


Abstract
When DNA replication is stalled at sites of DNA damage, a cascade of responses is activated in the cell to halt cell cycle progression and promote DNA repair. A pathway initiated by the kinase Ataxia teleangiectasia and Rad3 related (ATR) and its partner ATR interacting protein (ATRIP) plays an important role in this response. The Fanconi anemia (FA) pathway is also activated following genomic stress, and defects in this pathway cause a cancer-prone hematologic disorder in humans. Little is known about how these two pathways are coordinated. We report here that following cellular exposure to DNA cross-linking damage, the FA core complex enhances binding and localization of ATRIP within damaged chromatin. In cells lacking the core complex, ATR-mediated phosphorylation of two functional response targets, ATRIP and FANCI, is defective. We also provide evidence that the canonical ATR activation pathway involving RAD17 and TOPBP1 is largely dispensable for the FA pathway activation. Indeed DT40 mutant cells lacking both RAD17 and FANCD2 were synergistically more sensitive to cisplatin compared with either single mutant. Collectively, these data reveal new aspects of the interplay between regulation of ATR-ATRIP kinase and activation of the FA pathway.
INTRODUCTION
Fanconi anemia (FA) is a hereditary disorder characterized by cancer susceptibility and hypersensitivity to inducers of DNA interstrand cross-links (ICLs) (1,2). FA is caused by mutations in a genetically and biochemically complex set of proteins, including an FA core E3 ligase complex containing eight FA gene products (i.e. FANCA, B, C, E, F, G, L, M) and other associated proteins (i.e. FAAP100, FAAP24, FAAP20) (1,2). The FANCM–FAAP24 subcomplex is thought to load the rest of the core complex onto damaged chromatin (3,4). The core complex mediates monoubiquitination of the ID complex composed with FANCD2 and FANCI proteins. The monoubiquitinated ID complex in turn recruits the DNA repair nuclease FAN1 (2,5–7), and might function as histone chaperone during ICL repair (8). In addition, it has been suggested that the core complex might have other functions (9). Specific mutations of some additional FA genes (FANCD1/BRCA2, FANCJ/BRIP1, FANCN/PALB2, FANCO/RAD51C) are found in familial breast cancer (2,10–12), but these genes and a novel FA gene FANCP/SLX4 (13,14) do not affect the core signaling pathway, resulting in monoubiquitination of the ID complex. Slx4 is shown to be recruited by monoubiquitinated FANCD2 (15) and contributes to ICL repair mainly through regulation of XPF-ERCC1 nuclease (16).
A critical DNA damage response pathway is mediated by the checkpoint kinase ATR and its protein partner ATRIP. One connection between the FA pathway and ATRIP has been uncovered previously: the checkpoint kinase ATR-ATRIP controls multiple phosphorylation events on FANCI, which trigger FA pathway activation (17–20). ATR kinase activation proceeds in two largely independent steps (21–23): first, a stalled DNA replication fork generates a stretch of single-stranded DNA (ssDNA) covered by Replication protein A (RPA) complex, which in turn recruits ATRIP-ATR into distinct focal areas within cell nuclei. Interaction of RPA-bound ATRIP-ATR with the TOPBP1 protein leads to execution of the S-phase checkpoints. The latter step also involves the specialized RAD9-RAD1-HUS1 (9-1-1) checkpoint clamp and the RAD17-RFC clamp loader (21–23), but the molecular details of these processes are unclear.
We wished to clarify how ATR signaling and the FA pathway are coordinated. We examined the ATR signaling events in FA cell lines, and found that the FA core complex does not simply lie downstream of ATR, but functions in ATR kinase activation after replication stress by enhancing chromatin binding of ATRIP. Unexpectedly, we also found that the canonical ATR activation pathway involving RAD17 and TOPBP1 is largely dispensable for activation of the FA pathway. Taken together, our current data provide novel insights regarding the interplay between ATR-ATRIP kinase and activation of the FA pathway.
MATERIALS AND METHODS
Cell culture, gene targeting and transfection in DT40 cells
Wild-type (WT) and various mutant chicken DT40 cells were cultured in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), 1% chicken serum, 2 mM L-glutamine, 50 μM 2-mercaptoethanol and penicillin/streptomycin in a 5% CO2 incubator at 39.5°C. Generation of rad17, atm, fancd2, fancc, fancl, fancl complemented with Green fluorescence protein (GFP)-chFANCL cell lines has been described previously (24–28). FANCM-deficient DT40 cells (29), FANCM D203A ‘knock-in’ cells (30), usp1 (31) and ube2t knockout (32) cell lines were kindly provided by Dr K.J. Patel (Cambridge University).
Full-length chicken ATRIP cDNA was amplified by reverse transcriptase polymerase chain reaction (PCR) from DT40 RNA and cloned into pDONR vector (Invitrogen). After sequencing, the gateway system (Invitrogen) was used to transfer the cDNAs to the GFP expression vector (20). Targeting vectors were constructed by subcloning PCR-amplified genomic fragments on both sides of the resistance gene cassettes. All transfections in DT40 were done as described (17).
FANCI K525R ‘knock-in’ was achieved in a heterozygous FANCI knockout clone (17), and the resistance gene cassette flanked by the flippase recognition target (FRT) sites introduced into intron was removed by flippase (FLP) recombinase-mediated excision (Flp expression plasmid was provided by Dr Kyoji Horie, Osaka University). Briefly, cells were transiently transfected with a plasmid encoding FLP recombinase and IRES-puro, and excised cells were isolated by puromycin selection for 2 days followed by limiting dilution. While fanci-K525R cells could not monoubiquitinate FANCI, they showed a partial defect in FANCD2 monoubiquitination on DNA damage by mitomycin C (MMC), and displayed only mild sensitivity to cisplatin compared with fanci null cells. Conditional FANCD2 knockout cells were made by knock-in targeting of two FRT sites in FANCD2 heterozygous knockout clone. The FRT sites flanked a genomic segment that included the exon containing the monoubiquitination site. The resistance gene cassettes were removed by expression of Cre recombinase using plasmid-based transfection. In this conditional cell line, FANCD2 gene could be inactivated by FLP-mediated excision. RAD17 gene was targeted in the conditional FANCD2 knockout cells using RAD17 targeting vector previously described (25).
