

Abstract
The development of cisplatin drug resistance remains a chief concern in ovarian cancer chemotherapy. β-Elemene is a natural plant product with broad-spectrum antitumor activity towards many types of carcinomas. This study aimed to define the biological and therapeutic significance of β-elemene in chemoresistant ovarian cancer. In the present study, β-elemene significantly inhibited cell growth and proliferation of both the cisplatin-sensitive human ovarian cancer cell line A2780 and its cisplatin-resistant counterpart A2780/CP. β-Elemene also suppressed the growth of several other chemosensitive and chemoresistant ovarian cancer cell lines, including ES-2, MCAS, OVCAR-3, and SKOV-3, with the half maximal inhibitory concentration (IC50) values ranging from 54 to 78 μg/ml. In contrast, the IC50 values of β-elemene for the human ovarian epithelial cell lines IOSE-386 and IOSE-397 were 110 and 114 μg/ml, respectively, which are almost two-fold those for the ovarian cancer cell lines. Cell cycle analysis demonstrated that β-elemene induced a persistent block of cell cycle progression at the G2/M phase in A2780 and A2780/CP cells. This was mediated by alterations in cyclin and cyclin-dependent kinase expression, including the down-regulation of CDC2, cyclin A, and cyclin B1, and the up-regulation of p21WAF1/CIP1 and p53 proteins. Moreover, β-elemene triggered apoptosis and irreversible cell death in both sensitive and resistant ovarian cancer cells via the activation of caspase-3, -8 and 9; the loss of mitochondrial membrane potential (ΔΨm); the release of cytochrome c into the cytosol; and changes in the expression of BCL-2 family proteins. All of these molecular changes were associated with β-elemene-induced growth inhibition and cell death of ovarian cancer cells. Our results demonstrate that β-elemene has antitumor activity against both platinum-sensitive and resistant ovarian cancer cells, and thus has the potential for development as a chemotherapeutic agent for cisplatin-resistant ovarian cancer.
Keywords: β-Elemene, cisplatin resistance, cell cycle arrest, apoptosis, ovarian cancer, Chinese medicine, A2780, A2780/CP cells, p53, p21WAF/CIP1
Ovarian cancer is the most lethal gynecological malignancy. In 2010, the American Cancer Society reported 21,880 new cases of ovarian cancer and 13,850 deaths from ovarian cancer. The incidence of ovarian cancer in the U.S. is 1.4%, with a death rate of 1.1% (1). Advanced epithelial ovarian cancer is the fifth leading cause of cancer-related deaths in the U.S. (1). Despite recent improvements in surgery and adjuvant chemotherapy for advanced ovarian cancer, the majority of patients experience disease relapse, and most cases are incurable (1).
Platinum-based drugs coupled with paclitaxel have been the mainstay of chemotherapy for ovarian cancer (2–4). Cisplatin-based combination chemotherapy is associated with an initial clinical response rate of 40–60%. However, the overall 5-year survival rate for patients with advanced ovarian cancer is only around 20% (5) owing to the frequent development of drug resistance and the consequent failure of chemotherapy (4, 6–8). Chemoresistance, both intrinsic and acquired, has become a major obstacle in achieving sustainable responses in recurrent cancer. Thus, overcoming drug resistance is the key to successful treatment of ovarian cancer (9), and the development of new effective therapeutic modalities is urgently needed.
We initiated a series of studies to evaluate the antineoplastic activity and the molecular mechanism of action of β-elemene in the human epithelial ovarian cancer cell line A2780 and its cisplatin-resistant counterpart A2780/CP. β-Elemene was originally isolated from the Chinese medicinal herb Rhizoma zeodariae and may be a promising new anticancer drug. Significant antitumor effects of β-elemene were first reported in human and murine tumors that were not associated with severe bone marrow suppression or drug resistance (10). Our studies during the past several years have demonstrated antitumor activity of β-elemene in a broad range of human cancer cell lines (11–15). Importantly, the inhibition of cancer cell proliferation by β-elemene is associated with both cell cycle arrest and apoptosis induction in several cancer cell lines (11–15). Our findings for β-elemene are consistent with previous reports from other groups (16–18).
