infection (Fig. 1 B). Interestingly, by 16 weeks post-infection, both WT and arg− L. major-infected mice had comparable parasite burden despite the later having slightly (but not significantly) bigger lesion size (Fig. 1 A and B). The phenotypes seen were related strictly to loss of arg- alone rather than the process of genetic manipulation, because as previously shown in BALB/c mice (25), complementation of arg- L. major with ARG gene (arg−/+ARG; Add Back) fully restored the virulence of the parasites in C57BL/6 mice (Fig. 1 A and B).
Figure 1.
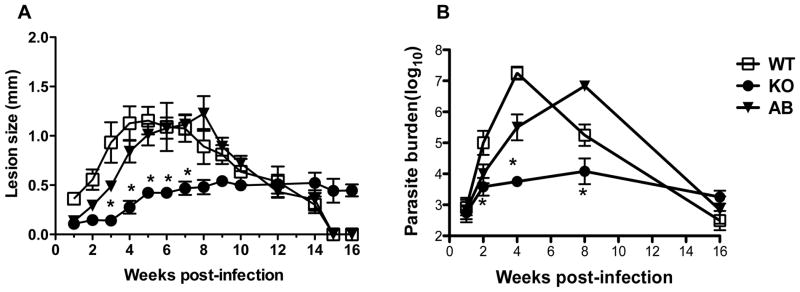
arg− Leishmania major parasites cause chronic infection in resistant mice. C57BL/6 mice were infected with 2 million (stationary phase promastigotes) wild type (WT), arg− (KO) and arg−+Arg+/+ (AB, add-back) and lesion sizes were monitored weekly with calipers (A). At different times after infection, mice were sacrificed and parasite burden in the infected footpad was determined by limiting dilution assay. Data are presented as means ± SE and are representative of 3 independent experiments (n = 5–6 mice) with similar results. *, p < 0.05.
Kinetics of immune response in C57BL/6 mice infected with arg− L. major
We next compared the immune response in C57BL/6 mice following infection with WT and arg− L. major. At 1 and 2 weeks post-infection, cells from lymph nodes draining the infection site (dLNs) were labeled with CFSE dye, stimulated with soluble leishmanial antigen (SLA) for 5 days and proliferation and intracellular cytokine (IFN-γ) production were assessed by flow cytometry. In some cases, the cell culture supernatant fluids were also assayed for IL-4 and IFN-γ by ELISA. At 1 week post-infection, there was no difference in proliferation and frequency of IFN-γ-producing CD4+ T cells (Fig. 2A and B; Fig. S1A) in mice infected with WT and arg− L. major and IL-4-producing cells were undetectable (data not shown). In contrast, antigen-specific proliferation and frequency of IFN-γ-producing CD4+ and CD8+ T cells were significantly (p < 0.05) lower in arg− L. major-infected mice at 2 weeks post-infection (Fig. 2C and D; Fig. S1B), which is consistent with the significantly lower parasite burden in these mice at this time point (see Fig. 1B). The impaired proliferation and IFN-γ production by T cells from arg− L. major-infected mice at 2 weeks was not related to reduction in the total numbers of CD4+ and CD8+ cells in their spleens or dLNs (data not shown).
Figure 2.

arg− and WT Leishmania major induce different immune response in vitro. At 1 (A and B) and 2 (C and D) weeks after infection, mice were sacrificed and draining lymph node cells were labeled with CFSE dye and cultured for 72 hr in the presence of soluble Leishmania antigen (SLA, 50 μg/ml). PMA (50 ng/ml), ionomycin (500 ng/ml) and Brefeldin A (BFA, 10 mg/ml) were added in the last 3–5 hr to enhance intracellular protein accumulation and the cells were surface stained for CD3, CD4 and CD8 and intracellularly for IFN-γ. The percentage of proliferating CD4+ (upper panels) and CD8+ (bottom panels) and CFSElow IFN-γ-secreting CD4+ and CD8+ (B and D) were determined by flow cytometry. Representative contour plots are presented in A and C, while means ± SE of IFN-γ+ proliferating (CFSEloIFN-γ+) cells in the group is presented as bar graphs (B and D). Representative of 3 independent experiments with similar results (n =3–4 mice) are shown. *, p < 0.05.
At 4 and 8 weeks post-infection, we found no significant difference in IL-4 production by ELISA (Fig. 3A), whereas the production of IFN-γ was still significantly (p < 0.05) lower in arg− L. major-infected than in WT-infected mice (Fig. 3B). Interestingly, despite having fewer parasite burden, the production of IL-10 was significantly (p < 0.05) higher in arg− L. major-infected than in WT-infected mice at 8 weeks post-infection (Fig. 3C). In addition, despite the higher production of IFN-γ, the levels of arginase activity in the footpads (lesion) of mice infected with WT parasites was higher at 4 and 8 weeks post-infection than in those infected with arg− L. major (Fig. 3D). Thus, unlike our findings in BALB/c mice where there were no differences in cytokine responses (25), the production of IFN-γ by splenic and draining lymph nodes cells from C57BL/6 mice infected with arg− L. major was significantly impaired later during infection despite the absence of progressive lesion development, parasite proliferation and higher arginase production.
Figure 3.
