

Abstract
Hydrogels actuators (HAs) that can reversibly respond to stimuli have applications in diverse fields. However, faster response rates and improved control over actuation timing and location are required to fulfill their potential. To address these criteria, we synthesized near-infrared light-driven HAs by interfacing genetically engineered elastin-like polypeptides with reduced-graphene oxide sheets. The resulting nanocomposites exhibited rapid and tunable motions controlled by light position, intensity, and path, including finger-like flexing and crawling. This work demonstrates the ability of rationally designed proteins to be combined with synthetic nanoparticles for the creation of macroscale functional materials.
Keywords: Elastin-like polypeptides, Graphene, Actuators, Hydrogels, Composites
Hydrogel actuators (HAs) are water swollen polymer networks that reversibly change their dimensions or shapes when external stimuli (e.g., solvent composition,1,2 temperature,1,3 and external fields4,5) cause local or global changes in network swelling. HAs with tunable speed and motion are highly sought after to fulfill applications in various fields, including biology,6 medicine,7,8 microfluidics,2,9 and robotics.10,11 To fulfill the bio-related applications, HAs which utilize materials and stimuli that are compatible with biomolecules and cells are also desired.
Light-driven HAs are particularly attractive systems but have limitations that must be addressed to expand their utility. Light is applied remotely, so solution-wide changes and the introduction of potentially invasive wires or electrodes are avoided.1,5 In addition, light is easily controlled with higher spatial and temporal resolution than other stimuli.12 Light drives actuation by causing the photoisomerization/ionization of chromophoresor by photothermal heating of chromophores or nanoparticles in thermoresponsive matrices.4,9,12–15 Examples of the former strategy were limited by slow actuation kinetics,16 the requirement for specific solution conditions17, or the use of potentially damaging ultraviolet wave lengths.12,16 The latter strategy is potentially better-suited to biological applications because it can use near infrared (nIR) wavelengths that are not significantly absorbed by most biomaterials.14 However, macroscale, photothermal HAs exhibit slow actuation kinetics (on the order of minutes)9,15,18, due to limited rates of water diffusion into and out of the networks19. In addition, previous photothermal HAs had isotropic structures that changed size relatively evenly in each dimension, making non-linear motions difficult to achieve.4,9,15 In contrast, actuation into more complex shapes can be achieved by creating hydrogel networks with anisotropic composition,1,20 crosslinking,21 or porosity.3 With these limitations in mind and with an eye towards future bio-related applications, we created light-driven HAs that could rapidly undergo non-linear motions by combining reduced graphene oxide (rGO) nanosheets and elastin-like polypeptides (ELPs).
rGO sheets are derived from the oxidation, exfoliation and subsequent reduction of graphite. They show great potential due to their unique combination of mechanical, electrical, and optical properties22. rGO efficiently generates heat by absorbing nIR light. Its photothermal efficiency is better than unreduced GO and it is easier to prepare than pristine graphene.14 Although still under investigation, rGO has also shown promising biocompatibility.23 The challenge of working with rGO in aqueous environments is that its hydrophobic nature makes it prone to aggregation without functionalization strategies.24
ELPs are recombinantly produced protein-based polymers (PBPs) based upon repeating sequences of the pentapeptide, Val-Pro-Gly-Val-Gly, a motif found in mammalian tropoelastin.25 ELPs, like all PBPs, have distinct advantages compared to chemically synthesized polymers and natural proteins26,27 typically used for creating hydrogels. Namely, we can modularly encode functional motifs of interest into their backbones using genetic engineering.28 This allows for fine control over their self-assembly;29,30 mechanical properties;31 and interactions with cells,32,33 molecules,34 ions,35 and surfaces.36 We chose ELPs, in particular, because they possess tunable thermoresponsiveness in aqueous solutions;25 below a transition temperature (Tt) they are soluble, and above it they phase separate. When crosslinked to form hydrogels, this inverse temperature transition (ITT) results in large changes in swelling over a broad range of temperatures.31 Furthermore, elastomeric proteins such as ELPs can undergo large, elastic deformations with little loss of energy.37 Finally, ELPs have exhibited excellent biocompatibility.38,39
Our three-step strategy for creating ELP-rGO composite HAs is shown schematically in Figure 1. First, we functionalized rGO with a rationally designed ELP to create hybrid nanoparticles (Figure 1a). Second, we crosslinked the hybrid nanoparticles into an ELP-based network in the presence of water vapor to create an anisotropic microstructure (Figure 1b). Third, we irradiated the hydrogels with nIR light to locally shrink the ELP network and induce bending motions (Figure 1c).