To ensure orderly cell division, the eukaryotic cell cycle comprises of highly regulated series of sequential molecular processes governed by the periodic activation of multiple cyclin-dependent kinases (CDKs) (19). There are two checkpoints in the cell cycle that are activated by cellular DNA damage, causing the temporary arrest of cell cycle progression. This process is regulated by cell cycle inhibitors and other proteins, which determine cell survival versus cell death depending on the proper repair of the DNA damage. The cell cycle checkpoints are the primary biological targets in the development of new therapeutic strategies for cancer (20–24). Cell death can result not only from cell cycle arrest but also through apoptosis. Frequent inactivation of apoptosis contributes to carcinogenesis in normal cells and to chemoresistance in cancer cells. The induction of apoptosis is currently a major focus for drug development in cancer therapy (25–28).
It has been nearly two decades since the anticancer activity of β-elemene was first reported. Nevertheless, the mechanism by which β-elemene causes cancer cell death is still not fully understood. There are few reports on the molecular mechanism underlying the antitumor effect of β-elemene, especially in drug-resistant carcinomas. In the present study, we examined the cytotoxic effect of β-elemene in human cisplatin-sensitive and cisplatin-resistant ovarian cancer cells, and investigated its possible molecular mechanism.
Materials and Methods
Chemicals and immunoreagents
(−)-β-Elemene (98% purity) was obtained from Yuanda Pharmaceuticals Ltd, Inc. (Dalian, P. R. China). Dimethylsufoxide (DMSO) and propidium iodide (PI) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Antibodies against cyclin-dependent kinase 1 (CDK1), also known as cell division control protein 2 homolog (CDC2), cyclin A, cyclin B1, p21WAF1/CIP1, p53, CDC25C, caspase-3, caspase-8, caspase-9, BCL-2–associated X protein (BAX), B-cell lymphoma 2 (BCL-2), BCL-XL, cytochrome c, β-actin, peroxidase-labeled anti-rabbit immunoglobulin G (IgG), Blotto B, and ECL western blotting system were all purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against phospho-CDC2 (Thr161) and phospho-CDC2 (Tyr15) were purchased from Cell Signaling Technology (Beverly, MA, USA).
Cell lines and cell culture conditions
The cisplatin-sensitive human ovarian cancer cell line A2780 and the cisplatin-resistant human ovarian cancer cell line A2780/CP have been described previously (29, 30). Other human ovarian carcinoma cell lines, including ES-2, OVCAR-3 and SKOV-3, and the cisplatin-resistant ovarian cancer cell line MCAS, were also used in this study. Cells were cultured in monolayer using RPMI-1640 medium (Invitrogen, Life Technologies, Gaithersburg, MD, USA), supplemented with 10% (v/v) fetal calf serum, 50 units/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). The human ovary epithelial cell lines IOSE-386 and IOSE-397 were a kind gift of Dr. Nelly Auersperg (University of British Columbia, Vancouver, BC, Canada), which were generated by transfecting normal human OSE cells with the immortalizing simian virus 40 virus early genes (31). These cell lines were maintained in a 1:1 mixture of Medium 199 and MCDB 105 (Sigma-Aldrich Chemical Co.) supplemented with 10% fetal bovine serum and 50 μg/ml gentamicin sulfate (Sigma-Aldrich Chemical Co.). All cell lines were grown in logarithmic growth phase at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air. Cells were routinely tested for mycoplasma infection using a commercial assay system (MytoTect; Invitrogen), and new cultures were established monthly from frozen stocks. All media and reagents contained <0.1 ng/ml endotoxin, as determined by Limulus polyphemus amebocyte lysate assay (Whittaker Bioproducts, Walkersiville, MD, USA). Before starting the experiments, cells were grown to 70–80% confluence after subculturing. β-Elemene was serially diluted in culture medium to obtain the desired concentrations.
