

Abstract
β-Elemene is a new anticancer compound extracted from the Chinese medicinal herb Rhizoma zedoariae. We have shown previously that β-elemene increases cisplatin cytotoxicity and enhances cisplatin sensitivity via blocking cell cycle progression at G2/M phase in resistant ovarian tumor cells. In the current study, we asked whether β-elemene-augmented cisplatin activity in ovarian carcinoma cells is mediated through the induction of apoptosis. Here, we show that β-elemene triggered apoptotic cell death in chemoresistant human ovarian cancer A2780/CP and MCAS cells in a dose- and time-dependent fashion, as assessed by six different apoptosis assays. Intriguingly, β-elemene was a stronger inducer of apoptosis than cisplatin in this model system, and a synergistic effect on induction of cell death was observed when the tumor cells were treated with both agents. Furthermore, β-elemene plus cisplatin exposure significantly disrupted the mitochondrial transmembrane potential (ΔΨm) and increased the release of cytochrome c from mitochondria into the cytoplasm. The combination treatment with both compounds also induced increases in caspase-3/8/9 activities and caspase-9 cleavage, enhanced protein expression of Bax and phosphorylation of Bcl-2 at Ser-70, and reduced the protein levels of Bcl-2 and Bcl-XL in the platinum-resistant ovarian cancer cells. Taken together, these data indicate that β-elemene sensitizes chemoresistant ovarian carcinoma cells to cisplatin-induced apoptosis and that the augmented effect of β-elemene on cisplatin cytotoxicity and sensitivity in resistant ovarian tumor cells is mediated through a mitochondria- and caspase-dependent cell death pathway.
Keywords: Apoptosis, Cisplatin resistance, β-Elemene, Chinese medicine, Ovarian cancer
Introduction
Human ovarian cancer is the leading cause of gynecological cancer-related mortality in the United States, with a high morbidity rate, poor prognosis, and low 5-year survival rate, especially in refractory cases [1, 2]. Cisplatin has been a frontline chemotherapeutic agent for a variety of cancers, including ovarian epithelial cancer. However, its effectiveness as a potent anticancer agent has been greatly limited by drug resistance and side effects, which eventually lead to treatment failure in clinical settings [3]. The molecular mechanisms of tumor resistance to cisplatin involve multiple factors, including impaired apoptosis, reduced DNA binding, and increased DNA repair [4]. It is necessary to pursue novel strategies to increase sensitization of cancer cells to cisplatin, minimize resistance to the drug, alleviate its side effects, and increase its effectiveness. To this end, we investigated the role of β-elemene as an adjuvant in sensitizing drug-resistant human ovarian cancer cells to cisplatin and the underlying mechanism in an in vitro study.
β-Elemene, a novel anticancer compound, was isolated from the Chinese medicinal herb Rhizoma zedoariae. It exhibits strong antitumor activity with minimal side effects. In the past several years, our laboratory has shown that β-elemene exerts potent antiproliferative and antitumor activities in a broad range of human cancer cell types, including lung cancer, prostate cancer, and ovarian cancer cells [5–10]. Importantly, we have demonstrated that β-elemene selectively increases cisplatin cytotoxicity and augments cisplatin sensitivity in chemoresistant human ovarian cancer cells. We have also reported that this enhanced chemosensitivity is attributable to the capacity of β-elemene to block cell cycle progression at G2/M phase [5].
Apoptosis, or programmed cell death, is a highly regulated cellular process between cell proliferation and cell death [11, 12]. During this process, cells undergo organized self-destruction as a protective biological response to maintain physiological homeostasis when cells meet harmful or abnormal stimuli [13–17]. Dysfunction of the apoptotic death process may promote the proliferation of abnormal cells and eventually lead to cancer. Apoptosis occurs by two pathways: the intrinsic and extrinsic pathways. The intrinsic pathway is centered on the release of cytochrome c from the mitochondrial outer membrane, in response to a disruption of the balance between pro-survival (e.g., Bcl-2 and Bcl-XL) and pro-apoptotic members of the Bcl-2 family (e.g., Bax and Bak). The extrinsic pathway is triggered by ligand binding to specific death receptors on the cell surface. Both pathways lead to the activation of caspase cascades. Apoptotic signaling pathways are the most promising therapeutic targets for cancer treatment [17–26].
Cisplatin induces a significant apoptotic response in cancer cells, and impaired apoptosis is one of the molecular mechanisms of chemoresistance to cisplatin in cancer cells. Considering that β-elemene blocks the cell cycle at G2/M phase and that cells accumulated in G2/M phase often enter the apoptotic process, we hypothesized that β-elemene sensitizes resistant human ovarian cancer cells to cisplatin through the induction of apoptosis. To test this hypothesis, we designed a series of experiments to detect apoptotic responses in cancer cells treated with either β-elemene or cisplatin alone, or the combination of both drugs. We found that β-elemene dramatically increased cisplatin anticancer activity in resistant human ovarian cancer cells by the induction of a remarkable apoptotic response mediated by a mitochondria- and caspase-dependent cell death pathway. These findings may have profound implications in ovarian cancer chemotherapy.
Materials and methods
Chemicals and immunoreagents
The (−)-β-elemene (98 % purity) was obtained from Yuanda Pharmaceuticals, Ltd, Inc. (Dalian, China). Cisplatin, dimethyl sulfoxide (DMSO), and propidium iodide (PI) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Antibodies against caspase-3, caspase-8, caspase-9, Bax, Bcl-2, Bcl-XL, cytochrome c, β-actin, peroxidase-labeled anti-rabbit immunoglobulin G (IgG), Blotto B, and ECL Western blotting system were all purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Phospho-Bcl-2 (Ser-70) was purchased from Cell Signaling Technology (Beverly, MA, USA).