Figure 1.
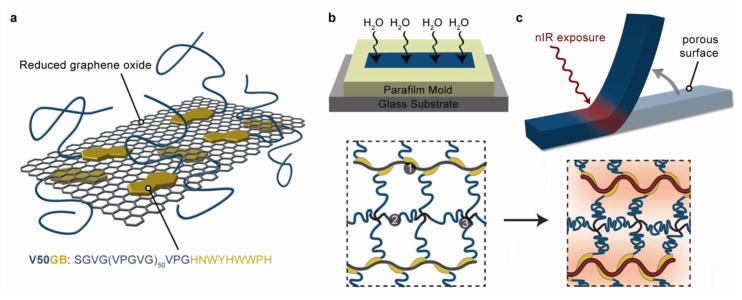
Strategy for creating ELP-rGO composite hydrogel actuators.(a) Schematic of V50GB binding to an idealized rGO sheet via the graphene-binding motif. The elastin-like and graphene-binding peptide sequences are shown. (b) Schematics of our method to introduce anisotropic porosity using water vapor exposure during the crosslinking process and of the resulting hydrogel network composed of (1) V50GB bound to rGO, (2) V50CK1 and (3) 4-arm crosslinkers. (c) Schematics of site-specific nIR-initiated bending of an anisotropic hydrogel and the corresponding local deswelling of the gel network.
We created a 50 pentapeptide long ELP, V50GB to functionalize the rGO sheets. V50GB displays a short graphene-binding (GB) peptide at its C-terminus to promote anchoring to rGO surfaces (Figure 1b, protein sequences in Supporting Information, Table S1). The GB sequence (HNWYHWWPH) was originally discovered by phage display against CNTs40 but we reasoned that it would also have affinity to hydrophobic rGO surfaces due to the presence of several hydrophobic, aromatic amino acid residues. It has the advantage of being much smaller than whole proteins previously used to functionalize graphene-based materials.24,41 We used bacterial expression to synthesize the ELP then verified its molecular weight and monodispersity by mass spectrometry (Supporting Information, Figure S1), and its thermoresponsiveness by measuring temperature-dependent light absorbance (Supporting Information, Figure S2).
V50GB bound to rGO sheets and modified their surface and colloidal properties. We confirmed binding by atomic force microscopy (AFM) imaging before and after ELP exposure (Figure 2a,b). The AFM height traces show a uniform increase in rGO thickness from ~1 nm to ~4 nm after V50GB exposure. The control ELP, V50, also increased the thickness but only by ~1 nm. Based on further solution binding studies, rGO was found to bind ~6 times its mass in V50GB (Supporting Information, Figure S3). V50GB-modified rGO nanoparticles were much less prone to aggregation in response to increased ionic strengths, and decreased pH, while V50-modifed rGO provided only modest changes (Supporting Information, Table S2). In addition, we could dry the hybrid V50GB-rGO nanoparticles and redisperse them in good organic solvents for ELPs, such as dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), and N-Methyl-2-pyrrolidone (NMP), at concentrations higher than achievable for rGO alone (Supporting Information, Figure S4).42 These results show that the ELP chains effectively suppress rGO aggregation when immobilized via the GB motif.
Figure 2.

Effects of ELP binding to rGO. (a) AFM images (2 Jm × 5 Jm) of rGO sheets (top), V50-treated rGO (middle) and V50GB-treated rGO (bottom). (b) AFM height profiles from (a) show the height increase due to ELP binding. (c) Schematic of hybrid nanoparticles going from a dispersed to aggregated state in response to temperature or nIR-light due to the ITT. (d) Images of reversible temperature induced aggregation of V50GB-treated rGO. (e) Images of aggregation induced by exposure to an 808 nm nIR laser. Aggregation occurred above the laser spot site (white asterisk) due to convection.
In terms of stimuli-responsiveness, we found that the thermoresponsive behavior of the ELPs persisted after binding to rGO. Hybrid nanoparticles heated and then cooled around the Tt reversibly aggregated and redispersed (Figure 2c–d). Aggregation also occurred when the hybrids were irradiated with an 808 nm nIR laser (Figure 2e), which demonstrated successful coupling of the photo and thermal responses of rGO and ELPs, respectively.