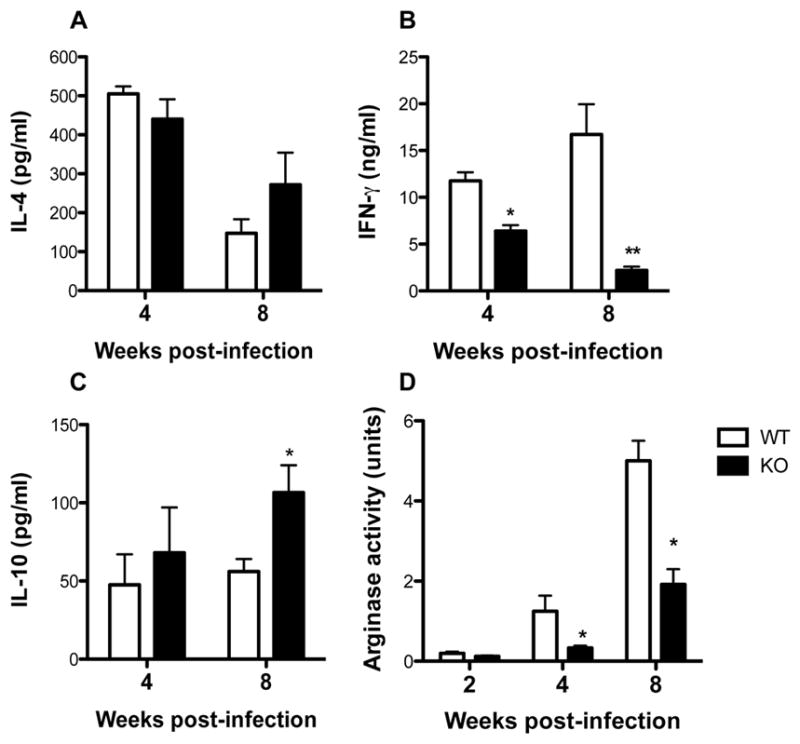
Cytokine and arginase responses in mice infected with arg− and WT Leishmania major. At various times after infection, infected mice were sacrificed and single cell suspensions of draining lymph nodes were cultured with SLA, (50μg/ml) and after 72 hr, the culture supernatant fluids were collected and assayed for IL-4 (A), IFN-γ (B) and IL-10 (C) by ELISA. Also, the infected footpads were homogenized in Triton X and the total arginase activity was determined as described in the materials and methods (C). Data are presented as means ± SE and are representative of 3 independent experiments (n = 3–4 mice) with similar results. *, p < 0.05; **, p < 0.01.
In vitro recall immune response in mice infected with arg− L. major is impaired
The preceding results show that the primary immune response to arg−-L. major is significantly altered in comparison to that of WT L. major. Not only did T cells from arg− L. major-infected mice show marked reduction in antigen-specific proliferation, their ability to produce IFN-γ production was also reduced. Therefore, we examined the quality of recall response in mice infected with arg−L. major in order to assess whether their ability to develop infection-induced immunity may also be altered. At 16 weeks post infection, antigen-specific proliferation of total CD3+ (data not shown) and CD4+ (Fig. 4A) T cells from the dLNs and spleens of arg− L. major-infected mice were significantly (p < 0.05) lower than those from WT-infected mice. In addition, the percentage of proliferating CD4+ T cells that express IFN-γ was also significantly (p < 0.05) lower in dLNs and spleens from arg− L. major-infected than those from WT L. major-infected mice (Fig. 4B). Consistent with the flow data, cells from arg− L. major-infected mice produced significantly (p < 0.01) less IFN-γ in culture supernatant fluids than cells from WT L. major-infected mice (Fig. 4C). In contrast, cells from arg− L. major-infected mice produced significantly (p < 0.05) more IL-10 than those from WT L. major-infected mice (Fig. 4D). Interestingly, co-culture of WT L. major-infected BMDCs with purified T cells from arg− L. major-infected mice did not restore their proliferation and IFN-γ production (Fig. 4E and F), suggesting that the impaired in vitro recall responses of cells from arg−L. major-infected mice is T cell intrinsic and not due to defects in antigen-presenting cells.
Figure 4.

Impaired in vitro recall response in arg− L. major-infected mice. Sixteen weeks after infection, dLN cells from WT and arg− L. major-infected mice were labeled with CFSE dye and restimulated in vitro with SLA (A-D), infected or uninfected bone marrow-derived dendritic cells (E and F) or plate-bound anti-CD3/anti-CD28 mAb (as positive controls) for 5 days as described in materials and methods. CD4+ T cell proliferation (A) and the frequency of proliferating and IFN-γ-producing CD4+ (CFSEloIFN-γ+, B and F) and CD3+ (E) were determined by flow cytometry. In addition, the production of IFN-γ (C) and IL-10 (D) in culture supernatant fluids was determined by sandwich ELISA. Data presented are representative of 3 independent experiments with similar results. *, p < 0.05,; **, p < 0.01.
Impaired in vivo recall immune response in mice infected with arg- L. major
To further rule out the effect of differences in antigen-presenting cells in the impaired recall response in mice infected with arg− L. major, we adoptively transferred highly purified T cells from Thy1.2 (CD90.2) mice infected with WT or arg− L. major into naïve congenic (Thy1.1, CD90.1) recipients and monitored the proliferative behavior of the donor (Thy1.2+) T cells following L. major challenge. Five days after challenge, the proliferation of donor CD3+ (Fig. 5A) or CD4+ (Fig. 5C) T cells from arg− L. major-infected mice was significantly (p < 0.05) lower than those from WT-infected mice. This was true whether donor cells were recovered from the draining lymph nodes or spleens (data not shown) of recipient mice. Furthermore, the frequency of IFN-γ-producing cells within the proliferating donor CD3+ or CD4+ T cells in mice that received cells from WT L. major-infected mice was significantly greater those cells were from arg− L. major-infected mice (Fig. 5B and D, left panels). In contrast, the frequency of IFN-γ negative cells within the proliferating donor cells (i.e. CFSElowIFN-γ −) in recipient mice were comparable, suggesting that only the IFN-γ-producing effectors cells are affected (Fig. 5B and D, right panels). Thus, similar to in vitro recall response, proliferation and IFN-γ production by cells from arg− L. major-infected mice are impaired in vivo following secondary encounter with Leishmania antigen.