Drug treatment and cell survival assay
The effects of β-elemene on cell survival were measured by the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay, carried out as described previously (11–15). In brief, cells were harvested using 0.25% trypsin-EDTA and resuspended to a final density of 5×104 cells/ml in fresh medium containing 10% fetal calf serum. Aliquots of 0.1 ml from each cell line suspension were distributed evenly into 96-well cell culture plates (Corning, Inc., Corning, NY, USA). One column from each plate containing medium alone was used as a blank control, and another column containing cells without drug exposure was used as an untreated control. After 24 h of incubation with fresh medium, serial dilutions of β-elemene (20 to 200 μg/ml) were added, and the cells were incubated for an additional 24 h, 48 h, 72 h and 96 h, respectively. The effects of β-elemene on cell growth in human ovary cells and human ovarian cancer cells were determined using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The absorbance (A) readings were taken using a 96-well Opsys MR™ Microplate Reader (ThermoLabsystems, Chantily, VA, USA) and Revelation™ QuickLink Software at 490 nm. The blank control wells were used for zeroing the absorbance. The percentage of cell survival was calculated using the background-corrected absorbance as follows: Cell viability (%)=100×(1-A of experimental well)/A of untreated control well. All experiments were performed at least three times with representative data presented.
Cell cycle analysis by flow cytometry
The effect of β-elemene on cell proliferation was evaluated by measuring the distribution of the cells in the different phases of the cell cycle by flow cytometry. This analysis was based on the measurement of the DNA content of nuclei labeled with propidium iodide (PI). Cells were cultured in 75-cm2 flasks and treated with β-elemene for 48 h at the indicated concentrations. Cell suspensions from either control cultures or from drug-treated cultures were prepared by trypsinization. Approximately 1×106 cells/condition were aliquoted, washed twice with ice-cold phosphate buffered saline (PBS), and pelleted in a centrifuge. Cell specimens were fixed in 70% ethanol overnight at −20°C. The cells were then washed again twice in PBS, permeabilized with 0.1% Triton X-100 in PBS containing 0.2 μg/ml RNase A (Boehringer Mannheim, Indianapolis, IN, USA) for 30 min on ice, and washed twice in PBS. A solution containing 10 μg/ml PI and 0.1% Triton X-100 in PBS was added, then cells were incubated for 30 min at room temperature to allow for maximum labeling of DNA. Finally, the cells were analyzed for cell cycle perturbation using a FACSCalibur instrument (Becton-Dickinson, San Diego, CA, USA). The CellQuest Pro software (Becton-Dickinson) and the ModFit LT software (Verity Software House, Inc., Topsham, MN, USA) were used to determine the distribution of cells in the various cell cycle compartments as G1, S, and G2/M.
Annexin V assay
Annexin V assay was used to detect apoptotic cells by staining cells with both annexin V-fluorescein isothiocyanate (FITC) and PI. Cells were treated with 0, 30, 50, 70 μg/ml β-elemene, respectively. After 48-h treatment, 1×106 cells/ml were washed twice with PBS and resuspended in 300 μl binding buffer (10 mM HEPES, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, pH 7.4), then 3 μl of annexin V-FITC (Caltag Laboratories, Burlingame, CA, USA) was added to the cells. Cells were incubated on ice in the dark for 1.5 h and washed with 4 ml HEPES buffer once, then 10 μl PI (50 μg/ml) were added to these cells. Cells were incubated for another 20 min and analyzed within 30 min by FACSCalibur (Becton-Dickinson). CellQuest Pro software (Becton-Dickinson) and ModFit LT software (Verity Software House, Inc.) were used to determine the distribution of apoptotic cells. Results were repeated for three times.
Terminal deoxynucleotidyltransferase-mediated uridine triphosphate end-labeling (TUNEL) assay
For in situ detection of apoptotic nuclei, the TUNEL assay was performed using the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany), according to the procedures described by the manufacturer. In brief, after the desired treatment, cells were washed with PBS and were then placed in Bouin’s fixative for at least 24 h, dehydrated in alcohol, and paraffin embedded using standard protocols. Sections of 5-μm thickness were mounted on glass slides and stained with hematoxylin and eosin and TUNEL assay. After rehydration, sections were post-fixed in 4% paraformaldehyde-PBS (pH 7.2), rinsed in PBS, and incubated for 1 h at room temperature with a mix containing fluoresceindeoxy-UTP and terminal deoxynucleotidyltransferase following the manufacturer’s instructions. Cells with brown granules in nuclei were TUNEL positive cells. Over 200 cells were counted under a light microscope to determine the percentage of positive cells.