Cell culture and cytotoxicity assay
The human cisplatin-resistant ovarian cancer cell lines A2780/CP and MCAS have been descried previously [27, 28]. Cells were cultured in monolayer using RPMI 1640 medium (Invitrogen, Gaithersburg, MD, USA) supplemented with 10 % (v/v) fetal calf serum, 50 units/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). Cells were grown in logarithmic growth at 37 °C in a humidified atmosphere consisting of 5 % CO2 and 95 % air. Cells were routinely tested for mycoplasma infection using a commercial assay system (MytoTect; Invitrogen), and new cultures were established monthly from frozen stocks. All media and reagents contained <0.1 ng/ml endotoxin as determined by Limulus polyphemus amebocyte lysate assay (Whittaker Bioproducts, Walkersville, MD, USA). Before starting the experiments, cells were grown to 70–80 % confluence after sub-culturing. Cisplatin was initially dissolved in phosphate-buffered saline (PBS) without Ca2+or Mg2+at 5 mM. Cisplatin and β-elemene were serially diluted in culture medium, respectively, to obtain the desired drug treatment concentrations. The drug treatment and cytotoxic effects of β-elemene, cisplatin, or the combination of both drugs on cell survival were described previously [5].
Annexin V assay
Annexin V assay was used to detect apoptotic cells by staining cells with both annexin V-FITC and PI. Cells were treated with either β-elemene or cisplatin alone, or combination of both drugs, respectively. After 48-h treatment, 1 ×106 cells/ml were washed twice with PBS and resuspended in 300 μl binding buffer [10 mM HEPES, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, pH 7.4]. Three microliters of annexin V-FITC (Caltag Laboratories, Burlingame, CA, USA) was added to the cells. Cells were incubated on ice in the dark for 1.5 h and washed with 4 ml HEPES buffer once. Ten microliters of PI (50 μg/ml) was added to these cells. Cells were incubated for another 20 min and then analyzed within 30 min by FACSCalibur (Becton–Dickinson, San Diego, CA, USA). The CellQuest Pro software (Becton–Dickinson) and the ModFit LT software (Verity Software House, Inc., Topsham, Maine, USA) were used to determine the distribution of apoptotic cells. The same experiments were repeated for three times.
TUNEL assay
For in situ detection of apoptotic nuclei in A2780/CP cells, the terminal deoxynucleotidyltransferase-mediated deoxy-UTP-fluorescein nick end-labeling (TUNEL) assay was performed using an In Situ Cell Death Detection kit (Roche, Germany) according to the procedures described by the manufacturer. In brief, after the desired treatments, cells were washed with PBS, placed in Bouin’s fixative for at least 24 h, dehydrated in alcohol, and paraffin-embedded using standard protocols. Sections of 5-μm thickness were mounted on glass slides and stained with hematoxylin and eosin and the TUNEL method. After rehydration, the sections were post-fixed in 4 % paraformaldehyde-PBS (pH 7.2), rinsed in PBS, and incubated for 1 h at room temperature with a mixture containing fluorescein-deoxy-UTP and terminal deoxynucleotidyltransferase following the manufacturer’s instructions. Cells with brown granules in the nucleus were TUNEL-positive cells. To determine the percentage of positive cells, more than 200 cells were counted under a light microscope.
DNA fragmentation was detected as a marker of apoptotic cell death in situ in MCAS ovarian cancer cells using an Apoptosis Cell Detection System (Roche Molecular Biochemicals, Indianapolis, IN, USA) in an established TUNEL assay in our laboratory. In brief, ovarian cancer cells were collected following β-elemene and/or cisplatin treatment, and the collected cell pellets were processed into formalin-fixed, paraffin-embedded cell blocks. Sections cut from the cell blocks were deparaffinized, washed three times with PBS, and incubated for 60 min at 37 °C in a humidified atmosphere in the dark with 50 μl of TUNEL reaction mixture. After three rinses with PBS, the sections were incubated with 50 μl of Converter-AP in a humidified chamber for 30 min at 37 °C. The sections were again rinsed three times with PBS and then incubated with 50 μl of substrate solution at 25 °C in the dark for a period of time adjusted for optimal staining. Finally, the slides were rinsed three times with PBS and mounted under a glass cover slip for analysis by light microscopy.
DNA fragment detection by enzyme-linked immunosorbent assay (ELISA)
Cells were seeded at density 1 ×104 cells/well in 96-well plates for overnight. Cells were then treated with β-elemene alone (50 μg/ml), cisplatin alone (20 μM), or cisplatin in combination with β-elemene for 24, 48, and 72 h. After desired treatment, cells were incubated in lysis buffer for 30 min to obtain cytoplasmic lysates. A cell death detection ELISA kit (Cell Death Detection ELISAplus; Boehringer Mannheim, Indianapolis, IN, USA) was used to quantitatively determine cytoplasmic histone-associated DNA oligonucleosome fragments associated with apoptotic cell death according to the manufacturer’s instructions. Briefly, samples were incubated in microtiter plates adsorbed with mouse anti-histone antibody (clone H11-4) to bind histone-associated DNA oligonucleosomes uncovered by endonuclease-mediated DNA nicking. Plates were washed, and nonspecific binding sites saturated with blocking buffer. Bound samples were then reacted with anti-mouse DNA monoclonal antibody (MCA-33) and then conjugated with peroxidase. To determine the amount of retained peroxidase, 2,2′-azino-di-(3-ethylbenzthiazoline sulfonate) (ABTS) was added as a substrate. The absorbance was read using a 96-well Opsys MR™ Microplate Reader (Thermo Labsystems, Chantilly, VA, USA) at 405 nm. The enrichment of mono- and oligonucleosomes released into the cytoplasm was calculated as absorbance of sample cells/absorbance of control cells. The apoptotic index was calculated as an enrichment factor, that is, the ratio of the result compared with the control set arbitrarily at 1.0. Each experiment was performed in triplicate, and means and standard deviations were calculated.
Caspases enzymatic activity assay
Caspase-3, caspase-8, and caspase-9 enzymatic activities were measured using the CasPASE™ Apoptosis Assay kit (Geno Technology, Inc., MO, USA) according to the manufacturer’s protocol. In brief, after being treated with either β-elemene or cisplatin alone, or the combination of both drugs at the indicated concentrations for 24 and 48 h, a total of 1 ×107 cells were harvested and washed twice with ice-cold PBS. Cytosolic extracts were prepared by repeated 5 cycles of freezing and thawing in 200 μl of lysis buffer, centrifuged at 12,000 rpm for 30 min at 4 °C. Cell lysates (50 μl) were diluted with 2 ×CasPASE™ buffer (50 μl) in 96-well plates in duplicate and incubated at 37 °C for 2 h with 5 μl 1 mM caspases substrates (Ac-LEHD-AFC for caspase-9, Ac-LETD-AFC for caspase-8, Ac-DEVD-AFC for caspase-3). The absorbance of the cleaved substrate was measured by a 96-well Opsys MR™ Microplate Reader (Thermo Labsystems) at a test wavelength of 405 nm. Nonapoptotic cell lysate was used as the negative control. The buffer without cell lysate was used as the blank.
