


Abstract
Human leukocyte antigen (HLA) typing at the allelic level can in theory be achieved using whole exome sequencing (exome-seq) data with no added cost but has been hindered by its computational challenge. We developed ATHLATES, a program that applies assembly, allele identification and allelic pair inference to short read sequences, and applied it to data from Illumina platforms. In 15 data sets with adequate coverage for HLA-A, -B, -C, -DRB1 and -DQB1 genes, ATHLATES correctly reported 74 out of 75 allelic pairs with an overall concordance rate of 99% compared with conventional typing. This novel approach should be broadly applicable to research and clinical laboratories.
INTRODUCTION
Human leukocyte antigens (HLAs) are highly polymorphic proteins that present peptides to T cell receptors to initiate adaptive immune response and set the boundaries between self and nonself. HLA typing at the allelic level determines mutations within coding sequences that alter the protein sequences. This is commonly performed by sequencing exons 2–4 of Class I genes (HLA-A, -B and -C) and exons 2 and/or 3 of Class II genes (HLA-DRB1 and -DQB1) (1). Due to the extreme diversity of HLA alleles in the population, sequence ambiguities frequently arise when the polymorphisms are outside the regions being typed and when different allelic combinations share the same sequence. Additional steps such as polymerase chain reaction (PCR) with sequence-specific primers (SSP) are necessary to resolve these ambiguities (2). Although this workflow determines the HLA genotypes at high resolution, it is laborious and expensive.
Next-generation sequencing has been applied to sequencing short-range amplicons of informative exons (3,4) with a recent transition to sequencing long-range amplicons of whole HLA genes on various platforms (5–7), suggesting a potential for parallel high-throughput HLA typing. Illumina sequencing of captured HLA genes is a cost-effective alternative that can bypass long-range PCRs. In fact, whole-exome sequencing (exome-seq) data, including those publicly available from the 1000 Genomes Project, should already contain adequate information for allelic HLA typing. However, this is challenging for several reasons: (i) reads specific to target HLA genes are not readily available, (ii) read coverage may vary substantially among different exons and between heterozygous alleles owing to capturing bias and (iii) the typical short read length and the level of polymorphism within the region increase the difficulty of differentiating near-identical alleles. Currently, there is no program to reliably accomplish this task given these challenges, and a recent report (8) demonstrated poor allelic HLA typing results from exome-seq data even at high coverage.
Here, we present a novel approach that includes an initial strategy to scout for target-specific reads and a core software named ATHLATES (Figure 1) for allelic HLA typing using Illumina exome-seq data with the typical 101 bp paired-end reads. Twenty such data sets were analyzed to predict the corresponding HLA genotypes at the allelic level. Fifteen of these data sets have adequate coverage for the target genes, and the in silico typing results of these samples were validated by conventional Sanger-based HLA-typing methods in a Clinical Laboratory Improvement Amendments licensed clinical laboratory that routinely performs typing in support of bone marrow and solid organ transplantation. With an overall concordance rate of 99%, ATHLATES outperforms HLAminer (8), the only other publicly available program that can derive HLA types from Illumina exome-seq data.
Figure 1.

Workflow of allelic HLA typing using exome-seq data. Exome-seq data are first filtered by comparison against all alleles of HLA genes obtained from IMGT/HLA database, and then fed into ATHLATES for in silico allelic HLA typing without human supervision.
MATERIALS AND METHODS
Nomenclature
The nomenclature of HLA alleles in this report follows the guidelines from the World Health Organization Nomenclature Committee for Factors of the HLA System (http://hla.alleles.org/nomenclature/naming.html). Allelic HLA typing refers to sequencing-based typing to determine variations in coding DNA sequences that alter the protein sequences. This is also commonly referred to as high-resolution typing or four-digit typing. There are also alleles that bear synonymous mutations and mutations within noncoding DNA, but resolution of these alleles is rarely necessary in clinical practice. In the IMGT/HLA database (9), the majority of HLA alleles are represented by full-length or partial complementary DNA (cDNA) sequences. Some HLA alleles have both cDNA and genomic DNA (gDNA) sequences deposited in the database. For simplicity, we also term a cDNA and/or gDNA sequence of an HLA gene an allele.