Human cell cultures and transfections
Human ATRIP-GFP or FANCL-Flag expression vector was constructed by cloning PCR product from HeLa cell cDNA into appropriate expression vectors. GFP-human FAAP100 was similarly made by PCR from a plasmid (a kind gift from Dr Weidong Wang).
Human HeLa S3 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS and penicillin/streptomycin in a 5% CO2 incubator at 37°C, and were introduced with retroviral bicistronic expression vector encoding FLAG-HA-tagged human ATRIP (FLAG-HA-ATRIP) and IRES-CD25 as described (33). The retrovirus was packaged using 293P cells, which were maintained in DMEM supplemented with 10% newborn calf serum and penicillin/streptomycin. The transduced cells were selected on the basis of CD25 expression using Dynabeads (33).
GM6914 (FA-A) with or without complementation with FANCA (a gift from Dr Takayuki Yamashita, Gunma University), and PD20 cells with or without complementation with GFP-hFANCD2 (a gift from Dr Toshiyasu Taniguchi, Fred Hutchinson Cancer Center) were maintained in α-minimum essential medium (MEM) supplemented with 20% FCS, 1% penicillin and streptomycin. Human ATRIP-GFP-expression vector was transfected into GM6914 and control cells using Lipofectamine LTX according to the manufacturer’s instructions. A549 cells expressing ATRIP-GFP have been described (34), and maintained in DMEM with 10% FCS. siRNA transfections were done with Lipofectamine RNAiMAX. The messenger RNA target sequences used for siRNAs were as follows: Rad17 (GAUGGGUCAACCCAGUCUGTT), TopBP1 (GUGGUUGUAACAGCGCAUC), FANCA (AAGGGUCAAGAGGGAAAAAUA), FANCD2 (CCAUGUCUGCUAAAGAGCGUUCAUU).
Antibodies
Anti-chicken FANCD2 (35) or anti-chicken FANCI (20) serum was described. Other antibodies were purchased from Santa Cruz Biotechnology (polyclonal anti-ATR: sc-1887, polyclonal anti-RAD17: sc-5613, monoclonal anti-Chk1: sc-8408), Bethyl Laboratories (polyclonal anti-FANCA: A301-980A, polyclonal anti-RPA32: A300-244A), Abcam (polyclonal anti-Histone H3: ab1791, polyclonal anti-FANCL: ab42639), Cell Signaling Technology (polyclonal anti-phospho-Chk1-Ser317: #2344), Novus (polyclonal anti-TopBP1: NB100-217, polyclonal human FANCD2: NB100-182, polyclonal anti-FANCG: NBP1-06035), Millipore (monoclonal anti-phospho-ATM-Ser1981: 05–740), Clontech (polyclonal anti-GFP: 632459), Sigma (monoclonal anti-tubulin: T5168) and Upstate Biotechnology (monoclonal anti-CD25: #05–170).
Fractionation of cells and western blotting
Cells were treated with indicated dose of DNA damaging agents and collected. Cell fractionation into soluble and chromatin fractions was done as described previously (36). Briefly, washed cells were resuspended in CSK buffer (10 mM PIPES, pH 7.0, 100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2, 1 mM EGTA, 10 mM NaF, 25 mM β-glycerophosphate, 0.2% Triton X-100, 0.25 mM ATP) containing protease inhibitor tablet (Roche), and centrifuged. Supernatants were saved as soluble fraction. Pellets were further washed using the same buffer twice, and the remained material was used as chromatin fraction. These samples were separated by polyacrylamide gel electrophoreses, transferred to a membrane and detected with indicated antibodies and ECL reagents (GE Healthcare). Phos-tag western blotting was done as described (17). Co-immunoprecipitation assay between overexpressed proteins in 293T cells was carried out as described (27).
Analysis of growth and cell sensitivity toward cisplatin
Cell proliferation rate was assessed as described using plastic microbeads. Cell viability in liquid culture containing cisplatin (Nippon Kayaku, Tokyo, Japan) was assessed after 48 h using FACSCalibur (BD Biosciences) and propidium iodide (PI) staining. Percentage of viable cells was calculated on the basis of forward scatter profile and exclusion of PI fluorescence among the acquired 10 000 events.
Subnuclear focus formation assay
After MMC (Kyowa-Hakkou-Kirin, Tokyo) exposure, cytospin slides or coverslips were fixed with 4% paraformaldehyde/phosphate buffered saline and stained with antibodies against FancD2 or RPA2. Then cells were stained with AlexaFluor-conjugated secondary antibody (Invitrogen) with 4′,6-Diamidino-2-Phenylindole Dihydrochloride (DAPI) counterstaining. Images were captured by fluorescent microscopy (DM5500B, Leica) or confocal laser scanning microscopy (TCS SP5, Leica).
RPA-ssDNA pull-down assay
This assay was carried out essentially as described (37). In brief, biotin-labeled 75 mer ssDNA (5 pmole) was attached to streptavidin-coated magnetic beads (Dynal), and incubated with 400 ng of purified recombinant RPA for 30 min in binding buffer A (10 mM Tris–HCl (pH 7.5), 100 mM NaCl, 10 ng/ml bovine serum albumin (BSA), 0.01% Nonidet P-40, 10% glycerol). The RPA complex was purified as described (38,39) from Escherichia coli carrying the RPA-expressing plasmids kindly provided by Dr Marc S. Wold (University of Iowa College of Medicine, Iowa City, Iowa, USA). Cell lines were lysed in 220 μl of lysis buffer (10 mM Tris–HCl (pH 7.5), 100 mM NaCl, 0.05% Nonidet P-40, 10 ng/ml BSA, 10% glycerol). Resultant lysate (100 μl) containing equal amount of protein was then mixed with the beads, and incubated for additional 30 min at room temperature. After washing, the bound material was eluted by addition of sodium dodecyl sulphate-polyacrylamide gel electrophoresis sample buffer and boiling, and examined by western blotting.
Yeast two-hybrid assay
The yeast strain EGY48 (Clontech) was used as a host to express fusion proteins with LexA or B42 transcription activation domain fusion. cDNAs encoding ATRIP, and human FA and FA-related proteins (FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, UBE2T, FAAP24 or FAAP100) were cloned into pLexA (bait) and pB42AD (prey) vectors (Clontech). Sources of the most cDNAs were previously described (17,27,40). FANCM cDNA was provided by Dr Weidong Wang. Human UBE2T or FAAP24 cDNA was amplified from HeLa cell cDNA. The transformants were selected on SD/-Ura/-His/-Trp plate, cultured in liquid of the same medium at 30° for 24 h and then spotted on to SD/-Ura/-His/-Trp/-Leu/+X-gal plates at 30° for 96 h.