Figure 5.

In vivo recall response in arg− L. major-infected mice is impaired. Naïve Thy1.1 (CD90.1) mice received (by i.v. injection) CFSE-labeled highly purified (> 98% pure) splenic T cells (purified by autoMACS) from uninfected (naïve) or WT and arg− L. major-infected (> 16 wk) Thy 1.2 (CD90.2) mice and challenged with 2×106 WT L. major the next day. At 7 days post-challenge, dLNs cells were stained for CD3 (A and B) and CD4 (C and D) and directly assessed for proliferation and IFN-γ production by flow cytometry, after gating on CD90.2+ donor population. Bar charts (B and D) represent the percentages of proliferating cells that are also positive (CFSEloIFN-γ+, left panels) or negative (CFSEloIFN-γ−, right panels) for IFN-γ. Data presented are representative of 2 independent experiments (n = 4–5 mice per group) with similar results. *, p < 0.05
Quantitative differences in memory T cells in mice infected with WT and arg− L. major
To determine whether there are quantitative differences in numbers of memory T cells generated in mice infected with WT and arg− parasites, we collected spleens and dLNs at 16 weeks post-infection and assessed the expression of CD62L and CD44 on CD3+ T cells directly ex vivo by flow cytometry. CD44 is a surface protein that is important for lymphocyte extravasation into inflammatory sites and its upregulation is a marker of previous T cell activation and hence is expressed by all memory T cells (45, 46). CD62L is a lymph node homing receptor for lymphocytes, which allows them to enter the high endothelial venules and it’s downregulation is a prerequisite for effector T cells to exit the lymph nodes into tissues to mediate effector functions. By using these markers, we could discriminate between central memory-like T cells (CD44hiCD62Lhi, Tcm) and effectors and/or effector memory-like T cells (CD44hiCD62Llo, Tem) [206, 210]. Both WT and arg− L. major-infected mice had higher Tcm- and Tem-like populations than naïve uninfected mice (data not shown). Interestingly, there was no significant difference in the percentage of Tcm-like population between WT and arg−-infected mice. However, the percentage of Tem-like cells in arg− L. major-infected mice was significantly (p < 0.05) lower than in WT-infected mice (Fig S2 A–C).
Infection with arg− L. major does protect against secondary virulent challenge
The preceding results suggest that infection with arg− L. major may not induce the same level of protection as WT L. major infection. To test this, we challenged mice infected with WT and arg− L. major at 16 weeks post-infection with virulent parasites in the contra-lateral footpad and at 3 days after challenge, assessed footpad thickness for delayed-type hypersensitivity (DTH) response. As shown in Fig. 6A, mice that healed from WT infections mounted strong DTH response following challenge infection that was significantly (p < 0.01) higher than infected naïve mice. In contrast, DTH response in mice infected with arg− L. major was significantly (p < 0.01) lower than that seen in mice that healed their WT L. major infection. Consistent with this low DTH response, arg− L. major-infected mice contain significantly (p < 0.01) higher parasite burden than their WT-L. major-infected counterpart mice at 3 weeks post-challenge (Fig. 6B). Thus, the resulting chronic and persistent infection generated in C57BL/6 mice by arg− L. major is not sufficient to stimulate protective immunity upon secondary re-challenge infections.
Figure 6.

Infection with arg− L. major does not protect against virulent L. major challenge. Uninfected (naïve) and C57BL/6 mice infected with WT or arg− L. major (> 16 wks) were challenge with 5 million WT parasites in their contralateral footpad and delayed type hypersensitivity (DTH) response was measured 72 hr post-challenge (A). After 3 wk post challenge, mice were sacrificed and parasite burden was determined by limiting dilution (B). In some experiments, CD3+ T cells were purified from spleens of WT or arg− L. major-infected mice and adoptively transferred into naïve mice that were then challenged with virulent L. major. Three weeks after challenge lesion size was determined (C) and mice were sacrificed to determine parasite burden (D). Data presented are representative of 4 (A and B) and 2 (C and D) independent experiments (n = 3–5 mice per group) with similar results. *, p < 0.05; **, p f 0.01.
To more carefully investigate the quality of secondary immunity in mice infected with arg− L. major and to exclude possible defects in non-T cell compartment, we adoptively transferred highly purified CD3+ T cells from spleens of naïve or mice infected with WT and arg− L. major for 16 wk into naive C57BL/6 mice and challenged them with virulent L. major. The lesion size in mice that received cells from WT-infected mice was significantly (p < 0.05) lower than those that received cells from arg− L. major-infected or naïve mice (Fig. 6C). Furthermore, parasite burden in mice that received cells from arg− L. major-infected or naive mice was over a 100-fold higher than those that received cells from WT L. major-infected mice (Fig. 6D). These results suggest that the inability of arg− L. major-infected mice to resist secondary virulent L. major challenge may be related to defects and/or impaired generation of cells that mediate infection-induced immunity (memory T cells) following primary infection.