Scouting for target-specific reads/read-pairs
The exome-seq data are aligned against a multi-FASTA file that consists of all known alleles of HLA genes available from the IMGT/HLA database (Supplementary Table S1). The inclusion of gDNA sequences enhances our ability to capture reads spanning intron–exon boundaries, although they are available for only a fraction of alleles in the database. To account for this, we allow soft-clipping during alignment. In other words, it is sufficient to retain a read when it has a high-quality suffix–prefix alignment with cDNA sequences. Novoalign (http://www.novocraft.com/main/index.php) was used as the aligner, where no more than one edit distance was allowed. We keep the information when a read or read pair is aligned to multiple HLA genes. The alignment result is recorded in a compressed BAM format, from which we extract reads/read pairs aligned to a target HLA gene (e.g. HLA-A) with a customized BED file registering all alleles of this gene. Likewise, reads/read pairs that aligned to nontarget genes (e.g. all nonHLA-A genes in the reference) are extracted in the same manner.
Assembly
Reads/read pairs uniquely aligned to the target gene are used in the assembly step. They are first oriented to be consistent with the alignment so that their reverse complementary strands need not be considered. Paired-end reads aligned to the same reference allele are merged to form single reads based on the following method: let a paired-end read 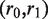 that aligned to the same allele be divided into substrings
that aligned to the same allele be divided into substrings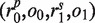 , where
, where 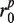 and
and 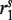 denote the prefix and suffix of
denote the prefix and suffix of 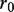 and
and 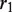 that do not overlap in the alignments, whereas
that do not overlap in the alignments, whereas 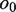 and
and 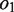 denote the overlapping substrings. Note that
denote the overlapping substrings. Note that 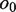 and
and 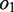 should be equal in length. When
should be equal in length. When 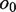 and
and 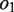 are not empty, they are merged to form string
are not empty, they are merged to form string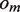 , where the IUPAC (International Union of Pure and Applied Chemistry) characters, or degenerate bases, are used to encode two different nucleotides merging at a position with one of them being a possible sequencing error. When
, where the IUPAC (International Union of Pure and Applied Chemistry) characters, or degenerate bases, are used to encode two different nucleotides merging at a position with one of them being a possible sequencing error. When 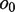 and
and 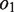 are empty, we encode the sequencing gap between
are empty, we encode the sequencing gap between 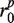 and
and 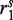 with degenerate bases ‘N’. Due to the existence of alleles that contain length polymorphisms (i.e. indels), we may not uniquely determine the fragment length for certain read pairs. The uncertainties regarding the degenerate bases and fragment length are resolved by identifying additional fragments that are likely to be obtained from the same genomic location during assembly (Supplementary Figure S1).
with degenerate bases ‘N’. Due to the existence of alleles that contain length polymorphisms (i.e. indels), we may not uniquely determine the fragment length for certain read pairs. The uncertainties regarding the degenerate bases and fragment length are resolved by identifying additional fragments that are likely to be obtained from the same genomic location during assembly (Supplementary Figure S1).
Reads containing sequencing errors typically contain low-frequency k-mers (substrings with length k set to be half of the read length) that occur only once or twice in the data (10). Because such reads may also belong to nontarget genes in exome-seq, we choose to exclude them from assembly rather than correcting them.
Next, each read is initialized to be a contig with the base frequency recorded at each position. Assuming that contigs sharing longer substrings are more likely to be from the same haplotype, ATHLATES prioritizes the comparison of contigs sharing longer substrings. ATHLATES also inspects high-frequency substrings first, as haplotype sequences such as exons with a higher read support can be recovered more reliably. Specifically, contigs are merged if they share common l-mers with l initially set to the value of the maximum read length and then iteratively decreased by a fixed amount until a minimum threshold (default 40) is reached. For each l value, contigs are decomposed into a set of l-mers, sorted in a decreasing order of frequency. For each l-mer in the sorted list, we assemble contigs sharing this l-mer through alignment, in which the relative positions of any two contigs can be determined in constant time by matching the l-mer. Because insertion/deletion errors are rare in Illumina sequencing, we disallow them when generating the full alignment. Two contigs are merged only if they are concordant at each alignment position. If degenerate bases in IUPAC code are present, their intersection should not be empty and the base present in this intersection is used in the assembled contig. Meanwhile, the base frequency for each position is accrued. This is used later to identify regions of a contig with low base support (≤2) that may result from insufficient exon capture or sequencing errors. As such regions do not provide reliable haplotype sequence information and may prevent further contig merging, they are replaced with degenerate base ‘N’. The prefix or suffix of a contig that consists of a string of Ns is trimmed, whereas internal and intermittent Ns are retained. We seek further contig merging when possible by repeating the above steps. To track the removal of existing contigs and the creation of new ones, we use a union-find algorithm (Supplementary Algorithm 1).