Protein extraction and Western blot analysis
Cells treated with β-elemene alone, cisplatin alone, or their combinations at the indicated concentrations were harvested by trypsinization following 48-h incubation. After washing with ice-cold PBS, the cells were lysed on ice for 30 min in a mammalian cell lysis buffer (Quality Biological, Inc., Gaithersburg, MD, USA), containing 10 μl/ml 200 mM phenylmethylsulfonyl fluoride (PMSF), 10 μl/ml 100 mM sodium orthovanadate, and 10 μg/ml aprotinin. Cellular extracts were clarified by centrifugation at 12,000 rpm at 4 °C for 30 min, and protein concentrations were determined using the Bradford assay (Bio-Rad, Richmond, CA, USA). Sixty micrograms of proteins from whole-cell lysates was mixed 1:1 with 2×sodium dodecyl sulfate (SDS) protein gel solution (Quality Biological), heated for 5 min at 95 °C, separated by 10 % SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto nitrocellulose membrane (Schleicher & Schuell BioScience, Inc., Keene, NH, USA). After blocking in Blotto B for 1 h at room temperature, membranes were incubated overnight at 4 °C with the specific primary antibodies (dilutions were 1:100–1:300). Filters were washed with TBS/0.1 % Tween 20 solution, incubated with anti-rabbit peroxidase-conjugated secondary antibody (dilution was 1:10,000), wash again, and developed with enhanced chemiluminescence substrate according to the manufacturer’s instructions. The protein bands were visualized using X-ray films (Eastman Kodak, Rochester, NY, USA). All blots were representative of three independent experiments.
Detection of cytochrome c release into the cytosol
The mitochondrial and cytosolic fractions were isolated using Mitochondrial Isolation Kit (Sigma-Aldrich). Briefly, 3 ×107 cells were harvested and washed with PBS. The cells were suspended in 10 volumes of mitochondrial extraction buffer A containing 2 mg/ml albumin and homogenized on ice by a Wheaton Dounce homogenizer. Unbroken cells and nuclei were removed by centrifugation at 600g for 5 min at 4 °C. The supernatant was further centrifuged at 11,000g for 10 min. The supernatant was saved as a cytosolic fraction while the precipitate was dissolved in storage buffer A and saved as the mitochondrial fraction. The cytosolic fraction was analyzed by Western blotting with an anti-cytochrome c monoclonal antibody.
Measurement of mitochondrial membrane potential by flow cytometry using BD MitoSensor™ reagent
Changes in mitochondrial membrane potential (ΔΨm) during the early stages of apoptosis were assayed using an ApoAlert™ Mitochondrial Membrane Sensor kit (BD Biosciences Clontech, Palo Alto, CA, USA). Briefly, 1 ×106 cells were harvested, suspended in 1 ml of Mito-Sensor reagent (final concentration, 5 μg/ml), and incubated at 37 °C in a 5 % CO2 incubator for 20 min. The cells were washed with 1 ml of incubation buffer and collected by centrifugation at 350×g for 5 min. The cell pellet was suspended in incubation buffer and analyzed by flow cytometry. The green fluorescence represented the geometric mean fluorescence of the cells. Higher geometric mean fluorescence indicated lower ΔΨm and a higher incidence of apoptosis.
Statistical data analysis
Data are presented as mean ± SD. Student’s t test was used to analyze the differences between the means of treatment groups and the control group. Differences with a P value of less than 0.05 were considered statistically significant.
Results
β-Elemene enhanced cisplatin-induced apoptotic membrane changes in ovarian cancer cells, as detected by annexin V binding
Translocation of phosphatidylserine to the outer surface of the cytoplasmic membrane is an early feature of apoptosis. Annexin V and propidium iodide (PI) binding was used to evaluate the surface expression of phosphatidylserine. Cells staining with annexin V alone have early apoptotic changes and intact cell membranes, whereas cells staining with annexin V and PI have membrane disintegration consistent with necrosis or a late stage of apoptosis. A2780/CP cells treated with both β-elemene and cisplatin for 48 h exhibited a significant increase in apoptosis and necrosis, as compared with untreated control cells, cells treated with β-elemene alone, or cells treated with cisplatin alone (Fig. 1a). The percentages of apoptosis plus necrosis in A2780/CP cells after each treatment were 1.35 % (untreated control cells), 20.17 % (β-elemene alone), 7.09 % (10 μM cisplatin alone), 10.41 % (20 μM cisplatin alone), 54.74 % (β-elemene plus 10 μM cisplatin), and 59.98 % (β-elemene plus 20 μM cisplatin). Similar data were obtained in MCAS cells (Fig. 1b). The percentages of early plus late apoptosis in MCAS cells after each treatment were 0.1 % (untreated control cells), 24.81 % (β-elemene alone), 8.54 % (cisplatin alone), and 58.15 % (β-elemene plus cisplatin). The increases in the surface expression of phosphatidylserine suggest that β-elemene augments cisplatin-induced apoptosis in resistant ovarian cancer cells.
Fig. 1.