RESULTS
Defective localization of ATRIP following MMC damage in cells lacking the FA core complex
At sites of DNA damage and replication fork stress, ATRIP-ATR concentrates in nuclear foci mediated by binding of ATRIP to RPA-coated ssDNA (41). To examine potential interplay between ATR signaling and the FA core complex, we examined focus formation of ATRIP following MMC-induced DNA damage in FA cells. Available anti-ATRIP antibodies did not allow convincing visualization of focus formation, and so we expressed a human ATRIP-GFP fusion in cells lacking FANCA. In MMC-untreated GFP-positive cells, ATRIP-GFP mostly distributed diffusely throughout the nuclei irrespective of the genotype. On MMC treatment, the complemented control cells displayed robust RPA foci formation that co-localized with ATRIP-GFP foci (Figure 1A). Strikingly, we found that ATRIP-GFP foci in MMC-stimulated FA-A cells were diffuse and greatly reduced, while RPA foci formed normally (Figure 1A). These data suggest that FANCA is required for MMC-induced ATRIP focus formation.
Figure 1.
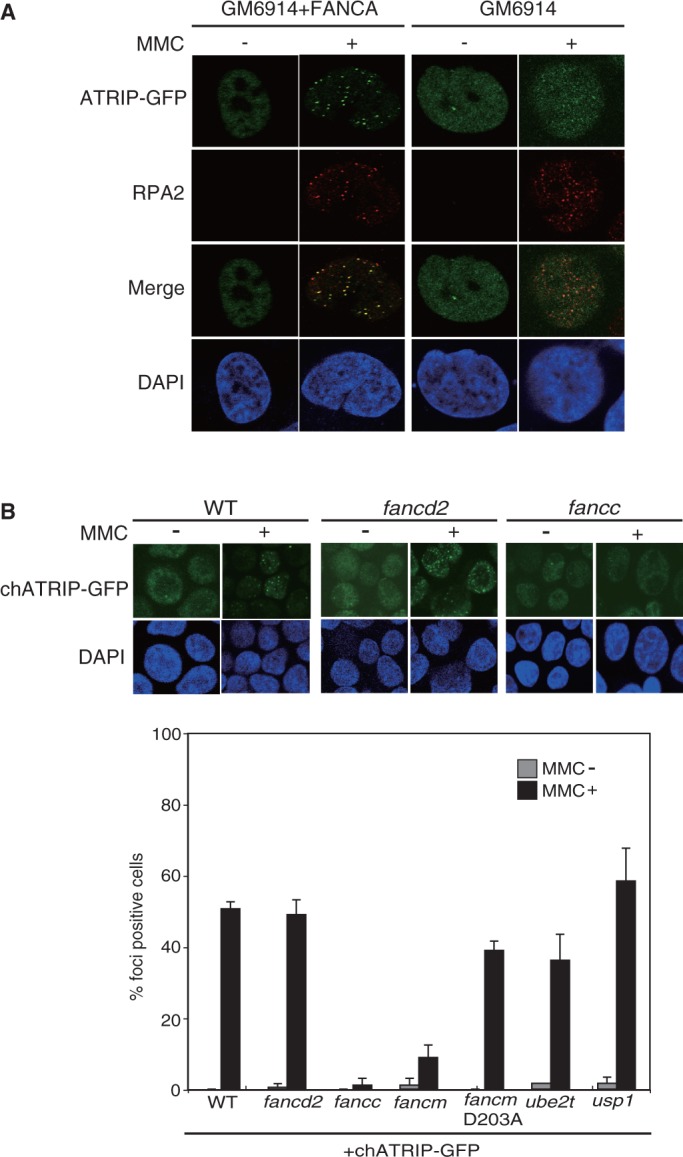
Defective ATRIP-GFP foci formation in FA cells. (A) MMC-induced human ATRIP-GFP foci formation in human FA-A (GM6914) and complemented control cells. Following transient transfection of the ATRIP-GFP expression vector, ∼10% of the cells became GFP-positive 24 h after transfection. Then the cells were treated with or without MMC (100 ng/ml for 24 h), fixed and stained with anti-RPA2 antibody, and the GFP was visualized directly. The images were captured by confocal laser scanning microscopy. (B) MMC-induced chicken ATRIP-GFP foci in chicken DT40 mutant cells. Indicated DT40 WT and FA mutant cells were stably transfected with chicken ATRIP-GFP expression vector. Clones selected on the basis of similar GFP expression levels were subjected to analysis. Following MMC treatment (500 ng/ml for 6 h), cells were fixed and observed by confocal laser scanning microscopy. The bar graph represents mean and SD of % GFP-foci positive cells in three independent experiments. Fifty nuclei were scored in each case, and nuclei containing more than four bright GFP foci were defined as foci positive.
To further study ATRIP localization in the absence of FA proteins, we stably expressed chicken ATRIP-GFP in chicken DT40 WT and FA knockout cell lines. Similar levels of GFP expression among transfectants were confirmed with FACS analysis (Supplementary Figure S1A). Consistent with the results with human FA-A cells, MMC-induced chicken ATRIP-GFP focus formation was abrogated in fancc and fancm cells (Figure 1B). In contrast, extensive foci formation was still evident in fancd2 cells, although observed foci were smaller and less intense (Figure 1B). The ATPase activity of the FANCM translocase domain was largely dispensable for focus formation, as shown in cells carrying a D203A knock-in mutation (Figure 1B) (30). The E3 ubiquitin ligase function of FANCL is unlikely to be important for chromatin localization of ATRIP because the E2 enzyme for FANCL, UBE2T, was dispensable for ATRIP focus formation (Figure 1B). The deubiquitinase for FANCD2, USP1, was also not required (Figure 1B). These data suggest that the FA core complex may play an important role in relocalization of ATRIP following DNA damage.