Identification of relevant alleles
With adequate coverage, we expect target exons to be well represented by assembled contigs. Next, we decompose each allele of a target gene into exons, for which we identify the best hit (i.e. a matching substring) among the contigs. The quality of a hit is determined by the length and similarity of matched substrings. We consider a shorter hit with a higher similarity to have a higher quality (Supplementary Figure S2). Hamming distance is used to quantify the differences between an exon and its hit. We only consider hits within a maximum Hamming distance of 2. Then an overall distance is calculated for each allele by summation of Hamming distances between all its exons and their best hits in the contigs. An ideal candidate allele should have a Hamming distance of zero. However, a true allele could have a nonzero distance for several reasons: (i) small exons and part of long exons may not be effectively captured in exome-seq (Supplementary Figure S2), and (ii) the subject may have a novel allele with a small number of mutations compared with a known allele. Therefore, it is reasonable to consider all alleles within a distance threshold (default 2). Although the threshold is a parameter adjustable by the user, a higher threshold is unlikely to be meaningful as many known alleles have almost identical protein coding sequences. Note that we exclude the following situations from calculation of the overall Hamming distance: (i) incompletely documented short exons (≤25 bp) and (ii) exons with no hits in the contigs. These exclusions may prevent unjustified penalties on partially sequenced/documented alleles in the database, or loss of novel alleles bearing inserted or deleted bases that inflate the Hamming distance dramatically when compared with a known allele. We consider an allele has adequate coverage if it satisfies the following criteria: (i) sufficient depth of sequencing (empirically we found that a minimum of 20-fold read coverage is necessary), (ii) the best hit for each of its exons, if identified, covers no less than a minimum percentage (de fault 85%) of the exon length and (iii) the summation of the exon lengths is no <70% of the overall cDNA length of this allele. The second criterion limits the tolerable partial miss of individual exons, whereas the third criterion limits the total amount of exons that can be missed (either not captured or not documented in the database). Note that the choice of these cutoff values is empirical and could be adjusted to be less or more stringent but it is not yet clear how to choose the optimal values. For each candidate allele identified, we report the following information: (i) exonic positions of mismatch compared with the best hit, termed U (stands for unsupported) positions, (ii) exonic positions not fully covered by the best hit, termed M (stands for missing) positions, (iii) total length of the covered region and (iv) the short exons excluded from distance calculation.
Inferring allelic pairs
Although we report all alleles conforming to the distance constraint, we choose a subset as the candidate list to represent the input data. An allele is excluded from the candidate list if it contains any exon with no hit. The candidate list consists of only alleles with distance zero on condition that they correspond to more than one type of protein coding sequences; otherwise, we include in the candidate list all alleles with distances less than or equal to one. Next, we choose two alleles with replacement from the candidate list and use a scoring scheme to measure the distance between the selected allelic pair and the remaining alleles. More specifically, let the candidate list consisting of n alleles be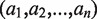 . The score of each allelic pair is initialized to be zero. Next, multiple sequence alignment (MSA) of these alleles is obtained. For alleles having the same protein coding sequence, only one of them is included in the MSA. Then the MSA is divided into different exons. Within each exon, we identify a set of variable positions, i.e. alignment positions with more than one type of nucleotides. Then, subsequences specified by the variable positions are obtained from all sequences in the MSA, which results in a list of strings
. The score of each allelic pair is initialized to be zero. Next, multiple sequence alignment (MSA) of these alleles is obtained. For alleles having the same protein coding sequence, only one of them is included in the MSA. Then the MSA is divided into different exons. Within each exon, we identify a set of variable positions, i.e. alignment positions with more than one type of nucleotides. Then, subsequences specified by the variable positions are obtained from all sequences in the MSA, which results in a list of strings 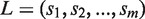 where
where  . L captures nonredundant haplotype information of the exonic region. For each allelic pair
. L captures nonredundant haplotype information of the exonic region. For each allelic pair 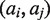 with
with 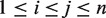 , we increment its score by
, we increment its score by 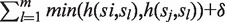 , where
, where 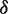 is initialized to be zero, and function