β-Elemene increased cisplatin-induced cell membrane changes during apoptosis in ovarian cancer cells, as detected by annexin V staining. A2780/CP or MCAS cells were treated with β-elemene alone, cisplatin alone, or β-elemene and cisplatin in combination at the indicated concentrations. After 48 h, the cells were harvested, stained using a Vybrant Apoptosis Assay kit, and analyzed by flow cytometry for annexin V-FITC and PI dual labeling. The results in a A2780/CP cells and b MCAS cells are shown. β-Elemene or cisplatin alone induced some change in the cell membrane. However, the combination of β-elemene and cisplatin resulted in considerable apoptotic membrane changes. Untreated cells were used as the negative control. UL upper left, represents necrotic cells (%); UR upper right, represents late apoptotic cells (%); LL lower left, represents viable cells (%); LR lower right, represents early apoptotic cells (%)
β-Elemene increased cisplatin-induced apoptotic nuclei in ovarian carcinoma cells, as detected by in situ TUNEL assay
A TUNEL assay was performed to detect apoptotic nuclei in A2780/CP and MCAS cells treated with β-elemene alone, cisplatin alone, or the combination of both agents for 24, 48, or 72 h. Representative light photomicrographs of apoptotic A2780/CP cells at 48 h are shown in Fig. 2a–d. At all three time points, the TUNEL-positive rates were significantly higher in cells treated with both β-elemene and cisplatin than in untreated cells (#P <0.01) or in cells treated with β-elemene alone or cisplatin alone (*P <0.05) (Fig. 2e). Similar results were observed in MCAS cells using a different apoptosis cell detection kit for the TUNEL assay (Fig. 3a–d). These findings demonstrate that β-elemene significantly increases cisplatin-induced apoptotic nuclei in resistant ovarian carcinoma cells.
Fig. 2.

β-Elemene promoted cisplatin-induced increases in apoptotic nuclei in A2780/CP cells, as detected by in situ TUNEL assay. Cells were treated with β-elemene (50 μg/ml) alone, cisplatin (20 μM) alone, or the combination of both agents as indicated for 24, 48, or 72 h. Cells were fixed with 4 % formaldehyde and labeled by TUNEL. In situ apoptotic responses were evaluated under a light microscope. a–d Apoptotic changes in A2780/ CP cell nuclei are evident after 48 h of drug treatment. e Bar graph showing the percentages of TUNEL-positive cells after 24, 48, and 72 h of drug treatment as indicated. Values are mean ± SD (bars) of triplicate samples. #P <0.01 compared with untreated control cells, and *P <0.05 compared with cells treated with either β-elemene or cisplatin alone. ELE 50, β-elemene alone; CDDP 20, cisplatin alone; ELE 50 + CDDP 20, the combination of β-elemene and cisplatin
Fig. 3.

Effect of β-elemene and cisplatin on apoptosis in human ovarian carcinoma MCAS cells, as assessed by the TUNEL assay. MCAS cells were treated with 50 μg/ml β-elemene alone (b), 20 μM cisplatin alone (c), or 50 μg/ml β-elemene plus 20 μM cisplatin (d) for 48 h. Untreated cells were used as the control (a). After treatment, the cells were collected, processed into paraffin-embedded cell blocks, sectioned at 5 μm, and evaluated by the TUNEL assay. TUNEL-positive cells were observed by light microscopy. Photomicrographs are representative of three independent experiments. DNA fragmentation consistent with apoptotic cell death is seen in MCAS cells treated with β-elemene (b) or cisplatin (c), and an additive effect is observed in cells treated with both cisplatin and β-elemene (d)
Effect of β-elemene and cisplatin on apoptosis in ovarian tumor cells as determined by the histological identification of nuclear karyorrhexis
Karyorrhexis is the destructive fragmentation of the nucleus of a dying cell, whereby its chromatin is distributed irregularly throughout the cytoplasm. It can occur as a result of either apoptosis or necrosis, although during apoptosis, the nucleus usually dissolves into apoptotic bodies after karyorrhexis. To confirm the effect of β-elemene and cisplatin on apoptotic cell death in chemoresistant ovarian carcinoma cells, we assessed nuclear karyorrhexis by histological identification in A2780/CP cells following exposure to the compounds (Fig. 4). The percentages of cells exhibiting karyorrhexis indicate that cisplatin or β-elemene independently induced apoptotic cell death in A2780/CP ovarian cancer cells in a concentration-dependent fashion and that β-elemene obviously promoted cisplatin-triggered apoptosis in this model system (Fig. 4a). Intact nuclei were observed in the untreated control cells (Fig. 4b), while typical cell morphology of karyorrhexis was observed in cells exposed to β-elemene and/or cisplatin (Fig. 4c).
Fig. 4.

Effect of β-elemene and cisplatin on apoptosis in A2780/CP ovarian tumor cells, as determined by the histological identification of nuclear karyorrhexis. Cells were treated with β-elemene (ELE) alone, cisplatin (CDDP) alone, or β-elemene plus cisplatin for 48 h at the indicated concentrations. Untreated cells were used as the control. Cells were harvested, processed into cell blocks, sectioned, and evaluated after staining with hematoxylin and eosin. Cell morphology was observed, and cells were counted under a light microscope. Cells with karyorrhectic nuclear features are reported as a percentage of the total cells counted (a). Cisplatin or β-elemene independently induced apoptosis in a dose-dependent manner, and an additive effect was observed in cells treated with both drugs. P <0.05 for cisplatin plus β-elemene versus cisplatin alone or β-elemene alone. Representative images from three separate experiments show intact nuclei stained with hematoxylin and eosin in untreated cells (b) and karyorrhectic nuclei in β-elemene- and cisplatin-treated cells (c)
β-Elemene increased cisplatin-induced DNA fragmentation in ovarian carcinoma cells
DNA degradation occurs several hours before plasma membrane breakdown during apoptotic responses. The enrichment of mono- and oligonucleosomes in the cell cytoplasm, signifying apoptosis, can be quantified by ELISA using an apoptosis detection kit. Compared with untreated control cells, the A2780/CP cells treated with either β-elemene or cisplatin alone showed a slight enrichment of nucleosomes (Fig. 5). In contrast, cells treated with both β-elemene and cisplatin exhibited a dramatic enrichment of nucleosomes (#P <0.01 and ##P <0.05 compared with untreated cells; *P <0.01 and **P <0.05 compared with β-elemene alone or cisplatin alone). Experiments with the other human ovarian tumor cell line gave similar results (data not shown). Thus, β-elemene significantly increases cisplatin-induced DNA fragmentation in resistant ovarian cancer cells.
Fig. 5.