Decreased chromatin localization of ATRIP in cells deficient in the FA core complex
We next determined whether endogenous ATR and ATRIP show defective relocalization to chromatin in human FA cell lines. FA-A (GM6914) and FA-D2 (PD20) cells, with isogenic complemented controls, were treated with MMC, biochemically fractionated and analyzed by western blotting. Consistent with the foci results, FA-A cells displayed a significant reduction (∼40%) in MMC-induced chromatin targeting of ATRIP relative to complemented control cells (Figure 2A). ATR localization in GM6914 cells may be deregulated because the ATR level in chromatin was increased in the untreated cells, and was not further increased following MMC treatment. In contrast, MMC-stimulated FA-D2 cells had only minor reduction of ATRIP-ATR levels in chromatin compared with the complemented control cells (Figure 2A). In line with the decreased chromatin binding of ATRIP, phosphorylation of ATRIP on Ser 68 and 72 was decreased to ∼50% in FA-A cells but not in FA-D2 cells (Figure 2A and Supplementary Figure S2B and S2C). This phosphorylation event occurs in chromatin and is dependent on ATR (34).
Figure 2.
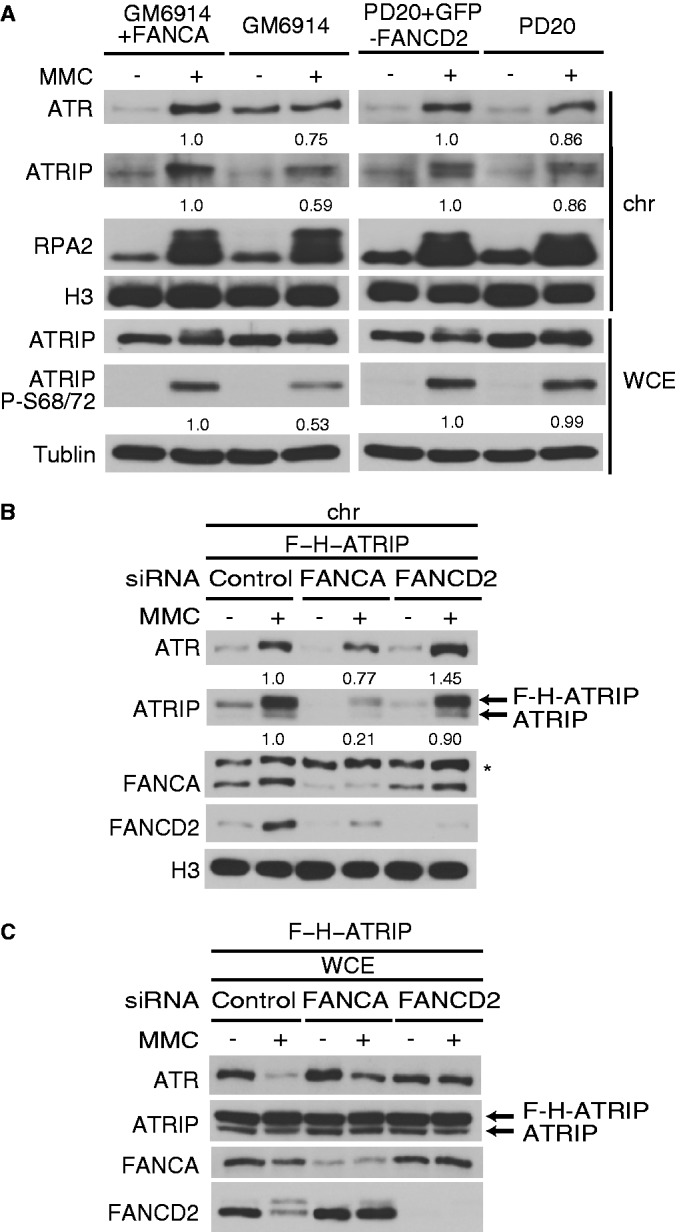
Chromatin localization of ATRIP is defective in human cells lacking the FA core complex. (A) FA-A (GM6914 fibroblast) or FA-D2 (PD20 fibroblast) and respective complemented control cells were treated with MMC (100 ng/ml for 24 h). The whole cell extract (WCE) and the chromatin fraction (chr) were prepared and analyzed by western blotting. (B and C) HeLa S3 cells expressing FLAG-HA-ATRIP (F-H-ATRIP) were transfected with siRNAs targeting FANCA or FANCD2. After 48 h, the cells were stimulated with MMC (100 ng/ml for 24 h). Chromatin fraction (B) or WCE (C) were analyzed by western blotting. The numbers indicate the ratio of the band intensity normalized to the control lane.
To extend these observations to cells acutely rendered defective in FA proteins, we also examined HeLa S3 cells expressing FLAG-HA-ATRIP treated with siRNAs against FANCA or FANCD2. This cell line expresses ∼2-fold more ectopically expressed tagged ATRIP than endogenous ATRIP. Cells depleted of FANCA reduced chromatin ATRIP levels by ∼5-fold on MMC damage compared with control cells. The ATR level was also modestly decreased (Figure 2B). This could be partially accounted for by DNA damage-induced decrease in total ATR levels as shown in Figure 2C, though the reduction was greatest in cells exposed to control siRNA. On the other hand, cells depleted of FANCD2 did not have decreased chromatin loading of both ATRIP and ATR (Figure 2B).
Collectively, we conclude that the FA core complex component FANCA promotes or stabilizes chromatin localization of ATRIP in human cells. Combined with the ATRIP-GFP foci results in DT40 cells, the data suggested that the FA core complex as a whole functions to regulate ATRIP levels in chromatin.
The FA core complex is required for ATRIP localization upon replication stress
To address whether the FA core complex plays a similar role following other types of DNA damage, we treated cells with ionizing radiation (IR) or the replication stress inducers MMC, hydroxy Urea (HU) and UV. ATRIP was less phosphorylated on Ser 68/72 in FA-A cells than in isogenic control cells on replication stress, particularly following HU treatment, while the phosphorylation was not decreased in FA-A cells compared with control following IR (Supplementary Figure S3A). Furthermore, the ATRIP-GFP foci formation in fancc DT40 cells treated with HU was drastically reduced, while the same cells treated with IR showed similar levels of ATRIP-GFP foci formation to WT cells (Supplementary Figure S3B and S3C). These observations indicate that the dependence of ATRIP localization in chromatin on the FA core complex is dependent on the type of genomic stress. Stalled DNA replication forks (MMC, HU, UV) require the FA core complex for ATRIP localization, but for direct double-strand breaks (IR), FA core complex is not needed.