is initialized to be zero, and function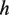 returns the Hamming distance between two strings except that (i) any U position in
returns the Hamming distance between two strings except that (i) any U position in 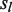 will be skipped for comparison, and (ii) for any M position in
will be skipped for comparison, and (ii) for any M position in 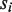 or
or 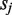 , when the base of
, when the base of 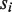 or
or 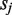 differs from the base in
differs from the base in 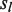 ,
, 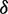 is increased by one. The former is to account for the fact that any unsupported base in the data should not be penalized and the latter is to penalize the bases in
is increased by one. The former is to account for the fact that any unsupported base in the data should not be penalized and the latter is to penalize the bases in 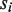 or
or 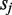 lacking read coverage when differing from bases having read support in
lacking read coverage when differing from bases having read support in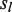 . In addition,
. In addition, 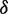 is increased by the number of U positions in
is increased by the number of U positions in 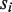 and
and 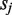 so that a correct homozygous allele pair should have a higher score compared with an alternative. Note that when allele
so that a correct homozygous allele pair should have a higher score compared with an alternative. Note that when allele 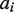 was not included in the MSA,
was not included in the MSA, 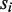 was obtained from the allele in the MSA that shares the same coding sequence with
was obtained from the allele in the MSA that shares the same coding sequence with 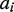 . After all exons have been examined, the pairs with the same minimum score are reported as the typing result.
. After all exons have been examined, the pairs with the same minimum score are reported as the typing result.
Evaluation measure
To measure the accuracy and to quantify the manual effort required to resolve ambiguities of the typing results, we used the measure of concordance rate: let the number of inferred allelic pairs for m HLA genes be 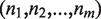 , among them
, among them 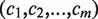 allelic pairs are consistent with the conventional typing results at four-digit level (same protein sequences from typed exons), then the concordance rate is given by
allelic pairs are consistent with the conventional typing results at four-digit level (same protein sequences from typed exons), then the concordance rate is given by 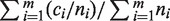 . Any ambiguity or wrong prediction would decrease this score. The calculation of concordance rate for ATHLATES is straightforward. However, because HLAminer does not infer allelic pairs but only assigns a likelihood score to each candidate allele, we choose the alleles with top two highest scores to be the predicted allelic pair. If more than two alleles share the same highest score, we consider all of the possible pairing among them. When only one allele is reported, a homozygous allelic pair is considered.
. Any ambiguity or wrong prediction would decrease this score. The calculation of concordance rate for ATHLATES is straightforward. However, because HLAminer does not infer allelic pairs but only assigns a likelihood score to each candidate allele, we choose the alleles with top two highest scores to be the predicted allelic pair. If more than two alleles share the same highest score, we consider all of the possible pairing among them. When only one allele is reported, a homozygous allelic pair is considered.
Configuration of other programs used
For HLAminer (v.1.0.5), the script HPTASRwgs_classI-II.sh was used with parameter ‘-i 1’ to obtain the best typing results. Novoalign (v.2.07.07) was run on nine cores with parameters ‘-t 30 -r all -l 80 -e 1 -i 230 140’. RAxML (v.7.3.3) was used to generate phylogenetic trees with parameters ‘-p 78960 -f a -x 12345 -# 1000 -m GTRGAMMA’.
Exome-seq data
The exome-seq data are from the 1000 Genomes Project (16 data sets; available at ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/data/) and an internal validation project at Genome Technology Access Center (GTAC) at Washington University (four data sets; available on request). Detailed information for these data sets is listed in Supplementary Table S2. The protocols for library preparation and exome capture were described previously (11). The protocols for the internal validation project at GTAC were similar, except that 3 μg of DNA was used as input, six cycles of PCR were performed to enrich for proper adaptor ligated fragments and the Agilent SureSelect Human All Exon v.2 kit was upgraded to v.3 kit. Sequencing was performed on Illumina platforms as 2 × 101 bp pair-end reads.