β-Elemene increased cisplatin-induced DNA fragmentation in A2780/CP cells, as quantified by cell death detection ELISA. Cells were treated with β-elemene (50 μg/ml) alone, cisplatin (20 μM) alone, or the combination of both drugs for 24, 48, or 72 h. Untreated cells were used as the control. After treatment, the cells were incubated in lysis buffer for 30 min to obtain cytoplasmic lysates. A cell death detection ELISA kit was used to quantitatively determine cytoplasmic histone-associated DNA oligonucleosome fragments associated with apoptotic cell death. ELE 50, β-elemene alone; CDDP 20, cisplatin alone; ELE 50 + CDDP 20, the combination of β-elemene and cisplatin. Data are representative of three independent experiments. Values are mean ± SD (bars) of triplicate samples. #P <0.01 and ##P <0.05 compared with untreated cells, and *P <0.01 and **P <0.05 compared with β-elemene alone or cisplatin alone
β-Elemene-promoted cisplatin-induced apoptosis in ovarian cancer cells was caspase dependent
Caspase activation is a critical signaling event in apoptotic pathways. To determine which caspases were involved in the apoptotic response to β-elemene and cisplatin, we measured the enzymatic activities of caspase-3, caspase-8, and caspase-9 in A2780/CP cells treated with both drugs. After cells were treated with the β-elemene plus cisplatin, the caspase-3, caspase-8, and caspase-9 activities were significantly increased, at both 24 and 48 h (#P <0.01 and ##P <0.05 compared with untreated cells; *P <0.01 and **P <0.05 compared with β-elemene alone or cisplatin alone) (Fig. 6a–c). The enhanced induction of apoptosis by the combination of β-elemene and cisplatin was blocked by the general caspase inhibitor Z-VAD-FMK (data not shown). Apoptosis triggered by β-elemene plus cisplatin was further confirmed by Western blot analysis showing the cleavage of caspase-9 in A2780/CP cells (Fig. 6d).
Fig. 6.

β-Elemene enhanced cisplatin-induced caspase activation in A2780/CP cells. Cells were treated with β-elemene alone (50 μg/ml), cisplatin alone (20 μM), or the combination of both drugs for 24 or 48 h. Untreated cells were used as the control. Cell lysates were prepared and assayed for caspases activities. The relative caspase activity was determined by calculating the fold increase relative to caspase activity in untreated cells. Caspase-3 (a), caspase-8 (b), and caspase-9 activities (c) are shown. ELE 50, β-elemene alone; CDDP 20, cisplatin alone; ELE 50 + CDDP 20, the combination of β-elemene and cisplatin. Data are the mean ± SD of three separate experiments. #P <0.01 and ##P <0.05 compared with untreated cells, and *P <0.01 and **P <0.05 compared with β-elemene alone or cisplatin alone. d Western blot analysis of caspase-9 cleavage. Cell lysates were prepared after 24 or 48 h of drug treatment at the indicated concentrations. Proteins (60 μg) were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and probed with appropriate primary and secondary antibodies. Proteins were visualized by enhanced chemiluminescence. Equal protein loading was verified by using an anti-β-actin antibody
β-Elemene-promoted cisplatin-induced apoptosis in ovarian cancer cells was mitochondria dependent
The reduction of mitochondrial transmembrane potential (ΔΨm) is a hallmark of apoptosis. The loss of ΔΨm has been shown to result in a mitochondrial permeability transition, resulting in the release of cytochrome c and other apoptogenic proteins from mitochondria [29]. To determine whether the mitochondria-dependent pathway was involved in the induction of apoptosis by β-elemene and cisplatin treatment, we examined the ΔΨm and cytochrome c release from the mitochondria during early stages of apoptosis in β-elemene- and cisplatin-treated A2780/CP cells. The ΔΨm was assessed by BD MitoSensor™ reagent staining. The geometric mean fluorescence, indicating loss of ΔΨm, was significantly higher in cells treated with β-elemene plus cisplatin than in untreated cells or in cells treated with β-elemene or cisplatin alone (Fig. 7a). The ΔΨm was reduced in A2780/CP cells after treatment with β-elemene and/or cisplatin for 48 h. We collected similar data from the other human ovarian carcinoma cells (data not shown). Thus, β-elemene and/or cisplatin treatment significantly disrupted ΔΨm. Western blot analysis performed with the cytosolic and mitochondrial fractions isolated from the cells at 48 h following drug exposure revealed that the cytosol of untreated cells contained no cytochrome c, whereas cytochrome c had accumulated in the cytosol of cells treated with the combination of β-elemene and cisplatin (Fig. 7b). The increase in cytosolic cytochrome c was consistent with the change in ΔΨm.
Fig. 7.

β-Elemene increased cisplatin-induced ΔΨm disruption and cytochrome c release in A2780/CP cells. Cells were treated with β-elemene (50 μg/ml) alone, cisplatin (10 or 20 μM) alone, or the combination of both drugs for 48 h. Untreated cells were used as the control. a The loss of ΔΨm was detected by flow cytometry as the geometric mean fluorescence of cells, where higher geometric mean fluorescence indicates lower ΔΨm and a higher incidence of apoptosis. Shown are the geometric means of cells after different treatments, indicating a reduction of ΔΨm in A2780/CP cells after exposure to β-elemene and/or cisplatin. b Treated A2780/CP cells were harvested and lysed. The mitochondrial and cytosolic fractions were prepared using a mitochondria isolation kit. Cytochrome c in the mitochondrial (M) and cytosolic (C) fractions was analyzed by Western blotting with anti-cytochrome c antibody. Equal protein loading was verified by using an anti-β-actin antibody
Alterations in Bcl-2 family protein levels in β-elemene-and cisplatin-treated ovarian carcinoma cells
Bcl-2 family proteins are important regulators of apoptosis [16, 30]. Western blot analysis of cells treated with the combination of β-elemene and cisplatin revealed significantly downregulated expression of Bcl-2 and Bcl-XL proteins, elevated levels of Bax protein, and increased phosphorylation of Bcl-2 at Ser-70 (Fig. 8).
Fig. 8.