Clonal exhaustion contributes to impaired T cell response and chronicity of arg− L. major infection in mice
Because arg− L. major-infection causes non-resolving infection, we investigated whether the significantly lower proliferation and effector cytokine (IFN-γ) production by T cells from these mice was related to increased expression of PD-1, a molecule that has been associated with immune cell exhaustion during chronic infections including Leishmania (36, 37, 47–49). As shown in Fig. 7A, CD4+ T cells from spleens and dLNs of mice infected with arg−L. major express significantly higher levels of PD-1 than those from WT L. major-infected mice. Interestingly, there was no difference in the percentage of Annexin-5+ cells (Fig. S3), suggesting that cells from arg− L. major-infected mice were not undergoing more apoptosis than those from WT L. major-infected mice as previously suggested in diffused cutaneous leishmaniasis (50).
Figure 7.

PD-1 expression mediates impaired immunity and disease chronicity in arg− L. major-infection. (A) Increased PD-1 expression on CD4+ T cells from arg−L. major-infected mice. Infected mice were sacrificed at 16 wk post-infection and the expression of PD-1 on splenic and dLN cells was assessed directly ex vivo by flow cytometry after gating on CD3+ cells. Numbers in parenthesis represent the mean fluorescence intensity of CD4+PD+ cells. (B) Blockade of PD-1 reverses impaired proliferation and IFN-γ production in vitro. Splenocytes from mice infected with WT and arg− L. major were labeled with CFSE dye, restimulated in vitro for 5 days with SLA in the presence or absence of anti-PD-1 mAb (10 μg/ml) and proliferation and IFN-γ production by CD4+ cells were assessed by flow cytometry by gating on CD4+ cells. Bar graph shows the percentages of proliferating IFN-γ+ (CFSEloIFN-γ+) cells (C). (D) Blockade of PD-1 interaction leads to resolution of chronic lesion. Mice infected with WT or arg− L. major were treated weekly (starting at week 14, arrow) with anti-PD-1 mAb or rat IgG (1 mg/mouse) and the lesion size was determined weekly. Data presented are representative of 2 independent experiments (n = 3–5 mice per group) with similar results.
To determine whether the observed increased PD-1 expression contributed to impaired proliferative response and lesion chronicity of arg− L. major infection, we assessed proliferative responses of cells from arg− L. major infected mice in the presence of anti-PD-1 mAb in vitro. Anti-PD-1 mAb significantly abolished the impaired CD4+ T cell proliferation and IFN-γ production by cells from arg− L. major-infected mice (Fig 7B and C). Consistent with the flow data, the level of IFN-γ in cell culture supernatant fluids of cells from arg− L. major-infected was similar to those from WT-infected mice (Fig. S4A). In contrast, IL-10 production was significantly downregulated (Fig. S4B). Strikingly, weekly treatment with anti-PD-1 mAb led to complete resolution of footpad lesions in arg− L. major-infected mice (Fig. 7D). Collectively, these results suggest that clonal exhaustion contributes to the poor proliferative and effector function of cells from arg− L. major-infected mice and this may be responsible for their inability to protect against secondary L. major challenge.
Discussion
We recently reported that infection of highly susceptible BALB/c mice with arg− L. major results in attenuated pathology and decreased parasite burden while generating comparable immune response to that seen following WT L. major infection (25). Surprisingly, we found that infection of resistant C57BL/6 mice with arg− L. major resulted in a chronic disease in which lesions developed slowly and non progressively and were maintained for over 16 weeks, several weeks after WT-infected mice were completely healed (Fig. 1A). The failure to completely resolve cutaneous lesion in arg− L. major-infected mice was associated with impaired antigen-specific CD4+ T cell proliferation and IFN-γ production and failure to resist secondary challenge infection. We showed that clonal exhaustion of CD4+ T cells due to increased PD-1 expression was responsible for impaired T cell response and non-healing disease observed in mice infected with arg− L. major. To our knowledge, this is the first demonstration of CD4+ T cell exhaustion in a parasitic infection.
Why does arg− L. major cause a chronic infection in B6 mice? In our previous study in the highly susceptible BALB/c mice, we proposed a “take rations for invasion” model whereby parasite-derived arginase plays a critical role during the early infection process (25). Thus, the de novo polyamine synthesis in Leishmania is of nutritional importance in the early colonization of the hostile macrophage environment. This model explains why the addition of exogenous polyamines to infected cells in vitro increases parasite proliferation but does not fully rescue the defect in arg− L. major (24). As parasites become established in the host cell, immune subversion mechanisms begin to direct the host response to allow a full-blown infection (51–53). In the susceptible BALB/c mice where a detrimental Th2 response predominates, proliferation of arg− L. major may eventually overcome the early nutritional impairment (39). We propose that the significantly lower Th1 response within the first two weeks post-infection in arg− L. major-infected C57BL/6 mice may only weaken parasite proliferation but insufficient to fully activate the immune system for effective parasite control.
Despite harboring comparable parasite burden at the site of infection after 16 weeks, mice with arg− L. major were not protected against virulent challenge. Recovery from primary infection with L. major usually results in concomitant immunity (referred to as infection-induced immunity), which is dependent on persistent parasites (2, 6, 40, 54). Manipulations that result in complete parasite clearance result in loss of infection-induced immunity and susceptibility to secondary challenge (6, 55). We previously reported that L. major parasites lacking the phosphoglycan coats (termed lpg2− L. major) are attenuated in vitro, persist indefinitely (56) and protect against virulent L. major challenge (57). The attenuation in virulence of phosphoglycan deficient parasites is related in part to the induction of skewed early IFN-γ and a concomitant blunted IL-4 and IL-10 responses when compared to WT infection (57, 58). Although infection with arg−L. major results in attenuated pathology, the early immune response was significantly reduced compared to WT-infection. Furthermore antigen-specific proliferation and IFN-γ production were greater in WT-infected mice than in arg−-infected mice and this reduced immune response may be implicated in the deficient memory response and lack of protection against secondary challenge observed in arg− L. major-infected mice. These observations indicate that persistence of parasites by itself is simply not enough for maintenance of immunity against secondary challenge. Consistent with this proposal, a recent report shows that sphingolipid deficient L. major parasites persist in infected mice without causing any overt pathology but fail to induce protection following virulent L. major challenge (59). Interestingly, deficiency of sphingolipids results in blunted T cell responses (59) akin to our observation in arg− L. major-infected mice. Collectively, these findings strongly suggest that the quality and/or magnitude of the early immune response may be critical in shaping the nature of the ensuing memory response.