Laboratory validation of HLA typing
Allelic HLA typing was performed using SeCore HLA Sequencing Reagents (Life Technologies, Brown Deer, WI) per the manufacturer’s instructions. Briefly, exon 2–4 of HLA-A, -B and -C, exon 2 of HLA-DRB1 and exon 2 and 3 of HLA-DQB1 were amplified in five PCR reactions followed by Sanger sequencing from both the 5′ and 3′ ends. Next, some cis/trans ambiguities from the first round of sequencing were resolved by sequencing one haplotype of a heterozygous allelic pair with allele-specific primers, called Z primers. For HLA-DRB1 gene, a variation at codon 86 (GGT/GTG) shows dichotomy among most HLA-DRB1 alleles, and a Z primer specific for this variation was used preemptively during the first round of sequencing to reduce cis/trans ambiguities. Data analysis was performed with uTYPE 6.0 (Life Technologies, Brown Deer, WI). Equivalent allele pairs were reported with letter strings specified by the National Marrow Donor Program (http://bioinformatics.nmdp.org/HLA/Allele_Codes/Allele_Code_Lists/Allele_Code_Lists.aspx) and the IMGT/HLA Database (9). Cases with unresolved ambiguities were further examined by the SSP method. This was performed using the Olerup-SSP kit (Olerup SSP AB, Stockholm, Sweden). Panels of allele-specific primers were selected for genes with ambiguous typing results, and the patterns of positive PCR amplifications were correlated with specific alleles using worksheets provided by the manufacturer. In addition, sequence-specific oligonucleotide probe hybridization was performed for all samples using the LabType SSO kit (One Lambda, Canoga Park, CA) on a Luminex platform (Luminex, Austin, TX).
RESULTS
Previous methods have relied on the assumption that more reads would align to the correct alleles (6,8). However, this assumption may not apply to exome-seq data as there could be large differences in the coverage of different exons (Figure 2A and Supplementary Figure S3). ATHLATES assembles a set of contigs from target-specific (e.g. HLA-A) reads to fully represent individual exons from each allele of the target gene. The target-specific reads are obtained using two stages of filtering (Figure 1). The exome-seq data are first filtered against all alleles of HLA genes obtained from the IMGT/HLA database (Supplementary Table S1) to narrow down the searching space. Due to homology between alleles of different HLA genes, ATHLATES further filters out reads that align equally well to target and off-target genes (e.g. HLA-A and nonHLA-A), as these reads may introduce ambiguities to the assembly. In our samples, we have observed that up to 9% of reads are multi-mapped to target and off-target genes.
Figure 2.

Coverage of target genes by exome-seq data and comparison of HLA typing results among conventional typing, ATHLATES and HLAminer. (A) Fold coverage at exons of each of the five target HLA genes. Paired-end reads are aligned to the typed alleles of individual samples, and the fold coverage data from all 15 samples are presented. Median coverage (gray lines) and range (gray areas) are plotted over alignment positions for individual HLA genes. Dotted lines are exon boundaries, and the exons are numbered below each plot. The disruptions in curves are caused by mapping the reads to an alignment coordinate based on a MSA of all alleles of a target gene. The insertions present in rare alleles make the curves appear to be discontinuous. The one or two exons toward the 3′ end are exceedingly short (e.g. 5 bp for exon 8 of HLA-A) and hence, may not be reliably aligned against. (B) Concordance rates of HLA typing results by ATHLATES and HLAminer as compared with conventional Sanger-based method for the 15 data sets with adequate coverage.
Unlike existing short read assemblers (10,12), ATHLATES adopts distinct strategies to facilitate sequence assembly in the highly polymorphic HLA region. First, ATHLATES converts a read pair to a haplotype sequence when both ends are aligned to the same reference allele to effectively elongate the input reads. Second, because many fragments may have sequencing gaps between both ends, ATHLATES uses degenerate bases as placeholders at uncertain positions in the contig, which may be resolved based on information from reads merged into the contig later (Supplementary Figure S1). Degenerate bases also serve as an indicator of insufficient read support, which ATHLATES will factor in during allelic pair inference. Third, ATHLATES prioritizes assembly of contigs sharing longer substrings with higher frequencies.