The FA core complex may physically interact with ATR-ATRIP kinase
Given the above data, we hypothesized that ATRIP may interact with the FA core complex. To test this idea, we carried out yeast two-hybrid interaction assay between ATRIP and various FA and FA-related proteins (including FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, UBE2T, FAAP24 or FAAP100) (Figure 3A). We could detect positive ATRIP interaction only with FANCL or FAAP100.
Figure 3.

Interplay between FA proteins and ATRIP. (A) Yeast two-hybrid interaction assay between ATRIP and various full-length FA or FA-related proteins. Selected yeast transformants were spotted onto SD/-Ura/-His/-Trp/-Leu/+X-gal plates at 30° for 96 h. ATRIP–ATRIP interaction served as positive control. (B) Co-immunoprecipitation assay between overexpressed human FANCL-GFP or GFP-FAAP100 and ATRIP-Flag in 293T cells. F or G, Flag or GFP vector without insert. (C) RPA-ssDNA pull-down assay from extracts of FA-A (GM6914) and complemented FA-A cells (+A). ssDNA-attached magnetic beads preincubated with or without recombinant RPA were mixed with cell extracts. After washing, the bound material was analyzed by western blotting.
To determine whether ATRIP could interact with FA core complex components in cells, we co-overexpressed ATRIP with FANCL or FAAP100. Only FANCL co-immunoprecipitated with ATRIP in this setting (Figure 3B). These results suggest that FANCL might be the core complex subunit that associates with ATRIP protein.
The FA core complex promotes binding of ATRIP to RPA-coated ssDNA
To gain further insight into the role of the core complex in the ATRIP recruitment to RPA-coated ssDNA, we assessed the amount of ATRIP protein that could be recovered from cell extracts following binding to RPA-coated ssDNA (Figure 3C). In the extracts lacking the core complex, significantly less ATRIP protein was pulled down compared with the matched control cell extracts (indicated as +A). Interestingly, the core complex components such as FANCA could be recovered from control cell extract by ssDNA beads without exogenous RPA complex. These data suggest that the core complex bound to RPA-coated ssDNA may facilitate or stabilize ATRIP binding to RPA through either direct or indirect interaction with ATRIP.
Loss of FANCI phosphorylation in cells lacking the FA core components
We wished to determine how the deficiency in the FA core complex affects signaling events through ATR because the altered ATRIP-ATR chromatin localization should have a consequence on downstream signaling events. Although mild S-phase checkpoint defects in FA cell lines have been reported (42–44), MMC-induced CHK1 phosphorylation was not significantly affected in cells deficient in the FA core components (Figure 4A, C and D). Interestingly, previous reports have indicated that CHK1 phosphorylation can occur without ATRIP focus formation, in cells harboring an ATRIP N-terminal deletion (45,46) or an LG mutant (47), which is incapable of binding to RPA or is deficient in dimerization (37,48). Further, stimulation of ATR activity by TOPBP1 can occur in the absence of RPA binding to ATRIP (49). Thus, an association of a residual amount of ATRIP-ATR kinase with TOPBP1 in chromatin might be sufficient to phosphorylate CHK1.
Figure 4.

FANCI phosphorylation mediated by ATR is defective in cells lacking FANCD2 or the FA core complex. (A) Cells were treated with MMC (100 or 500 ng/ml for 6 h) and analyzed with western blotting. (B) Samples were prepared as in A, and analyzed with Phos-tag western blotting. (C) Phos-tag western blotting of FancI protein in fancl cells and cells complemented with GFP-chFANCL. (D) WT and fancm cells were stimulated as indicated and analyzed by western blotting with or without Phos-tag. Numbers indicate ratio of phosphorylated CHK1 or FANCI to total CHK1 or FANCI, respectively.
Next, we investigated FANCI phosphorylation in DT40 mutants because FANCI is the critical ATR substrate in the FA pathway (20) following DNA damage. We used the Phos-tag western blotting (17) to detect phosphorylated FANCI. Phospho-proteins could be detected as slower migrating bands in a Phos-tag-containing polyacrylamide gel electrophoresis gel. FANCI K525R knock-in DT40 cells were used as a control, in which the FANCI monoubiquitination site (Lysine) was replaced with Arginine by gene targeting (Supplementary Figure S4). This mutant was defective in FANCI monoubiquitination, similarly to fancl, fancc and fancd2 mutants, yet FANCI phosphorylation was clearly observed (Figure 4B).
Strikingly, fancl, fancc and fancd2 mutant cells were completely deficient in FANCI phosphorylation (Figure 4B). Furthermore, fancl cells complemented with GFP-chFANCL (27) could phosphorylate FANCI at a level comparable with WT cells (Figure 4C). fancm cells showed an intermediate phenotype, with FANCI phosphorylation ∼20–30% of normal observed following MMC treatment (Figure 4D). This is consistent with the weakened but still clearly detectable FANCD2 monoubiquitination in fancm cells (Figure 4D). A stimulatory role of FANCM in FANCD2 monoubiquitination has been previously described (29,50–52).
FA pathway activation is independent of RAD17 or TOPBP1
The results described above and our previous studies (17,20) clearly indicated that FANCI phosphorylation is mediated by ATR kinase in a manner dependent on the FA core complex and FANCD2 protein. This phosphorylation event then triggers FA pathway activation, leading to FANCD2 monoubiquitination and DNA repair. It has been well accepted that the phosphorylation of the ATR substrates requires the kinase activator TOPBP1 as well as its recruiter, the 9-1-1 complex, which is loaded by the RAD17-RFC complex (21–23). To gain more insight into the interplay between ATR and the FA pathway, we wished to test whether the activation of the FA pathway is affected by the absence of the canonical ATR activators RAD17 or TOPBP1. Surprisingly, MMC-induced FANCD2 and FANCI monoubiquitination were only slightly affected by the loss of RAD17 gene (Figure 5A), while CHK1 phosphorylation was severely diminished in the knockout DT40 cell lines (Figure 5A). Consistently, FANCI phosphorylation was not defective in rad17 cells (Figure 5A). FANCD2 focus formation was also not affected (Figure 5B). These data indicate that the FA pathway activation occurs independently of RAD17.
Figure 5.