We fabricated ELP-rGO nanocomposite hydrogels with anisotropic porosity. We mixed V50GB-rGO hybrid nanoparticles; V50CK1, an ELP having one primary amine at its N-terminal and one near its C-terminus (characterization and sequence in Supplementary Information); and N-hydroxysuccinimide (NHS) ester-functionalized 4-armed poly (ethylene glycol) molecules in DMSO/DMF (ELPs are not thermoresponsive in water-free solvents). In this mixture, crosslinks form from the reaction of the primary amines on the ELPs with the NHS esters (Figure 1b). This method allows for seamless incorporation of the rGO sheets and may suppress their segregation from the surrounding matrix after synthesis.43 To introduce anisotropic porosity, we dispensed the pre-gel mixtures into glass-bottom molds and exposed the top surface to water vapor as it crosslinked at 37 °C (Figure 1b). Water absorption allowed an ITT-induced phase separation to occur and resulted in a localized porous structure at the exposed surface (Figure 3a–c). The porous region accounted for approximately 20% of each gel’s thickness but absorbs over four times more water per unit of dry mass than the non-porous region (Supporting Information, Table S3). Water could diffuse more quickly into and out of the gel at the porous surface. Consequently, the entire gel curled in response to solution-wide heating and cooling due to faster deswelling and swelling of the porous layer, respectively (Figure 3d, Supporting Information, Movie S1).
Figure 3.

Microstructure and consequent behavior of ELP-rGO nanocomposite hydrogel actuators.(a) Cross-sectional SEM image showing the sharp microstructural contrast between the top and bulk of the hydrogel. (b) SEM images of the highly porous top surface and (c) relatively non-porous bottom surface. (d) Images of an anisotropic hydrogel reversibly curling in response to cooling or heating of the surrounding solution.
While whole hydrogel curling resulted from changes in solution temperature, site-specific, hinge-like bending occurred in response to nIR-laser irradiation (Figure 4a, Supporting Information, Movie S2). The hydrogels always bent such that the porous surface was on the concave side, regardless of which surface was exposed to the beam. We determined that bending rates and angles increased with increasing laser intensity and rGO concentration by analyzing videos of bending hydrogels (Figure 4b–c). The gels unbent upon removal of the nIR light, recovering ~74–84% within 10 seconds (Figure S5). We also followed the trajectories of the hydrogel during 100 repeated cycles of bending and unbending and found that there was little change over the course of the experiment (Supporting Information, Figure S6). Similar motions were not observed in composite hydrogels synthesized with isotropic networks. For such gels, even after one minute of nIR irradiation, only small changes in shape were evident (Supporting Information, Movie S3). This is consistent with the slow kinetics observed with previous macroscale photothermally-driven hydrogels.15,18 Therefore, introducing porosity using our vapor diffusion method was critical to achieving photomechanical responses that do not require extended heating times.
Figure 4.

Light-controlled bending of ELP-rGO composite hydrogels.(a) Images of site-specific nIR-induced bending of an anisotropic hydrogel. Measurement of bending angle, θ, as a function of exposure time, t. θ(0) = ∠LOL = 0. θ(m) = ∠LOM. θ(n) = ∠LON.(b) Change in bending angle in response to nIR laser exposure for hydrogels with varying rGO concentrations. Weight percent values correspond to the mass of V50GB-rGO hybrids divided by the total mass of V50GB-rGO hybrids, V50CK1, and 4-arm PEG crosslinker. (Error bars represent one standard deviation from the mean).(c) Change in bending angle of 2.6 wt% gels in response to nIR laser exposure of different power densities.
We created nanocomposite HAs that exhibit diverse mechanical motions by controlling their shape and surface patterns and by modulating laser positioning, timing, and movement. As a demonstration, we patterned a hand-shaped matrix (Figure 5a) and sequentially irradiated its “fingers” to create joint-like flexing motions (Supporting Information, Movie S4). We could bend the fingers in arbitrary order and any position along each finger could serve as a “joint”. Therefore, unlike previous polymeric actuators, bending positions did not need to be predetermined and fixed during the synthesis process.44 In addition to bending site, bending directions could also be manipulated. By rastering the laser in a line across a hydrogel in different directions, we could influence the resulting bending axis (Figure 5b, Supporting Information, Movie S5). We also showed that the preferred bending direction could be changed by surface patterning. Specifically, we blocked water vapor from entering specific regions of our molds which made those regions stiffer than the exposed regions (Supporting Information, Figure S7a). Typically, the bending axis corresponded to a hydrogel’s shorter dimension at the irradiation site; however, in our patterned gels, the increased stiffness of the blocked regions was sufficient to make bending along the longer dimension more favorable (Supporting Information, Figure S7b).