In order to become memory T cells, naive T cells must first become activated, proliferate and take on effector phenotype upon encountering their cognate antigens (60). Following antigen clearance and the subsequent immune contraction, a small number of these effector T cells acquire memory phenotype and remain in circulation as central and effector memory cells (60). Since the quality and magnitude of memory T cell response is directly proportional to the magnitude of primary response, the primary infection must generate sufficient clonal T cell expansion and contraction to ensure an effective memory T cell response (61). We found that the early immune response (proliferation and IFN-γ production) to arg− L. major was remarkably reduced compared to those induced by WT infection. This defect in early priming response following arg− L. major infection could contribute to impaired memory T cell generation leading to impaired protection upon secondary WT challenge.
A third possibility that might contribute to failure of T cells from arg− L. major-infected mice to protect against secondary virulent challenge is T cell exhaustion, a dysfunction that is characterized by high expression of inhibitory receptors such as PD-1 and poor function at both effector and memory T cells levels (32). T cell exhaustion occurs during many chronic infections (47–49) and is a hallmark of many protozoan diseases including malaria (33), toxoplasmosis (34, 35) and leishmaniasis (36, 37). Exhaustion is characterized by increased expression of PD-1 molecule, a transmembrane receptor of the Ig superfamily that is expressed on thymocytes, mature T and B cells following activation (62, 63). Interaction of PD-1 with its ligand PD-L1 or PD-L2, negatively regulates cytokine production and T cell proliferation. Recently, PD-1-mediated exhaustion of CD8+ T cells has been linked to increased susceptibility to L. donovani (37) and L. mexicana infections in humans (50) and mice (36). Although T-cell exhaustion in leishmaniasis has been described for CD8+ T cells, CD4+ T cell exhaustion has not been demonstrated in this disease. We found that the expression of PD-1 on CD4+ T cells from mice infected with arg− L. major is significantly higher than those infected with WT parasites (Fig. 7A). We propose that the chronic nature of arg- L. major infection in C57BL/6 mice and the concomitant increase in PD-1 expression leads to T cell exhaustion (poor proliferation and IFN-γ production) resulting in impaired effector function and failure to protect against secondary L. major challenge. In line with this, we found that treatment of arg- L. major-infected mice with anti-PD-1 mAb leads complete resolution of chronic cutaneous lesion, increased recall response and resistance to secondary L. major challenge.
We were puzzled by the striking effects of in vivo anti-PD1- mAb treatment given the relatively small (although significant) difference in PD-1 expression on CD4+ T cells from WT and arg− L. major- infected mice. In addition to blocking the interaction between CD4+PD-1+ cells with their ligands (PDL1/PDL2 expressing cells) thereby restoring CD4+ T cell proliferation and cytokine release (Fig. 7B and C), it is possible that this striking outcome (complete resolution of chronic lesion) may also be related to other indirect effects associated with PD-1 signaling. For example, a recent report showed that PD-1 triggering on monocytes enhances IL-10 production by these cells, which in turn inhibits CD4+ T cell response (64). Another report showed that CD4+PD-1+ cells produce large amounts of IL-10, and uses this cytokine to mediate its immunoregulatory properties (65). Since IL-10 is known to enhance susceptibility to cutaneous leishmaniasis (66–68), it is conceivable that anti-PD-1 mAb treatment may globally block IL-10 production by monocytes/macrophages and CD4+PD-1+ cells resulting in enhanced resistance to L. major infection. In line with this, we found that anti-PD-1 mAb treatment significantly reduced IL-10 production by cells from both WT and arg− infected mice (Fig. S4) leading to enhanced proliferation and IFN-γ production (Fig. 7B and C). In addition, in vivo anti-PD-1 mAb treatment was associated with transient increase in swelling of infected footpads in both WT and arg− L. major-infected mice (Fig. 7D), an effect that could be due to increased inflammation resulting from more effector T cell response presumably related in part to decreased IL-10 regulatory activities.
In summary, we have shown that proliferation of arg− L. major is impaired in C57BL/6 mice. Although the early immune response is able to contain the infection, the persistent nature of the parasite results in a chronic, low-level infection resulting in non-resolving (chronic) lesion at the site of infection. We propose that the impaired early immune response to infection with arg− L. major (manifested as decreased proliferation and IFN-γ production), creates a poor priming environment for memory cell generation. This, combined with CD4+ T cell exhaustion due to chronic infection-induced expression of PD-1 molecule, results in failure of T cells from arg− L. major-infected mice to protect against secondary virulent L. major challenge. Intriguingly, these observations show that secondary immunity in L. major-infected mice does not only depend on the presence of persistent parasites. Our studies therefore present an interesting question as to what parasite factors determine a successful memory response capable of controlling L. major infection.
Supplementary Material
Acknowledgments
We thank members of the Parasite Vaccine Development Laboratory for their technical assistance, insightful comments and constructive criticisms.