DNA fragment detection by enzyme-linked immunosorbent assay (ELISA)
Cells were seeded at a density of 104 cells/well in 96-well plates overnight. After the desired treatment of cells with 0, 30, 50, 70 μg/ml β-elemene for 24, 48, and 72 h, cells were incubated in lysis buffer for 30 min to obtain cytoplasmic lysates. A cell death detection ELISA kit (Cell Death Detection ELISAplus; Boehringer Mannheim, Indianapolis, IN, USA) was used to quantitatively determine cytoplasmic histone-associated DNA oligonucleosome fragments associated with apoptotic cell death, according to the manufacturer’s instructions. Briefly, samples were incubated in microtiter plates coated with mouse anti-histone antibody (clone H11-4) to bind histone-associated DNA oligonucleosomes uncovered by endonuclease-mediated DNA nicking. Plates were washed, and nonspecific binding sites were saturated with blocking buffer. Bound samples were then reacted with anti-mouse DNA monoclonal antibody (MCA-33) and then conjugated with peroxidase. To determine the amount of retained peroxidase, 2, 2′-azino-di-(3-ethylbenzthiazoline sulfonate) (ABTS) was added as a substrate. The absorbance was read using a 96-well Opsys MR™ Microplate Reader (ThermoLabsystems) at 405 nm. The enrichment of mono- and oligonucleosomes released into the cytoplasm was calculated as absorbance of sample cells/absorbance of control cells. The apoptotic index was calculated as an enrichment factor, that is, the ratio of the result compared with the control set arbitrarily at 1.0. Each experiment was performed in triplicate, and means and standard deviations were calculated.
Measurement of changes in mitochondrial membrane potential (Δψm) by flow cytometry using BD MitoSensor™ Reagent
Since reduction of mitochondrial membrane potential is a hallmark of apoptosis, we studied Δψ during the early stages of apoptosis with BD ApoAlert™ Mitochondrial Membrane Sensor Kit containing 3,3′-dihexyloxacarbocyanine iodide (DiOC6) (BD Biosciences Clontech, Palo Alto, CA, USA). DiOC6 is a cyanine dye that accumulates in the mitochondrial matrix under the influence of the mitochondrial membrane potential. Briefly, 1×106 cells were incubated in 1 ml BD MitoSensor Reagent (final concentration, 5 μg/ml) containing DiOC6 at 37°C in an incubator with 5% CO2 for 20 min. Cells were then washed with 1 ml incubation buffer and centrifuged at 350 ×g for 5 min. Pelletized cells were resuspended in incubation buffer and analyzed by flow cytometry.
Caspase enzymatic activity assay
Caspase-3, caspase-8, and caspase-9 enzymatic activities were measured using the CasPASE™ Apoptosis Assay Kit (Geno Technology, Inc., MO, USA) according to the manufacturer’s protocols. In brief, after treatment with β-elemene for 48 h, a total of 1×107 cells were harvested and washed twice with ice-cold PBS. Cytosolic extracts were prepared by five repeated cycles of freezing and thawing in 200 μl of lysis buffer, then centrifuged at 12,000 rpm for 30 min at 4°C. Cell lysates (50 μl) were diluted with 2×CasPASE™ buffer (50 μl) in 96-well plates, and incubated at 37°C for 2 h with 5 μl 1 mM caspase substrates (Ac-DEVD-AFC for caspase-3, Ac-LETD-AFC for caspase-8, Ac-LEHD-AFC for caspase-9). The absorbance of the cleaved substrate was measured by a 96-well Opsys MRTM Microplate Reader (ThermoLabsystems) at a test wavelength of 405 nm. Nonapoptotic cell lysates were used as a control. The buffer without cell lysate was used as blank. Each experiment was performed in triplicate, and means and standard deviations were calculated.