Figure 5.
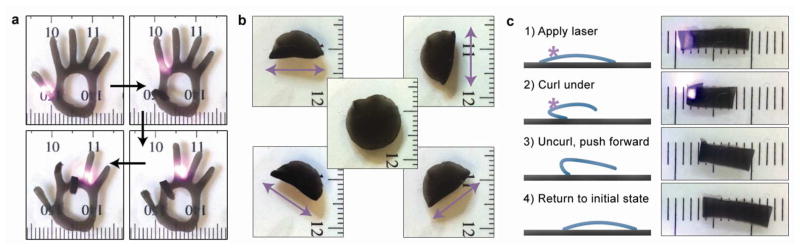
Controlled motion of hydrogel actuators. (a) Images of the fingers of a hand-shaped hydrogel bending and unbending in response to the location of a nIR laser spot. (b) Images of the laser raster direction-dependent folding axis of a circular composite hydrogel (center). The raster directions are indicated by double-headed arrows (1 mm tick marks). (c) Schematic and images of a light-driven crawler. A hydrogel molded with a slight curvature is placed with porous side facing down. The laser is applied so as to induce gel curling. Subsequent uncurling during recovery after the laser is removed pushes the gel forward (1 mm tick marks).
We created light-driven crawlers by exploiting the photomechanical response of the ELP-rGO composite hydrogels (Figure 5c and Supporting Movie S6). Hydrogels were synthesized on curved substrates to create arch-shaped gels (vapor exposed face on concave side). After crosslinking and exchange into water, we placed them porous side down onto glass slides. We initiated each stepping cycle by exposing the “rear” edge of the hydrogel to the nIR laser. This caused the rear edge to fold underneath, at which point we stopped the laser. Afterwards the front edge rose and the back edge generated a forward-directed force by pushing against the glass as it uncurled. This caused the entire gel to move forward ~3 mm each cycle. Unlike previous motile hydrogels, our hydrogels do not require additional chemical reactants nor ratcheted substrates.5,20
In summary, we showed that rationally designed ELPs could be used to non-covalently functionalize and regulate the colloidal properties of rGO. The modified rGO nanosheets were seamlessly integrated into an isotropically porous ELP-based hydrogels, and these hydrogels showed rapid, reversible and tunable bending motions at specific positions that could be determined on the fly. More broadly, our actuators demonstrate the synergistic properties that can be obtained by rationally combining PBPs with synthetic nanomaterials. The control of PBP properties afforded by genetic engineering will allow this strategy to be applied to other nanoparticles of interest and for simple modular incorporation of other functional peptides, such as different graphene-binding sequences45 or specific cell/molecule binding motifs.28 Therefore, a wide variety of technological applications can be explored in the future, including light-controlled dynamic cell-culture, drug delivery, microfluidics, and soft robotics.
Supplementary Material
Acknowledgments
This work was supported by the U. S. Army Engineering Research Development Center (W912HZ-11-2-0047), National Science Foundation Center of Integrated Nanomechanical Systems (EEC-0832819), and NIH ARRA supplement to an NIDCR R21 grant (DE 018360-02). The authors thank Dr. Jin-Woo Oh and Dong Shin Choi for helping with laser setup and video capture.