Abbreviations used in this paper
- arg− L. major
arginase gene deficient Leishmania major
- BMDC
Bone marrow-derived dendritic cell
- CFSE
carboxy fluorescin succinimidyl ester
- DCs
dendritic cells
- dLN
lymph node draining the infection site
- DTH
delayed-type hypersensitivity
- SLA
soluble leishmanial antigen
- Tcm
central memory T cell
- Tem
effector memory T cell
- WT
wild-type
Footnotes
Funding for this study was provided by the Canadian Institutes of Health Research, the Manitoba Health Research Council (to JEU) and National Institutes of Health (AI29103 to SB).
References
- 1.Mendonca MG, de Brito ME, Rodrigues EH, Bandeira V, Jardim ML, Abath FG. Persistence of leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure? J Infect Dis. 2004;189:1018–1023. doi: 10.1086/382135. [DOI] [PubMed] [Google Scholar]
- 2.Okwor I, Uzonna J. Persistent parasites and immunologic memory in cutaneous leishmaniasis: implications for vaccine designs and vaccination strategies. Immunol Res. 2008;41:123–136. doi: 10.1007/s12026-008-8016-2. [DOI] [PubMed] [Google Scholar]
- 3.Scott P, Artis D, Uzonna J, Zaph C. The development of effector and memory T cells in cutaneous leishmaniasis: the implications for vaccine development. Immunol Rev. 2004;201:318–338. doi: 10.1111/j.0105-2896.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 4.Aebischer T, Moody SF, Handman E. Persistence of virulent Leishmania major in murine cutaneous leishmaniasis: a possible hazard for the host. Infect Immun. 1993;61:220–226. doi: 10.1128/iai.61.1.220-226.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uzonna JE, Wei G, Yurkowski D, Bretscher P. Immune elimination of Leishmania major in mice: implications for immune memory, vaccination, and reactivation disease. J Immunol. 2001;167:6967–6974. doi: 10.4049/jimmunol.167.12.6967. [DOI] [PubMed] [Google Scholar]
- 6.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 7.Carnauba D, Jr, Konishi CT, Petri V, Martinez IC, Shimizu L, Pereira-Chioccola VL. Atypical disseminated leishmaniasis similar to post-kala-azar dermal leishmaniasis in a Brazilian AIDS patient infected with Leishmania (Leishmania) infantum chagasi: a case report. Int J Infect Dis. 2009;13:e504–507. doi: 10.1016/j.ijid.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhary RG, Bilimoria FE, Katare SK. Diffuse cutaneous leishmaniasis: co-infection with human immunodeficiency virus (HIV) Indian J Dermatol Venereol Leprol. 2008;74:641–643. doi: 10.4103/0378-6323.45111. [DOI] [PubMed] [Google Scholar]
- 9.Iniesta V, Gomez-Nieto LC, Corraliza I. The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J Exp Med. 2001;193:777–784. doi: 10.1084/jem.193.6.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iniesta V, Gomez-Nieto LC, Molano I, Mohedano A, Carcelen J, Miron C, Alonso C, Corraliza I. Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol. 2002;24:113–118. doi: 10.1046/j.1365-3024.2002.00444.x. [DOI] [PubMed] [Google Scholar]
- 11.Kropf P, Fuentes JM, Fahnrich E, Arpa L, Herath S, Weber V, Soler G, Celada A, Modolell M, Muller I. Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. Faseb J. 2005;19:1000–1002. doi: 10.1096/fj.04-3416fje. [DOI] [PubMed] [Google Scholar]
- 12.Stenger S, Thuring H, Rollinghoff M, Bogdan C. Tissue expression of inducible nitric oxide synthase is closely associated with resistance to Leishmania major. J Exp Med. 1994;180:783–793. doi: 10.1084/jem.180.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. Journal of Immunology. 1998;160:5347–5354. [PubMed] [Google Scholar]
- 14.Modolell M, I, Corraliza M, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25:1101–1104. doi: 10.1002/eji.1830250436. [DOI] [PubMed] [Google Scholar]
- 15.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–3777. [PubMed] [Google Scholar]
- 16.Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun. 1995;206:667–673. doi: 10.1006/bbrc.1995.1094. [DOI] [PubMed] [Google Scholar]
- 17.Kropf P, Herath S, Weber V, Modolell M, Muller I. Factors influencing Leishmania major infection in IL-4-deficient BALB/c mice. Parasite Immunol. 2003;25:439–447. doi: 10.1111/j.1365-3024.2003.00655.x. [DOI] [PubMed] [Google Scholar]
- 18.Kepka-Lenhart D, Mistry SK, Wu G, Morris SM., Jr Arginase I: a limiting factor for nitric oxide and polyamine synthesis by activated macrophages? Am J Physiol Regul Integr Comp Physiol. 2000;279:R2237–2242. doi: 10.1152/ajpregu.2000.279.6.R2237. [DOI] [PubMed] [Google Scholar]
- 19.Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. British Journal of Pharmacology. 2009;157:9. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boucher JL, Custot J, Vadon S, Delaforge M, Lepoivre M, Tenu JP, Yapo A, Mansuy D. N omega-hydroxyl-L-arginine, an intermediate in the L-arginine to nitric oxide pathway, is a strong inhibitor of liver and macrophage arginase. Biochem Biophys Res Commun. 1994;203:1614–1621. doi: 10.1006/bbrc.1994.2371. [DOI] [PubMed] [Google Scholar]
- 21.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 22.Roberts SC, Tancer MJ, Polinsky MR, Gibson KM, Heby O, Ullman B. Arginase plays a pivotal role in polyamine precursor metabolism in Leishmania. Characterization of gene deletion mutants. J Biol Chem. 2004;279:23668–23678. doi: 10.1074/jbc.M402042200. [DOI] [PubMed] [Google Scholar]