Next, ATHLATES identifies relevant alleles whose exons match the contigs assembled at the previous step (Figure 1). The quality of each allele is measured by summing the Hamming distances between each individual exon and its best match in the contigs. During the matching process, ATHLATES prioritizes sequence similarity over the length of matching substrings (Supplementary Figure S2) for two reasons. First, alleles with almost identical sequences need to be differentiated; second, exons may be only partially sequenced resulting in degenerate bases in the contig. ATHLATES identifies alleles within a distance of 2, which allows inclusion of potential novel alleles closely related to known alleles. If no alleles within a distance of two are identified, ATHLATES will report no typing result for the gene.
Finally, assuming one or more alleles are identified, ATHLATES infers homozygous or heterozygous allelic pairs by considering all possible allelic pairs and checking if the allelic pairs are consistent with the data as represented by contigs. To achieve this, ATHLATES uses the principle of parsimony, which ensures allelic pairs fully supported by the data are prioritized over pairs consistent with but not fully supported by the data (Supplementary Figure S4A). The final allelic pairs should also explain as much of the information present in the data as possible (Supplementary Figure S4B). Meanwhile, the phasing information among discrete exons may be inferred (Supplementary Figure S4B) because exome-seq may not fully capture introns.
We applied the above approach to 20 Illumina exome-seq data sets (18 subjects of 5 ethnicities; two data sets are duplicates) from sources including the 1000 Genomes Project (13). Currently, according to the criteria described previously (section ‘Identification of relevant alleles’), 15 of these data sets are of adequate coverage, and ATHLATES generated allelic HLA typing from them followed by laboratory validation. The characteristics of these data sets and diversity of included HLA types are summarized in Supplementary Table S2 and Supplementary Figure S5, respectively. The remaining five data sets have insufficient coverage of the target HLA regions (Supplementary Figure S6). It is our algorithmic choice to avoid reporting any typing results in case of insufficient coverage instead of reporting probable results. Hence, these samples were rejected by ATHLATES and excluded from further analysis.
For the 15 data sets with adequate coverage, the median coverage of different exons of individual genes ranges approximately from 10 - to a 1000-fold after read filtering (Figure 2A). The coverage decays toward exon boundaries and varies significantly among exons (Supplementary Figure S3); the variation among exons is likely due to bias toward certain exons during exome capturing.
ATHLATES efficiently identified allelic pairs without human supervision. At a resolution that tolerates synonymous mutations in coding sequences, ATHLATES reported the correct allelic pairs for 74 out of 75 genes tested, an overall concordance rate of 99% compared with conventional typing (Supplementary Table S3). The only case of discordance occurred to HLA-A of HG01872, where in addition to the correct allelic pair, one more pair with one base difference was reported. Interestingly, read support for both exist in the exome-seq data. ATHLATES harnessed the advantage of clonal sequencing to distinguish cis/trans ambiguities encountered during Sanger sequencing (which are traditionally resolved by additional sequencing or PCR with SSPs). ATHLATES also detected polymorphisms in exons not covered by conventional methods, and was able to exclude additional allelic pairs (Supplementary Figure S7 and Supplementary Table S3).
The reporting style of ATHLATES is intuitive and straightforward. A sample report is shown in Supplementary Table S4, which demonstrates the typing of HLA-DQB1 gene for sample HG01757. Twenty-one alleles are initially identified to be within a distance of two. Four out of the 21 alleles have exact hits within the contigs assembled from the reads, and are included in the candidate list. Exhaustive comparison of the 16 possible allelic pairs and the contigs suggests that DQB1*02:01:01 has to be included because pairing among DQB1*03:03:02:01-03 cannot account for all the variants present in the contigs. On the other hand, pairing of DQB1*02:01:01 with any one of DQB1*03:03:02:01-03 can explain the contigs equally well, and are collectively reported as the final genotypes. The lack of resolution among DQB1*03:03:02:01-03 indicates that they could not be distinguished by inspecting exons alone. An example of inferring the phase of different exons while pairing candidate alleles to determine the final genotype is shown in Supplementary Figure S4B for HLA-B gene, sample HG01873. Among the four candidate alleles, two different haplotypes, designated as A and B, are present for each exon of exons 1–4. Sharing of the same exons among the candidate alleles and the lack of introns make it difficult to determine the phase relationship of these exons. However, the information within the assembled contigs suggests that all four exons must be heterozygous. Only one allelic pair, B*55:02:01 and B*35:03:01, results in heterozygosity at all four exons, which turns out to be the correct genotype.