RAD17 and TOPBP1 are dispensable for FA pathway activation. (A) Cells were treated with MMC (100 or 500 ng/ml for 6 h) and analyzed with western blotting with or without Phos-tag. Numbers indicate ratio of phosphorylated CHK1 to total CHK1. (B) FANCD2 foci formation following MMC treatment (500 ng/ml for 6 h) was analyzed in DT40 WT and rad17 mutant cells by fluorescent microscopy. The bar graph represents mean and SD of % FANCD2-foci positive cells in three independent experiments. Fifty nuclei were scored in each case, and nuclei containing more than four bright foci were defined as foci positive. (C) Cell growth rate (left panel) and % survival of the cells cultured for 48 h in the medium containing cisplatin (right panel). Cell number or cell viability was assessed by flow cytometry using plastic microbeads or PI staining, respectively. The error bar represents SD in three independent experiments. (D and E) HeLa S3 cells (D) or A549 cells expressing ATRIP-GFP (E) were transfected with indicated siRNAs. After 48 h, cells were treated with MMC (100 ng/ml) for additional 24 h. WCE was analyzed by western blotting.
To verify this conclusion by a genetic test, we generated a double knockout DT40 cell line lacking both RAD17 and FANCD2 genes (Supplementary Figure S5). The double knockout cells grew slower and were much more sensitive to cisplatin compared with either single mutant cell line (Figure 5C), suggesting a synergistic relationship. We also used siRNA knockdown in HeLa S3 or A549 cells expressing ATRIP-GFP. FANCD2 monoubiquitination was affected only slightly by siTOPBP1 treatment in HeLa S3 (L/S ratio 1.32 in control versus 0.97 in TOPBP1-depleted cells), whereas CHK1 phosphorylation was mostly abrogated (Figure 5D). In siRAD17- or siTOPBP1-treated A549 cells (Figure 5E), FANCD2 monoubiquitination was only slightly diminished compared with control. Furthermore, FANCD2 still efficiently accumulated in subnuclear foci and in chromatin following MMC damage similarly to cells transfected with control siRNA (Supplementary Figure S6A and B).
DISCUSSION
ATRIP-ATR kinase is essential for checkpoint signaling via CHK1 phosphorylation (21–23), and it is also required for DNA repair by activating the FA pathway through FANCI phosphorylation (17,20). It has been well accepted that ATR signaling proceeds in two steps: a localization step via ATRIP-RPA interaction, and then enzymatic activation by ATR-TOPBP1 interaction. In this study, we attempted to clarify interplay between ATR signaling and the FA pathway. Our data indicate that the FA core complex (including FANCM) regulates the former step, and also connects ATR and the FA pathway activation. On the other hand, the latter step is not critical for the FA pathway. There have been several reports that demonstrate roles of the FANCM/FAAP24 complex in the ATR signaling independent of the FA core complex (52–56). For example, FANCM has a role for chromatin accumulation of RPA (55,56) or TOPBP1 (54) following DNA damage. Our results complement these previous studies and indicate that other components of the FA core complex have a role in the ATR signaling.
In the current study, we provide evidence that the FA core complex affects the localization step by facilitating ATRIP interaction with RPA-ssDNA. Of note, FANCL has been reported to play an upstream role in RPA-ATR-Chk1 signaling in Xenopus egg extracts stimulated by plasmids carrying an ICL (57). The present results extend these studies to deletion of specific components of the FA core complex in mammalian and avian somatic cells, and reveal that FA core complex defects are associated with impaired intracellular and chromatin localization of ATRIP.
The mechanisms for enhancing ATRIP-RPA interaction remain unknown. ATRIP-FANCL interaction was detected using yeast two-hybrid assay and by co-immunoprecipitation of the transfected proteins. However, whether the endogenous proteins actually interact is currently unclear. Because the E2 enzyme UBE2T is not required for this process, the FA core complex stabilizes the interaction independently of the ubiquitination activity. The ATPase activity of FANCM is also dispensable. A previous study indicated that in vitro binding of ATRIP-ATR to RPA-ssDNA is enhanced by an unknown human protein (37). It will be interesting to test whether this unknown factor is a component of the FA core complex.
We showed that, in cells lacking the FA core complex, phosphorylation of ATRIP as well as FANCI protein is defective. MMC-induced CHK1 phosphorylation occurs normally in FA knockout cells. Loss of FANCI phosphorylation is functionally important because it is the triggering event in activation of the FA pathway (17). However, it is obscure whether defective ATRIP-ATR chromatin localization is the direct cause of the loss of FANCI phosphorylation. The FA core complex may function downstream of ATRIP-ATR as an essential link to connect FANCI with ATR, resulting in FANCI phosphorylation. It is possible that ATR phosphorylates FANCI when it is in a complex with FANCD2. We have previously reported that efficiency of FANCI phosphorylation by ATR in vitro is enhanced by the presence of FANCD2 (20). Interestingly, a recent report suggests that chromatin binding of FANCI depends on FANCD2 in Xenopus egg extracts (58). Collectively, these results indicate the defective core complex abolishes the upstream phosphorylation events on FANCI in addition to its ubiquitin E3 ligase activity, thus blocking the FA pathway signaling at two levels.
Our data also indicate that TOPBP1 and RAD17 do not play a major role in FA pathway activation. This is consistent with our previous observation that ATRIP does not require TOPBP1 interaction to activate the FA pathway (20). An in vitro study has shown that at high concentration of substrate the influence of TOPBP1 on ATR kinase activity is less pronounced (59). In FA pathway activation, the phosphorylation substrate FANCI may accumulate in damaged chromatin, perhaps though the interaction with FANCD2 and/or the core complex (60,61), achieving concentrations that reduce the requirement for TOPBP1. Alternatively, the locally accumulated ATRIP-ATR might have sufficient activity to phosphorylate FANCI without TOPBP1 stimulation. Interestingly, auto-phosphorylation of ATR on Thr 1989 is not dependent on the presence of TOPBP1 (62).
Given these findings, we propose two distinct subpathways in ATR signaling (Figure 6). One is dependent on RAD17-TOPBP1 pathway in which the important substrate is CHK1. This pathway is also affected by the FANCM-FAAP24 subcomplex. The other pathway is dependent on the FA core complex, and critical for phosphorylating ATRIP, FANCI and perhaps FANCD2 and BLM (63). Other substrates in this subpathway of ATR signaling may be identified.
Figure 6.