- 23.da Silva ER, Castilho TM, Pioker FC, de Paula Silva C, Floeter-Winter LM. Genomic organization and transcription characterization of the gene encoding Leishmania (Leishmania) amazonensis arginase and its protein structure prediction. International Journal of Parasitology. 2002;32:11. doi: 10.1016/s0020-7519(02)00002-4. [DOI] [PubMed] [Google Scholar]
- 24.Gaur U, Roberts SC, Dalvi RP, Corraliza I, Ullman B, Wilson ME. An effect of parasite-encoded arginase on the outcome of murine cutaneous leishmaniasis. J Immunol. 2007;179:8446–8453. doi: 10.4049/jimmunol.179.12.8446. [DOI] [PubMed] [Google Scholar]
- 25.Muleme HM, Reguera RM, Berard A, Azinwi R, Jia P, Okwor IB, Beverley S, Uzonna JE. Infection with arginase-deficient Leishmania major reveals a parasite number-dependent and cytokine-independent regulation of host cellular arginase activity and disease pathogenesis. J Immunol. 2009;183:8068–8076. doi: 10.4049/jimmunol.0803979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modolell M, Choi BS, Ryan RO, Hancock M, Titus RG, Abebe T, Hailu A, Muller I, Rogers ME, Bangham CR, Munder M, Kropf P. Local suppression of T cell responses by arginase-induced L-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis. 2009;3:e480. doi: 10.1371/journal.pntd.0000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109:1568–1573. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vercelli D. Arginase: marker, effector, or candidate gene for asthma? J Clin Invest. 2003;111:1815–1817. doi: 10.1172/JCI18908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruch-Gerharz D, Schnorr O, Suschek C, Beck KF, Pfeilschifter J, Ruzicka T, Kolb-Bachofen V. Arginase 1 overexpression in psoriasis: limitation of inducible nitric oxide synthase activity as a molecular mechanism for keratinocyte hyperproliferation. Am J Pathol. 2003;162:203–211. doi: 10.1016/S0002-9440(10)63811-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zea AH, Culotta KS, Ali J, Mason C, Park HJ, Zabaleta J, Garcia LF, Ochoa AC. Decreased expression of CD3zeta and nuclear transcription factor kappa B in patients with pulmonary tuberculosis: potential mechanisms and reversibility with treatment. J Infect Dis. 2006;194:1385–1393. doi: 10.1086/508200. [DOI] [PubMed] [Google Scholar]
- 31.Fu H, Khan A, Coe D, Zaher S, Chai JG, Kropf P, Muller I, Larkin DF, George AJ. Arginine depletion as a mechanism for the immune privilege of corneal allografts. Eur J Immunol. 2011;41:2997–3005. doi: 10.1002/eji.201141683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 33.Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, Waldschmidt TJ, Crompton PD, Harty JT. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2012;13:188–195. doi: 10.1038/ni.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gigley JP, Bhadra R, Khan IA. CD8 T Cells and Toxoplasma gondii: A New Paradigm. J Parasitol Res. 2011;2011:243796. doi: 10.1155/2011/243796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhadra R, Gigley JP, Weiss LM, Khan IA. Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc Natl Acad Sci U S A. 2011;108:9196–9201. doi: 10.1073/pnas.1015298108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang SC, Greenwald RJ, Latchman YE, Rosas L, Satoskar A, Freeman GJ, Sharpe AH. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur J Immunol. 2006;36:58–64. doi: 10.1002/eji.200535458. [DOI] [PubMed] [Google Scholar]
- 37.Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stager S. B7-H1 blockade increases survival of dysfunctional CD8(+) T cells and confers protection against Leishmania donovani infections. PLoS Pathog. 2009;5:e1000431. doi: 10.1371/journal.ppat.1000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hernandez-Ruiz J, Salaiza-Suazo N, Carrada G, Escoto S, Ruiz-Remigio A, Rosenstein Y, Zentella A, Becker I. CD8 cells of patients with diffuse cutaneous leishmaniasis display functional exhaustion: the latter is reversed, in vitro, by TLR2 agonists. PLoS Negl Trop Dis. 2010;4:e871. doi: 10.1371/journal.pntd.0000871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reguera RM, Balana-Fouce R, Showalter M, Hickerson S, Beverley SM. Leishmania major lacking arginase (ARG) are auxotrophic for polyamines but retain infectivity to susceptible BALB/c mice. Mol Biochem Parasitol. 2009;165:48–56. doi: 10.1016/j.molbiopara.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaph C, Uzonna J, Beverley SM, Scott P. Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat Med. 2004;10:1104–1110. doi: 10.1038/nm1108. [DOI] [PubMed] [Google Scholar]
- 41.Uzonna JE, Joyce KL, Scott P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med. 2004;199:1559–1566. doi: 10.1084/jem.20040172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buffet PA, Sulahian A, Garin YJ, Nassar N, Derouin F. Culture microtitration: a sensitive method for quantifying Leishmania infantum in tissues of infected mice. Antimicrob Agents Chemother. 1995;39:2167–2168. doi: 10.1128/aac.39.9.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Titus RG, Marchand M, Boon T, Louis JA. A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunology. 1985;7:545–555. doi: 10.1111/j.1365-3024.1985.tb00098.x. [DOI] [PubMed] [Google Scholar]