ABBREVIATIONS
- HA
hydrogel actuator
- nIR
near infrared
- rGO
reduced graphene oxide
- ELP
elastin-like polypeptide
- PBP
protein-based polymer
- Tt
transition temperature
- ITT
inverse temperature transition
- GB
graphene-binding
- AFM
atomic force microscopy
- DMSO
dimethyl sulfoxide
- DMF
N,N-Dimethylformamide (DMF)
- NMP
N-Methyl-2-pyrrolidone
- NHS
N-hydroxysuccinimide
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ASSOCIATED CONTENT
Supporting Information. ELP sequences, characterization, and binding data; hybrid nanoparticle colloidal stability and organic solvent dispersability data; hydrogel fatigue and patterning data; hydrogel actuation movies; and detailed experimental methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hu Z, Zhang X, Li Y. Science. 1995;269:525–527. doi: 10.1126/science.269.5223.525. [DOI] [PubMed] [Google Scholar]
- 2.Beebe DJ, Moore JS, Bauer JM, Yu Q, Liu RH, Devadoss C, Jo BH. Nature. 2000;404:588–590. doi: 10.1038/35007047. [DOI] [PubMed] [Google Scholar]
- 3.Asoh T-a, Matsusaki M, Kaneko T, Akashi M. Adv Mater. 2008;20:2080–2083. [Google Scholar]
- 4.Fujigaya T, Morimoto T, Niidome Y, Nakashima N. Adv Mater. 2008;20:3610–3614. [Google Scholar]
- 5.Osada Y, Okuzaki H, Hori H. Nature. 1992;355:242–244. [Google Scholar]
- 6.Pelah A, Seemann R, Jovin TM. J Am Chem Soc. 2006;129:468–469. doi: 10.1021/ja067171+. [DOI] [PubMed] [Google Scholar]
- 7.Hoare T, Timko BP, Santamaria J, Goya GF, Irusta S, Lau S, Stefanescu CF, Lin D, Langer R, Kohane DS. Nano Lett. 2011;11:1395–1400. doi: 10.1021/nl200494t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X, Kim J, Cezar CA, Huebsch N, Lee K, Bouhadir K, Mooney DJ. Proc Natl Acad Sci U S A. 2011;108:67–72. doi: 10.1073/pnas.1007862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sershen SR, Mensing GA, Ng M, Halas NJ, Beebe DJ, West JL. Adv Mater. 2005;17:1366–1368. doi: 10.1002/adma.200401239. [DOI] [PubMed] [Google Scholar]
- 10.Chan V, Park K, Collens MB, Kong H, Saif TA, Bashir R. Sci Rep. 2012:2. doi: 10.1038/srep00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bassik N, Abebe BT, Laflin KE, Gracias DH. Polymer. 2010;51:6093–6098. [Google Scholar]
- 12.Tomatsu I, Peng K, Kros A. Adv Drug Delivery Rev. 2011;63:1257–1266. doi: 10.1016/j.addr.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki A, Tanaka T. Nature. 1990;346:345–347. [Google Scholar]
- 14.Robinson JT, Tabakman SM, Liang Y, Wang H, Casalongue HS, Vinh D, Dai H. J Am Chem Soc. 2011;133:6825–6831. doi: 10.1021/ja2010175. [DOI] [PubMed] [Google Scholar]
- 15.Lo CW, Zhu D, Jiang H. Soft Matter. 2011;7:5604. [Google Scholar]
- 16.Takashima Y, Hatanaka S, Otsubo M, Nakahata M, Kakuta T, Hashidzume A, Yamaguchi H, Harada A. Nat Commun. 2012;3:1270. doi: 10.1038/ncomms2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satoh T, Sumaru K, Takagi T, Kanamori T. Soft Matter. 2011;7:8030–8034. doi: 10.1039/c8sm00865e. [DOI] [PubMed] [Google Scholar]
- 18.Zhu CH, Lu Y, Peng J, Chen JF, Yu SH. Adv Funct Mater. 2012;22:4017–4022. [Google Scholar]
- 19.Imran AB, Seki T, Takeoka Y. Polym J. 2010;42:839–851. [Google Scholar]
- 20.Maeda S, Hara Y, Sakai T, Yoshida R, Hashimoto S. Adv Mater. 2007;19:3480–3484. [Google Scholar]
- 21.Kim J, Hanna JA, Byun M, Santangelo CD, Hayward RC. Science. 2012;335:1201–1205. doi: 10.1126/science.1215309. [DOI] [PubMed] [Google Scholar]
- 22.Huang X, Yin Z, Wu S, Qi X, He Q, Zhang Q, Yan Q, Boey F, Zhang H. Small. 2011;7:1876–1902. doi: 10.1002/smll.201002009. [DOI] [PubMed] [Google Scholar]