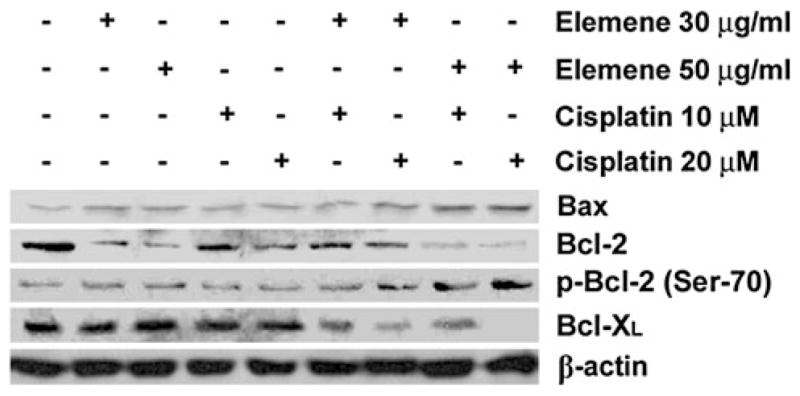
β-Elemene enhanced cisplatin-induced changes in Bcl-2 family protein levels in A2780/CP cells. Cells were treated with β-elemene (30 or 50 μg/ml) alone, cisplatin (10 or 20 μM) alone, or the combination of both drugs for 48 h. The cells were harvested, and whole-cell protein lysates were prepared as described in the “Materials and methods.” The levels of Bax, Bcl-2, and Bcl-XL proteins, and p-Bcl-2 (Ser-70) phosphorylation were analyzed by Western blotting. Equal protein loading was verified using an anti-β-actin antibody
Discussion
β-Elemene is a new antitumor compound with potent anti-growth and antiproliferative activities in a broad range of cancer cell types [5–10]. However, the molecular mechanisms underlying its anticancer activity are not fully known. Our previous study has shown that β-elemene significantly enhances the cytotoxicity of cisplatin and sensitizes cancer cells to cisplatin in the chemoresistant human ovarian cancer A2780/CP cell line and that this sensitization is mediated via cell cycle arrest at G2/M phase [5]. In the present study, we demonstrated that β-elemene strongly promotes cisplatin-induced apoptotic responses in A2780/CP and MCAS ovarian carcinoma cells. This suggests that the antitumor activity of β-elemene may involve multiple molecular targets. This hypothesis is supported by several lines of evidence from the present study.
First, β-elemene increased cisplatin-induced cell membrane changes during apoptosis in A2780/CP and MCAS cells, as shown with an annexin V binding assay. Translocation of phosphatidylserine to the outer surface of the cytoplasmic membrane is an early feature of apoptosis. The surface expression of phosphatidylserine was markedly increased after treatment of A2780/CP or MCAS cells with both β-elemene and cisplatin, compared with treatment with either agent alone.
Second, β-elemene increased cisplatin-induced apoptotic nuclei in A2780/CP and MCAS cells, as assessed by in situ TUNEL assay. The number of TUNEL-positive cells at 24, 48, and 72 h after drug treatment was significantly higher in the β-elemene plus cisplatin group than in any other group, suggesting that β-elemene synergistically increases cisplatin-induced apoptotic nuclei in ovarian cancer cells.
Third, β-elemene increased cisplatin-induced DNA fragmentation in A2780/CP cells, as determined with an apoptosis detection ELISA. DNA degradation and consequently the enrichment of mono- and oligonucleosomes in the cytoplasm occur several hours before plasma membrane breakdown during the apoptotic response. Compared with untreated cells, cells treated with β-elemene or cisplatin alone showed slight enrichment, and cells treated with both drugs showed a marked enrichment of cytoplasmic nucleosomes. Furthermore, A2780/CP ovarian tumor cells exposed to β-elemene and/or cisplatin exhibited cell morphology typical of karyorrhexis. These data indicate that β-elemene substantially increases cisplatin-induced DNA fragmentation in resistant ovarian cancer cells.
Taken together, these results clearly demonstrate that β-elemene considerably increases cisplatin cytotoxicity and cisplatin sensitivity by augmenting apoptosis induction in ovarian carcinoma cells. The initiation of apoptosis is among the most important therapeutic targets in cancer chemotherapy [21, 23, 31–33]. Apoptosis is a well-organized and well-characterized cell death process with significant morphological and biochemical features [11, 12]. This process is finely gene-directed and can be divided into three different stages: (1) the initiation stage: cells receive a stimulus and respond to this stimulus by engagement of multiple possible apoptotic pathways; (2) the effector stage: the initiated apoptotic signals are integrated each other and eventually decide the cells’ fates, either live or death; and (3) the execution stage: this is the last and irreversible cell death stage, in which some critical intra-cellular proteins are auto-digested and DNAs are cleaved [11, 12].
The initiation of apoptosis is mediated at least by two distinct pathways: the intrinsic or mitochondrial pathway and the extrinsic or death receptor pathway, involving either mitochondria or death receptors [34–38]. In the mitochondria pathway, apoptotic signals lead to permeability changes in mitochondrial membrane and the subsequent release of cytochrome c into cytosol [24, 39–43]. Cytochrome c, a component of the mitochondrial electron-transfer chain that is present in the inter-membrane space, is released into the cytosol during the early phases of apoptosis [44]. Mitochondrial transmembrane potential (ΔΨm, MTP) plays a critical role in controlling apoptotic responses. Loss of MTP can lead to opening of the permeability transition pore [45]; the pore opening has been seen as a key stage in apoptosis [42]. Our results revealed that the combination of β-elemene and cisplatin treatment significantly disrupted MTP and increased cytosolic cytochrome c. These data suggest that β-elemene augmented cisplatin-induced apoptotic response in A2780/CP cells is mitochondria dependent.
During apoptosis, a cascade of intracellular cysteine proteases, known as caspases, is activated [46, 47]. In general, activation of the caspase cascade requires both initiator caspases such as caspase-8, caspase-9, and caspase-10 and effector caspases such as caspase-3 and caspase-7 [35, 48–50]. Caspase-3, a downstream caspase, is the most important member in caspase family. Caspase-3 plays a pivotal role in the execution phase of apoptosis which was induced by diverse stimuli [47]. Cytochrome c and other different factors recruit and process procaspase-9 and lead to the activation of caspase-9, which then processes and activates the effector caspases to orchestrate the biochemical execution of apoptosis [51, 52].