Our proposed model for two distinct pathways in ICL-induced ATR signaling. RPA complex accumulates on exposed ssDNA region surrounding the ICL (middle). RPA-ATRIP binding, which is facilitated by the FA core complex, promotes ATR recruitment. In the left side of the pathway, which is dependent on the FA core complex but not on the RAD17-TOPBP1 pathway, FANCI is phosphorylated, triggering FANCD2 monoubiquitination. In the right side, CHK1 is phosphorylated depending on the RAD17-TOPBP1 pathway, but the FA core complex is largely dispensable for the CHK1 activation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–6.
FUNDING
Ministry of Education, Science, Sports and Culture of Japan [JSPS KAKENHI, 21390094 and MEXT KAKENHI, 23114010] (in part); The Uehara Memorial Foundation (to M.T.); Takeda foundation (to M.T.); the Ichiro Kanehara Foundation (to M.I.); the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to M.I.). Funding for open access charge: Grants-in aid from the Ministry of Education, Science, Sports and Culture of Japan.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs Richard D. Wood (MD Anderson Cancer Center) and Agata Smogorzewska (Rockefeller University) for critical reading of the manuscript; Drs Kyoji Horie, Toshiyasu Taniguchi, K.J. Patel, Weidong Wang, Marc S. Wold, Takayuki Yamashita for reagents; Drs Hitoshi Kurumizaka and Hiroshi Kimura for FANCD2 siRNA sequence; Drs Kenshi Komatsu, Junya Kobayashi, Nigel Jones for advice and help; Ms Seiko Arai for secretarial assistance.
REFERENCES
- 1.Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 3.Kim JM, Kee Y, Gurtan A, D'Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani el H, Joenje H, McDonald N, de Winter JP, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang M, D'Andrea AD. A new nuclease member of the FAN club. Nat. Struct. Mol. Biol. 2010;17:926–928. doi: 10.1038/nsmb0810-926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato K, Ishiai M, Toda K, Furukoshi S, Osakabe A, Tachiwana H, Takizawa Y, Kagawa W, Kitao H, Dohmae N, et al. Histone chaperone activity of Fanconi anemia proteins, FANCD2 and FANCI, is required for DNA crosslink repair. EMBO J. 2012;31:3524–3536. doi: 10.1038/emboj.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsushita N, Kitao H, Ishiai M, Nagashima N, Hirano S, Okawa K, Ohta T, Yu DS, McHugh PJ, Hickson ID, et al. A FancD2-monoubiquitin fusion reveals hidden functions of Fanconi anemia core complex in DNA repair. Mol. Cell. 2005;19:841–847. doi: 10.1016/j.molcel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–105. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Levy-Lahad E. Fanconi anemia and breast cancer susceptibility meet again. Nat. Genet. 2010;42:368–369. doi: 10.1038/ng0510-368. [DOI] [PubMed] [Google Scholar]
- 12.Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat. Genet. 2010;42:406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- 13.Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011;43:142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, Jiricny J, Takeda S, Hirota K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc. Natl Acad. Sci. USA. 2011;108:6492–6496. doi: 10.1073/pnas.1018487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, McIntyre RE, Gallagher F, Kettunen MI, Lewis DY, Brindle K, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 2011;43:147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2008;15:1138–1146. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andreassen PR, D'Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang LC, Stone S, Hoatlin ME, Gautier J. Fanconi anemia proteins stabilize replication forks. DNA Repair (Amst.) 2008;7:1973–1981. doi: 10.1016/j.dnarep.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shigechi T, Tomida J, Sato K, Kobayashi M, Eykelenboom JK, Pessina F, Zhang Y, Uchida E, Ishiai M, Lowndes NF, et al. ATR-ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway. Cancer Res. 2012;72:1149–1156. doi: 10.1158/0008-5472.CAN-11-2904. [DOI] [PubMed] [Google Scholar]
- 21.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burrows AE, Elledge SJ. How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes Dev. 2008;22:1416–1421. doi: 10.1101/gad.1685108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2010;36:133–140. doi: 10.1016/j.tibs.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takao N, Kato H, Mori R, Morrison C, Sonada E, Sun X, Shimizu H, Yoshioka K, Takeda S, Yamamoto K. Disruption of ATM in p53-null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene. 1999;18:7002–7009. doi: 10.1038/sj.onc.1203172. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi M, Hirano A, Kumano T, Xiang SL, Mihara K, Haseda Y, Matsui O, Shimizu H, Yamamoto K. Critical role for chicken Rad17 and Rad9 in the cellular response to DNA damage and stalled DNA replication. Genes Cells. 2004;9:291–303. doi: 10.1111/j.1356-9597.2004.00728.x. [DOI] [PubMed] [Google Scholar]
- 26.Hirano S, Yamamoto K, Ishiai M, Yamazoe M, Seki M, Matsushita N, Ohzeki M, Yamashita YM, Arakawa H, Buerstedde JM, et al. Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. EMBO J. 2005;24:418–427. doi: 10.1038/sj.emboj.7600534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seki S, Ohzeki M, Uchida A, Hirano S, Matsushita N, Kitao H, Oda T, Yamashita T, Kashihara N, Tsubahara A, et al. A requirement of FancL and FancD2 monoubiquitination in DNA repair. Genes Cell. 2007;12:299–310. doi: 10.1111/j.1365-2443.2007.01054.x. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto K, Hirano S, Ishiai M, Morishima K, Kitao H, Namikoshi K, Kimura M, Matsushita N, Arakawa H, Buerstedde JM, et al. Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol. Cell. Biol. 2005;25:34–43. doi: 10.1128/MCB.25.1.34-43.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosedale G, Niedzwiedz W, Alpi A, Perrina F, Pereira-Leal JB, Johnson M, Langevin F, Pace P, Patel KJ. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat. Struct. Mol. Biol. 2005;12:763–771. doi: 10.1038/nsmb981. [DOI] [PubMed] [Google Scholar]