- 44.Corraliza IM, Campo ML, Soler G, Modolell M. Determination of arginase activity in macrophages: a micromethod. J Immunol Methods. 1994;174:231–235. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 45.DeGrendele HC, Estess P, Picker LJ, Siegelman MH. CD44 and its ligand hyaluronate mediate rolling under physiologic flow: a novel lymphocyte-endothelial cell primary adhesion pathway. J Exp Med. 1996;183:1119–1130. doi: 10.1084/jem.183.3.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeGrendele HC, Estess P, Siegelman MH. Requirement for CD44 in activated T cell extravasation into an inflammatory site. Science. 1997;278:672–675. doi: 10.1126/science.278.5338.672. [DOI] [PubMed] [Google Scholar]
- 47.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 48.Smith P, Walsh CM, Mangan NE, Fallon RE, Sayers JR, McKenzie AN, Fallon PG. Schistosoma mansoni worms induce anergy of T cells via selective up-regulation of programmed death ligand 1 on macrophages. J Immunol. 2004;173:1240–1248. doi: 10.4049/jimmunol.173.2.1240. [DOI] [PubMed] [Google Scholar]
- 49.Jurado JO, I, Alvarez B, Pasquinelli V, Martinez GJ, Quiroga MF, Abbate E, Musella RM, Chuluyan HE, Garcia VE. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol. 2008;181:116–125. doi: 10.4049/jimmunol.181.1.116. [DOI] [PubMed] [Google Scholar]
- 50.Hernandez-Ruiz J, Becker I. CD8+ cytotoxic lymphocytes in cutaneous leishmaniasis. Salud Publica Mex. 2006;48:430–439. doi: 10.1590/s0036-36342006000500009. [DOI] [PubMed] [Google Scholar]
- 51.Kane MM, Mosser DM. Leishmania parasites and their ploys to disrupt macrophage activation. Curr Opin Hematol. 2000;7:26–31. doi: 10.1097/00062752-200001000-00006. [DOI] [PubMed] [Google Scholar]
- 52.Carrera L, Gazzinelli RT, Badolato R, Hieny S, Muller W, Kuhn R, Sacks DL. Leishmania promastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. J Exp Med. 1996;183:515–526. doi: 10.1084/jem.183.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mosser DM, Karp CL. Receptor mediated subversion of macrophage cytokine production by intracellular pathogens. Curr Opin Immunol. 1999;11:406–411. doi: 10.1016/s0952-7915(99)80068-5. [DOI] [PubMed] [Google Scholar]
- 54.Kebaier C, Uzonna JE, Beverley SM, Scott P. Immunization with persistent attenuated Delta lpg2 Leishmania major parasites requires adjuvant to provide protective immunity in C57BL/6 mice. Infect Immun. 2006;74:777–780. doi: 10.1128/IAI.74.1.777-780.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okwor I, Kuriakose S, Uzonna J. Repeated inoculation of killed Leishmania major induces durable immune response that protects mice against virulent challenge. Vaccine. 2010;28:5451–5457. doi: 10.1016/j.vaccine.2010.05.077. [DOI] [PubMed] [Google Scholar]
- 56.Spath GF, Lye LF, Segawa H, Sacks DL, Turco SJ, Beverley SM. Persistence Without Pathology in Phosphoglycan-Deficient Leishmania major. Science. 2003;301:1241–1243. doi: 10.1126/science.1087499. [DOI] [PubMed] [Google Scholar]
- 57.Uzonna JE, Spath GF, Beverley SM, Scott P. Vaccination with phosphoglycan-deficient Leishmania major protects highly susceptible mice from virulent challenge without inducing a strong Th1 response. J Immunol. 2004;172:3793–3797. doi: 10.4049/jimmunol.172.6.3793. [DOI] [PubMed] [Google Scholar]
- 58.Liu D, Kebaier C, Pakpour N, Capul AA, Beverley SM, Scott P, Uzonna JE. Leishmania major phosphoglycans influence the host early immune response by modulating dendritic cell functions. Infect Immun. 2009;77:3272–3283. doi: 10.1128/IAI.01447-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu W, Xin L, Soong L, Zhang K. Sphingolipid degradation by Leishmania major is required for its resistance to acidic pH in the mammalian host. Infect Immun. 2011;79:3377–3387. doi: 10.1128/IAI.00037-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 61.Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. 2007;19:309–314. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 63.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 64.Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, Hill BJ, Noto A, Ancuta P, Peretz Y, Fonseca SG, Van Grevenynghe J, Boulassel MR, Bruneau J, Shoukry NH, Routy JP, Douek DC, Haddad EK, Sekaly RP. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. 2010;16:452–459. doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meng Q, Yang P, Li B, Zhou H, Huang X, Zhu L, Ren Y, Kijlstra A. CD4+PD-1+ T cells acting as regulatory cells during the induction of anterior chamber-associated immune deviation. Invest Ophthalmol Vis Sci. 2006;47:4444–4452. doi: 10.1167/iovs.06-0201. [DOI] [PubMed] [Google Scholar]
- 66.Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol. 2001;166:1141–1147. doi: 10.4049/jimmunol.166.2.1141. [DOI] [PubMed] [Google Scholar]
- 67.Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, Sacks DL. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med. 2001;194:1497–1506. doi: 10.1084/jem.194.10.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Noben-Trauth N, Lira R, Nagase H, Paul WE, Sacks DL. The relative contribution of IL-4 receptor signaling and IL-10 to susceptibility to Leishmania major. J Immunol. 2003;170:5152–5158. doi: 10.4049/jimmunol.170.10.5152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.