- 23.Jastrzebska AM, Kurtycz P, Olszyna AR. J Nanopart Res. 2012;14 doi: 10.1007/s11051-012-1320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C, Adamcik J, Mezzenga R. Nature Nanotech. 2012;7:421–427. doi: 10.1038/nnano.2012.62. [DOI] [PubMed] [Google Scholar]
- 25.Urry DW. J Phys Chem B. 1997;101:11007–11028. [Google Scholar]
- 26.Ehrick JD, Deo SK, Browning TW, Bachas LG, Madou MJ, Daunert S. Nature Mat. 2005;4:298–302. doi: 10.1038/nmat1352. [DOI] [PubMed] [Google Scholar]
- 27.Miyata T, Asami N, Uragami T. Nature. 1999;399:766–769. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 28.DiMarco RL, Heilshorn SC. Adv Mater. 2012;24:3923–3940. doi: 10.1002/adma.201200051. [DOI] [PubMed] [Google Scholar]
- 29.Yu SM, Conticello VP, Zhang G, Kayser C, Fournier MJ, Mason TL, Tirrell DA. Nature. 1997;389:167–170. doi: 10.1038/38254. [DOI] [PubMed] [Google Scholar]
- 30.MacKay AJ, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Nature Mat. 2009;8:993–999. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trabbic-Carlson K, Setton LA, Chilkoti A. Biomacromolecules. 2003;4:572–580. doi: 10.1021/bm025671z. [DOI] [PubMed] [Google Scholar]
- 32.Girotti A, Reguera J, Rodríguez-Cabello J, Arias F, Alonso M, Testera A. J Mater Sci: Mater Med. 2004;15:479–484. doi: 10.1023/b:jmsm.0000021124.58688.7a. [DOI] [PubMed] [Google Scholar]
- 33.Charati MB, Ifkovits JL, Burdick JA, Linhardt JG, Kiick KL. Soft Matter. 2009;5:3412–3416. doi: 10.1039/b910980c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Straley KS, Heilshorn SC. Adv Mater. 2009;21:4148–4152. [Google Scholar]
- 35.Currie HA, Deschaume O, Naik RR, Perry CC, Kaplan DL. Adv Funct Mater. 2011;21:2889–2895. doi: 10.1002/adfm.201100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang E, Lee SH, Lee SW. Biomacromolecules. 2011;12:672–680. doi: 10.1021/bm101322m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Eldijk MB, McGann CL, Kiick KL, van Hest JC. Top Curr Chem. 2012;310:71–116. doi: 10.1007/128_2011_205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urry DW, Parker TM, Reid MC, Gowda DC. J Bioact Compat Polym. 1991;6:263–282. [Google Scholar]
- 39.Sallach RE, Cui WX, Balderrama F, Martinez AW, Wen J, Haller CA, Taylor JV, Wright ER, Long RC, Chaikof EL. Biomaterials. 2010;31:779–791. doi: 10.1016/j.biomaterials.2009.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Humphreys ES, Chung SY, Delduco DF, Lustig SR, Wang H, Parker KN, Rizzo NW, Subramoney S, Chiang YM, Jagota A. Nature Mat. 2003;2:196–200. doi: 10.1038/nmat833. [DOI] [PubMed] [Google Scholar]
- 41.Laaksonen P, Kainlauri M, Laaksonen T, Shchepetov A, Jiang H, Ahopelto J, Linder MB. Angew Chem, Int Ed. 2010;49:4946–4949. doi: 10.1002/anie.201001806. [DOI] [PubMed] [Google Scholar]
- 42.Park S, An J, Jung I, Piner RD, An SJ, Li X, Velamakanni A, Ruoff RS. Nano Lett. 2009;9:1593–1597. doi: 10.1021/nl803798y. [DOI] [PubMed] [Google Scholar]
- 43.Haberl JM, Sánchez-Ferrer A, Mihut AM, Dietsch H, Hirt AM, Mezzenga R. Adv Mater. 2013 doi: 10.1002/adma.201204406. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Pint CL, Lee MH, Schubert BE, Jamshidi A, Takei K, Ko H, Gillies A, Bardhan R, Urban JJ, Wu M, Fearing R, Javey A. Nano Lett. 2011;11:3239–3244. doi: 10.1021/nl201503e. [DOI] [PubMed] [Google Scholar]
- 45.Cui Y, Kim SN, Jones SE, Wissler LL, Naik RR, McAlpine MC. Nano Lett. 2010;10:4559–4565. doi: 10.1021/nl102564d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.