- 30.Rosado IV, Niedzwiedz W, Alpi AF, Patel KJ. The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res. 2009;37:4360–4370. doi: 10.1093/nar/gkp365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oestergaard VH, Langevin F, Kuiken HJ, Pace P, Niedzwiedz W, Simpson LJ, Ohzeki M, Takata M, Sale JE, Patel KJ. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol. Cell. 2007;28:798–809. doi: 10.1016/j.molcel.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alpi A, Langevin F, Mosedale G, Machida YJ, Dutta A, Patel KJ. UBE2T, the Fanconi anemia core complex, and FANCD2 are recruited independently to chromatin: a basis for the regulation of FANCD2 monoubiquitination. Mol. Cell. Biol. 2007;27:8421–8430. doi: 10.1128/MCB.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 34.Itakura E, Umeda K, Sekoguchi E, Takata H, Ohsumi M, Matsuura A. ATR-dependent phosphorylation of ATRIP in response to genotoxic stress. Biochem. Biophys. Res. Commun. 2004;323:1197–1202. doi: 10.1016/j.bbrc.2004.08.228. [DOI] [PubMed] [Google Scholar]
- 35.Yoshikiyo K, Kratz K, Hirota K, Nishihara K, Takata M, Kurumizaka H, Horimoto S, Takeda S, Jiricny J. KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc. Natl Acad. Sci. USA. 2010;107:21553–21557. doi: 10.1073/pnas.1011081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P, Barkley LR, Day T, Bi X, Slater DM, Alexandrow MG, Nasheuer HP, Vaziri C. The Chk1-mediated S-phase checkpoint targets initiation factor Cdc45 via a Cdc25A/Cdk2-independent mechanism. J. Biol. Chem. 2006;281:30631–30644. doi: 10.1074/jbc.M602982200. [DOI] [PubMed] [Google Scholar]
- 37.Namiki Y, Zou L. ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc. Natl Acad. Sci. USA. 2006;103:580–585. doi: 10.1073/pnas.0510223103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- 39.Masuda Y, Suzuki M, Piao J, Gu Y, Tsurimoto T, Kamiya K. Dynamics of human replication factors in the elongation phase of DNA replication. Nucleic Acids Res. 2007;35:6904–6916. doi: 10.1093/nar/gkm822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato K, Toda K, Ishiai M, Takata M, Kurumizaka H. DNA robustly stimulates FANCD2 monoubiquitylation in the complex with FANCI. Nucleic Acids Res. 2012;40:4553–4561. doi: 10.1093/nar/gks053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 42.Sala-Trepat M, Rouillard D, Escarceller M, Laquerbe A, Moustacchi E, Papadopoulo D. Arrest of S-phase progression is impaired in Fanconi anemia cells. Exp. Cell Res. 2000;260:208–215. doi: 10.1006/excr.2000.4994. [DOI] [PubMed] [Google Scholar]
- 43.Centurion SA, Kuo HR, Lambert WC. Damage-resistant DNA synthesis in Fanconi anemia cells treated with a DNA cross-linking agent. Exp. Cell Res. 2000;260:216–221. doi: 10.1006/excr.2000.4995. [DOI] [PubMed] [Google Scholar]
- 44.Pichierri P, Rosselli F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004;23:1178–1187. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ball HL, Myers JS, Cortez D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Itakura E, Takai KK, Umeda K, Kimura M, Ohsumi M, Tamai K, Matsuura A. Amino-terminal domain of ATRIP contributes to intranuclear relocation of the ATR-ATRIP complex following DNA damage. FEBS Lett. 2004;577:289–293. doi: 10.1016/j.febslet.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 47.Itakura E, Sawada I, Matsuura A. Dimerization of the ATRIP protein through the coiled-coil motif and its implication to the maintenance of stalled replication forks. Mol. Biol. Cell. 2005;16:5551–5562. doi: 10.1091/mbc.E05-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ball HL, Cortez D. ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J. Biol. Chem. 2005;280:31390–31396. doi: 10.1074/jbc.M504961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell. Biol. 2007;27:3367–3377. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, van der Wal A, van der Valk M, Joenje H, te Riele H, et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 2009;18:3484–3495. doi: 10.1093/hmg/ddp297. [DOI] [PubMed] [Google Scholar]
- 51.Singh TR, Bakker ST, Agarwal S, Jansen M, Grassman E, Godthelp BC, Ali AM, Du CH, Rooimans MA, Fan Q, et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood. 2009;114:174–180. doi: 10.1182/blood-2009-02-207811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Leung JW, Jiang Y, Lowery MG, Do H, Vasquez KM, Chen J, Wang W, Li L. FANCM and FAAP24 Maintain Genome Stability via Cooperative as Well as Unique Functions. Mol. Cell. 2013;49:997–1009. doi: 10.1016/j.molcel.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC, Boulton SJ. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the fanconi anemia core complex. Mol. Cell. 2008;32:313–324. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 54.Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010;29:806–818. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang M, Kim JM, Shiotani B, Yang K, Zou L, D'Andrea AD. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol. Cell. 2010;39:259–268. doi: 10.1016/j.molcel.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duquette ML, Zhu Q, Taylor ER, Tsay AJ, Shi LZ, Berns MW, McGowan CH. CtIP is required to initiate replication-dependent interstrand crosslink repair. PLoS Genet. 2012;8:e1003050. doi: 10.1371/journal.pgen.1003050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo CJ, Gottesman ME, Gautier J. Checkpoint signaling from a single DNA interstrand crosslink. Mol. Cell. 2009;35:704–715. doi: 10.1016/j.molcel.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sareen A, Chaudhury I, Adams N, Sobeck A. Fanconi anemia proteins FANCD2 and FANCI exhibit different DNA damage responses during S-phase. Nucleic Acids Res. 2012;40:8425–8439. doi: 10.1093/nar/gks638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mordes DA, Cortez D. Activation of ATR and related PIKKs. Cell Cycle. 2008;7:2809–2812. doi: 10.4161/cc.7.18.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pace P, Johnson M, Tan WM, Mosedale G, Sng C, Hoatlin M, de Winter J, Joenje H, Gergely F, Patel KJ. FANCE: the link between Fanconi anaemia complex assembly and activity. EMBO J. 2002;21:3414–3423. doi: 10.1093/emboj/cdf355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cole AR, Lewis LP, Walden H. The structure of the catalytic subunit FANCL of the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2010;17:294–298. doi: 10.1038/nsmb.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, Zou L. ATR Autophosphorylation as a Molecular Switch for Checkpoint Activation. Mol. Cell. 2011;43:192–202. doi: 10.1016/j.molcel.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 2004;23:3154–3163. doi: 10.1038/sj.emboj.7600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.