ATHLATES significantly outperforms HLAminer (8), the only other publicly available program capable of HLA typing using exome-seq data. Without read filtering, which was not originally described as a prerequisite for its input, HLAminer could not finish the computation even after 10 days of running (three data sets tested). Using filtered reads as input (the input reads to ATHLATES as shown in Figure 1), HLAminer reported candidate alleles ranked by likelihood without inferring the final allelic pair(s) (Supplementary Table S3). After testing all data sets, the estimated overall concordance rate was 46% for HLAminer compared with conventional typing results, consistent with the previous report (8). The performance comparison between ATHLATES and HLAminer for individual genes is shown in Figure 2B. All of the above experiments were executed at a workstation with six-core 880 and 2400 MHz AMD processors with 250 GB RAM running GNU/Linux x86_64. ATHLATES typically finishes within 2 min, while HLAminer requires 10 min to complete.
As sequencing of captured exons is susceptible to allelic bias (14), we compared the coverage of heterozygous alleles at positions where they differ from each other (Supplementary Figure S8 and Supplementary Table S5). Statistically significant allelic biases are most frequently observed at exons 2 through 4 of Class I genes and exon 2 of Class II genes, and neighboring exons can also exhibit preference to different haplotypes. The patterns of allelic bias are highly reproducible in the two replicates examined. Almost all the bias at individual variant positions (99%) is within 80% of the total coverage (Supplementary Figure S9), which does not affect the HLA typing by ATHLATES.
DISCUSSION
Distinct from previous allelic HLA typing approaches (6,8) that mainly rely on read alignment, we proposed an alternative method, ATHLATES, which mainly relies on accurate recovery of exon sequences via assembly. While HLAminer (8) reports a list of candidate alleles for each target HLA gene, ATHLATES automatically infers the homozygous or heterozygous allelic pair that best explains the read data. Our method achieved accurate typing in exome-seq data with a minimum of 175 million paired-end reads and 10-fold coverage of at least 96% of the exome, whereas the five samples rejected by ATHLATES have up to 152 million paired-end reads and 10-fold coverage of 80–96% of the exome (Supplementary Table S2). However, the read count does not directly reflect whether the exons of HLA genes are sufficiently captured, which is dictated by the capturing method used including the conditions for target enrichment and the design of probes. We do not expect a computational method to overcome this problem, but the capacity of target enrichment and depth of sequencing will only improve as the technology continues to mature. We anticipate that the current method can also be adapted to other types of sequencing data sets, including those obtained from amplicons of HLA genes, whole genome sequencing, and RNA-seq (8,15).
It is worth noting that exome-seq data may not provide sufficient coverage in intronic regions; while this information is rarely necessary in current practice, it could be obtained if capture probes for HLA introns are included. In addition, adequate coverage is a prerequisite to obtain accurate results for exome-based HLA typing. Without satisfying this criterion, homozygous alleleic pairs may be inferred if only one of the heterozygous alleles is sufficiently captured; on par with the traditional typing, such cases require additional attention to verify the homozygosity. The detection of novel alleles by ATHLATES using exome-seq data remains challenging, although ATHLATES has features to tolerate a small number of substitutions and indels to prevent premature exclusion of possible novel alleles. In cases where no allelic pairs are reported from data sets with adequate coverage, users are advised to follow the clues from reported relevant alleles and perform further investigation by Sanger sequencing.
ATHLATES overcomes a bioinformatic hurdle in applying targeted Illumina sequencing to allelic HLA typing. Qualified exome-seq data sets from research projects can be analyzed to revisit HLA-disease associations with allelic resolution. In addition, the workflow outlined in this report paves the way to deep sequencing of captured HLA genes alone rather than the whole exome, which will allow multiplexing samples for high-throughput typing of bone marrow donors at reduced cost. The HLA genes can also be bundled with existing oncogene panels (16) for targeted Illumina sequencing to prepare leukemic patients for transplantation while characterizing the molecular profiles of their diseases.