In the current study, the activities of caspase-3, caspase-8, and caspase-9 were significantly elevated and the auto-cleavage of caspase-9 was increased in A2780/CP cells after treatment with both β-elemene and cisplatin. Moreover, the caspase inhibitor Z-VAD-FMK appreciably suppressed apoptosis induced by the combination of β-elemene and cisplatin (data not shown). These data suggest that the activation and auto-cleavage of caspases are involved in the β-elemene-enhanced apoptotic response induced by cisplatin.
Bcl-2 family proteins play important roles in regulating the sensitivity of cancer cells to chemotherapeutic drug-induced apoptosis [53, 54]. Bcl-2 and Bcl-XL act as anti-apoptotic factors, and Bax acts as a pro-apoptotic factor. The anti-apoptotic activity of Bcl-2 is inhibited by phosphorylation of Ser-70 [55]. In the present study, treatment of A2780/CP cells with β-elemene plus cisplatin markedly downregulated Bcl-2 and Bcl-XL expression, upregulated Bax expression, and increased Bcl-2 phosphorylation at Ser-70 in cells. These observations suggest that the apoptotic process induced by β-elemene and cisplatin in ovarian tumor cells is regulated by Bcl-2 family genes.
Altogether, the present study has demonstrated that β-elemene strongly sensitizes chemoresistant ovarian carcinoma cells to cisplatin through potent induction of apoptosis. The enhanced apoptosis was mediated by a mitochondria- and caspase-dependent cell death pathway. The precise mechanisms linking these two signaling events remain to be experimentally determined. On the other hand, our previous studies have revealed that β-elemene also sensitizes resistant ovarian tumor cells to cisplatin by blocking cell cycle progression at G2/M phase. It is unclear whether induced apoptosis is a secondary event resulting from cell cycle arrest or a novel mechanism of β-elemene in cancer cells. Further studies are necessary to delineate the precise signaling pathways linking cell cycle arrest and apoptosis induction by β-elemene and cisplatin in human ovarian cancer cells.
Acknowledgments
This publication was made possible by grants from the Natural Science Foundation of Science and Technology Department of Guangxi Province (No. 0991294) and the Guangxi Scientific Research and Technological Development Program (No. 200901059), and by grants from the National Institutes of Health (Nos. P20RR16440-010003, P20RR16440-020003, P20RR16440-030003, P20RR16440-040003) and West Virginia University School of Medicine Research Grant (to Q. Q. Li).
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Qingdi Quentin Li, Email: liquenti@mail.nih.gov, quentinli2004@yahoo.com, Beihai Institute of Endocrine and Metabolic Diseases, Beihai 536000, Guangxi, China. West Virginia University Cancer Center, Morgantown, WV 26506, USA. National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Rebecca X. Lee, West Virginia University Cancer Center, Morgantown, WV 26506, USA
Huasheng Liang, Beihai Institute of Endocrine and Metabolic Diseases, Beihai 536000, Guangxi, China.
Yuhua Zhong, Email: zhongyh111@163.com, Beihai Institute of Endocrine and Metabolic Diseases, Beihai 536000, Guangxi, China.
Eddie Reed, West Virginia University Cancer Center, Morgantown, WV 26506, USA.
References
- 1.Sfakianos GP, Havrilesky LJ. A review of cost-effectiveness studies in ovarian cancer. Cancer Control. 2010;18:59–64. doi: 10.1177/107327481101800109. [DOI] [PubMed] [Google Scholar]
- 2.Murphy G. Cancer statistics. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 3.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–20. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 4.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–84. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Wang G, Zhao J, Ding H, Cunningham C, Chen F, Flynn DC, Reed E, Li QQ. Antiproliferative effect of beta-elemene in chemoresistant ovarian carcinoma cells is mediated through arrest of the cell cycle at the G2-M phase. Cell Mol Life Sci. 2005;62:894–904. doi: 10.1007/s00018-005-5027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li QQ, Wang G, Zhang M, Cuff CF, Huang L, Reed E. beta-Elemene, a novel plant-derived antineoplastic agent, increases cisplatin chemosensitivity of lung tumor cells by triggering apoptosis. Oncol Rep. 2009;22:161–70. doi: 10.3892/or_00000420. [DOI] [PubMed] [Google Scholar]
- 7.Li QQ, Wang G, Huang F, Banda M, Reed E. Antineoplastic effect of beta-elemene on prostate cancer cells and other types of solid tumour cells. J Pharm Pharmacol. 2010;62:1018–27. doi: 10.1111/j.2042-7158.2010.01135.x. [DOI] [PubMed] [Google Scholar]
- 8.Li QQ, Wang G, Reed E, Huang L, Cuff CF. Evaluation of Cisplatin in Combination with beta-Elemene as a Regimen for Prostate Cancer Chemotherapy. Basic Clin Pharmacol Toxicol. 2010;107:868–76. doi: 10.1111/j.1742-7843.2010.00592.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Li QQ, Zou B, Wang G, Li X, Kim JE, Cuff CF, Huang L, Reed E, Gardner K. In vitro combination characterization of the new anticancer plant drug beta-elemene with taxanes against human lung carcinoma. Int J Oncol. 2007;31:241–52. [PubMed] [Google Scholar]
- 10.Wang G, Li X, Huang F, Zhao J, Ding H, Cunningham C, Coad JE, Flynn DC, Reed E, Li QQ. Antitumor effect of beta-elemene in non-small-cell lung cancer cells is mediated via induction of cell cycle arrest and apoptotic cell death. Cell Mol Life Sci. 2005;62:881–93. doi: 10.1007/s00018-005-5017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 12.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 13.Ellis M, Yuan J, Horvitz H. Mechanisms and function of cell death. Annu Rev Cell Biol. 1991;7:663–98. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 14.Thompson C. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 15.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–9. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Feng Q, Kim J-M, Li M, Fung FK, Tsang BK. Human ovarian cancer and cisplatin resistance: possible role of inhibitor of apoptosis proteins. Endocrinology. 2001;142:370–80. doi: 10.1210/endo.142.1.7897. [DOI] [PubMed] [Google Scholar]
- 17.MacFarlane M. Cell death pathways–potential therapeutic targets. Xenobiotica. 2009;39:616–24. doi: 10.1080/00498250903137990. [DOI] [PubMed] [Google Scholar]
- 18.Wyllie A. Apoptosis and carcinogenesis. Eur J Cell Biol. 1997;73:189–97. [PubMed] [Google Scholar]
- 19.Selzner M, Bielawska A, Morse M, Rudiger H, Sindram D, Hannun Y, Clavien P. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001;61:1233–40. [PubMed] [Google Scholar]
- 20.Serrano M, Sanchez-Rovira P, Algarra I, Jaen A, Lozano A, Gaforio J. Evaluation of a gemcitabine-doxorubicin-paclitaxel combination schedule through flow cytometry assessment of apoptosis extent induced in human breast cancer cell lines. Jpn J Cancer Res. 2002;93:559–66. doi: 10.1111/j.1349-7006.2002.tb01291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv Drug Deliv Rev. 2009;61:1250–75. doi: 10.1016/j.addr.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 22.Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E, Michaud M, Zischka H, Castedo M, Kroemer G. Mitochondrial gateways to cancer. Mol Aspects Med. 2010;31:1–20. doi: 10.1016/j.mam.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Xu CX, Jin H, Cho MH. Apoptosis and apoptosis-based therapy in lung cancer. Anticancer Agents Med Chem. 2009;9:952–7. doi: 10.2174/187152009789377682. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 26.Fraser M, Leung B, Jahani-Asl A, Yan X, Thompson W, Tsang B. Chemoresistance in human ovarian cancer: the role of apoptotic regulators. Reprod Biol Endocrinol. 2003;1:66. doi: 10.1186/1477-7827-1-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young RC, Ozols RF. Characterization of a cis-diamminedichloroplatinum (II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987;47:414–8. [PubMed] [Google Scholar]
- 28.Parker RJ, Eastman A, Bostick-Bruton F, Reed E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced DNA repair of cisplatin-DNA lesions and reduced drug accumulation. J Clin Invest. 1991;87:773–7. doi: 10.1172/JCI115080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitabayashi A, Hirokawa M, Hatano Y, Lee M, Kuroki J, Miura A. Granulocyte colony-stimulating factor down-regulates allogeneic immune responses by post-transcriptional inhibition of tumor necrosis factor- production. Blood. 1995;86:2220. [PubMed] [Google Scholar]
- 30.Sasaki H, Sheng Y, Kotsuji F, Tsang B. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 2000;60:5659–66. [PubMed] [Google Scholar]
- 31.Ho YS, Duh JS, Jeng JH, Wang YJ, Liang YC, Lin CH, Tseng CJ, Yu CF, Chen RJ, Lin JK. Griseofulvin potentiates antitumorigenesis effects of nocodazole through induction of apoptosis and G2/M cell cycle arrest in human colorectal cancer cells. Int J Cancer. 2001;91:393–401. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1070>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 32.Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los M. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510. doi: 10.1136/jmg.2009.066944. [DOI] [PubMed] [Google Scholar]
- 33.Ledgerwood EC, Morison IM. Targeting the apoptosome for cancer therapy. Clin Cancer Res. 2009;15:420–4. doi: 10.1158/1078-0432.CCR-08-1172. [DOI] [PubMed] [Google Scholar]
- 34.Ashkenazi A, Dixit V. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 35.Nunez G, Benedict M, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17:3237–45. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- 36.Stennicke H, Jurgensmeier J, Shin H, Deveraux Q, Wolf B, Yang X, Zhou Q, Ellerby H, Ellerby L, Bredesen D, Green D, Reed J, Froelich C, Salvesen G. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem. 1998;273:27084–90. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- 37.Deng Y, Lin Y, Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16:33–45. doi: 10.1101/gad.949602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grunewald S, Sharma R, Paasch U, Glander HJ, Agarwal A. Impact of caspase activation in human spermatozoa. Microsc Res Tech. 2009;72:878–88. doi: 10.1002/jemt.20732. [DOI] [PubMed] [Google Scholar]
- 39.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–90. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colin J, Gaumer S, Guenal I, Mignotte B. Mitochondria, Bcl-2 family proteins and apoptosomes: of worms, flies and men. Front Biosci. 2009;14:4127–37. doi: 10.2741/3517. [DOI] [PubMed] [Google Scholar]
- 41.Jourdain A, Martinou JC. Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol. 2009;41:1884–9. doi: 10.1016/j.biocel.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 42.Zamzami N, Susin S, Marchetti P, et al. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–44. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green D, Reed J. Mitochondria and apoptosis. Science. 1998;281:1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Kim C, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 45.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–76. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 46.Jacobson M, Weil M, Raff M. Programmed cell death in animal development. Cell. 1997;88:347–54. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 47.Mandlekar SYR, Tan TH, Kong AN. Activation of caspase-3 and c-Jun NH2-terminal kinase-1 signaling pathways in tamoxifen-induced apoptosis of human breast cancer cells. Cancer Res. 2000;60:5995–6000. [PubMed] [Google Scholar]
- 48.Fulda S. Caspase-8 in cancer biology and therapy. Cancer Lett. 2009;281:128–33. doi: 10.1016/j.canlet.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 49.Sun S, Yue P, Hong W, Lotan R. Augmentation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by the synthetic retinoid 6-[3-(1-adamantyl)-4-hydroxy-phenyl]-2-naphthalene carboxylic acid (CD437) through up-regulation of TRAIL receptors in human lung cancer cells. Cancer Res. 2000;60:7149–55. [PubMed] [Google Scholar]
- 50.Thornberry N, Lazebnik Y. Caspases. Enemies within. Science. 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 51.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols: increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–42. [PubMed] [Google Scholar]
- 52.Kuo HM, Tsai HC, Lin YL, Yang JS, Huang AC, Yang MD, Hsu SC, Chung MC, Gibson Wood W, Chung JG. Mitochondrial-dependent caspase activation pathway is involved in baicalein-induced apoptosis in human hepatoma J5 cells. Int J Oncol. 2009;35:717–24. doi: 10.3892/ijo_00000384. [DOI] [PubMed] [Google Scholar]
- 53.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–32. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941–53. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 55.Srivastava R, Mi Q, Hardwick J, Longo D. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci USA. 1999;96:3775–80. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]