ATHLATES is free for academic noncommercial use, and an academic licensing will be provided along with the software package on request.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Algorithm 1, Supplementary Tables 1–5 and Supplementary Figures 1–9.
FUNDING
Laboratory and Genomic Medicine Director discretionary fund from the Department of Pathology and Immunology, Washington University School of Medicine, and in part by Federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Department of Health and Human Services [contract no.: HHSN272200900018C]. Funding for open access charge: Department of Pathology and Immunology, Washington University School of Medicine and NIAID [HHSN272200900018C].
Conflict of interest statement. The authors have filed for provisional patent on the ATHLATES algorithm.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Bruce Birren and Barry Sleckman for their support of this project, and thank Eric Tycksen, Savita Shrivastava, Nicole Rockweiler and Maggie O’Guin for their technical assistance.
REFERENCES
- 1.Lind C, Ferriola D, Mackiewicz K, Heron S, Rogers M, Slavich L, Walker R, Hsiao T, McLaughlin L, D'Arcy M, et al. Next-generation sequencing: the solution for high-resolution, unambiguous human leukocyte antigen typing. Hum. Immunol. 2010;71:1033–1042. doi: 10.1016/j.humimm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 2.Erlich H. HLA DNA typing: past, present, and future. Tissue Antigens. 2012;80:1–11. doi: 10.1111/j.1399-0039.2012.01881.x. [DOI] [PubMed] [Google Scholar]
- 3.Gabriel C, Danzer M, Hackl C, Kopal G, Hufnagl P, Hofer K, Polin H, Stabentheiner S, and Proll J. Rapid high-throughput human leukocyte antigen typing by massively parallel pyrosequencing for high-resolution allele identification. Hum. Immunol. 2009;70:960–964. doi: 10.1016/j.humimm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Bentley G, Higuchi R, Hoglund B, Goodridge D, Sayer D, Trachtenberg EA, Erlich HA. High-resolution, high-throughput HLA genotyping by next-generation sequencing. Tissue Antigens. 2009;74:393–403. doi: 10.1111/j.1399-0039.2009.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erlich RL, Jia X, Anderson S, Banks E, Gao X, Carrington M, Gupta N, DePristo MA, Henn MR, Lennon NJ, et al. Next-generation sequencing for HLA typing of class I loci. BMC Genomics. 2011;12:42. doi: 10.1186/1471-2164-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C, Krishnakumar S, Wilhelmy J, Babrzadeh F, Stepanyan L, Su LF, Levinson D, Fernandez-Vina MA, Davis RW, Davis MM, et al. High-throughput, high-fidelity HLA genotyping with deep sequencing. Proc. Natl Acad. Sci. USA. 2012;109:8676–8681. doi: 10.1073/pnas.1206614109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiina T, Suzuki S, Ozaki Y, Taira H, Kikkawa E, Shigenari A, Oka A, Umemura T, Joshita S, Takahashi O, et al. Super high resolution for single molecule-sequence-based typing of classical HLA loci at the 8-digit level using next generation sequencers. Tissue Antigens. 2012;80:305–316. doi: 10.1111/j.1399-0039.2012.01941.x. [DOI] [PubMed] [Google Scholar]
- 8.Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, Holt RA. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012;4:95. doi: 10.1186/gm396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson J, Mistry K, McWilliam H, Lopez R, Parham P, Marsh SG. The IMGT/HLA database. Nucleic Acids Res. 2011;39:D1171–D1176. doi: 10.1093/nar/gkq998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe D. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boegel S, Lower M, Schafer M, Bukur T, de Graaf J, Boisguerin V, Tureci O, Diken M, Castle JC, Sahin U. HLA typing from RNA-Seq sequence reads. Genome Med. 2013;4:102. doi: 10.1186/gm403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spencer DH, Abel HJ, Lockwood CM, Payton JE, Szankasi P, Kelley TW, Kulkarni S, Pfeifer JD, Duncavage EJ. Detection of FLT3 internal tandem duplication in targeted, short-read-length, next-generation sequencing data. J. Mol. Diagn. 2013;15:81–93. doi: 10.1016/j.jmoldx.2012.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.