Protein extraction and western blotting
Cells treated with β-elemene were harvested by trypsinization following a 48-h incubation. After washing with ice-cold PBS, the cells were lysed on ice for 30 min in a mammalian cell lysis buffer (Quality Biological, Inc., Gaithersburg, MD, USA), containing 10 μl/ml 200 mM phenylmethylsulfonyl fluoride (PMSF), 10 μl/ml 100 mM sodium orthovanadate, and 10 μg/ml aprotinin. Cellular extracts were clarified by centrifugation at 12,000 rpm at 4°C for 30 min, and protein concentrations were determined using the Bradford Assay (Bio-Rad, Richmond, CA, USA). Protein (60 μg) from whole cell lysates was mixed 1:1 with 2× sodium dodecyl sulfate (SDS) protein gel solution (Quality Biological, Inc.), heated for 5 min at 95°C, separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane (Schleicher & Schuell BioScience, Inc., Keene, NH, USA). After blocking in Blotto B (Santa Cruz) for 1 h at room temperature, membranes were incubated overnight at 4°C with the specific primary antibodies (dilutions were 1:100-1:300). Membranes were washed with TBS/0.1% Tween 20 solution, incubated with anti-rabbit peroxidase-conjugated secondary antibody (the dilution was 1:1000), washed again, and developed with enhanced chemiluminescence substrate (Santa Cruz Biotechnology) according to the manufacturer’s instructions. The protein bands were visualized using X-ray films (Eastman Kodak, Rochester, NY, USA). All blots are representative of three independent experiments.
Detection of cytochrome c release into the cytosol
Cell cytosolic fraction was isolated using the Mitochondrial Isolation Kit (Sigma-Aldrich). Briefly, 3×107 cells were harvested and washed with PBS. The cells were suspended in 10 volumes of mitochondrial extraction buffer A containing 2 mg/ml albumin and homogenized on ice with a wheaton dounce homogenizer. Unbroken cells and nuclei were removed by centrifugation at 600 ×g for 5 min at 4°C. The supernatant was further centrifuged at 11,000 ×g for 10 min. The supernatant was saved as a cytosolic fraction, while the precipitate was dissolved in storage buffer A and saved as the mitochondrial fraction. The cytosolic fractions were analyzed by western blotting with a monoclonal antibody against cytochrome c.
Statistical data analysis
Data are presented as the mean±SD. The Student’s t-test was used to analyze the difference between the means of the treatment groups and the control group. Differences with a p-value of less than 0.05 were considered statistically significant.
Results
β-Elemene significantly inhibited cell growth in both cisplatin-sensitive and cisplatin-resistant human ovarian cancer cell lines
Six human ovarian cancer cell lines (A2780, A2780/CP, ES-2, OVCAR-3, SKOV-3, and MCAS) and two human ovarian epithelial cell lines (IOSE-386 and IOSE-397) were treated with β-elemene for 24, 48, 72, and 96 h. Based on MTT assays of each cell line at each time point, β-elemene significantly inhibited both cisplatin-sensitive and cisplatin-resistant human ovarian cancer cell growth compared with untreated control cells (Table I, Figure 1A and 1B). The IC50 values of β-elemene for A2780, A2780/CP, ES-2, OVCAR-3, SKOV-3, and MCAS cancer cells were 65, 75, 54, 57, 67, and 78 μg/ml, respectively. The average IC50 of β-elemene for the normal ovary control IOSE-386 and IOSE-397 cells was about 112 μg/ml, which is nearly two-fold those for the ovarian cancer cells (p<0.01). However, the β-elemene IC50 did not differ significantly between the cisplatin-sensitive (A2780) and cisplatin-resistant (A2780/CP) ovarian cancer cells (p>0.05) (Figure 1C). These data indicate that β-elemene has differential cytotoxicity in normal versus carcinoma cells, but has similar antitumor activity towards both sensitive and resistant ovarian cancer cells.
Table I.
In vitro cytotoxicity of β-elemene towards human ovarian cancer and epithelial cell lines.
| Cell line | β-Elemene IC50 (μg/ml)* |
|---|---|
| ES-2 | 54±2.5 |
| OVCAR-3 | 57±2.7 |
| SKOV-3 | 67±2.8 |
| MCAS | 78±3.1 |
| IOSE-386 | 110±3.8 |
| IOSE-397 | 114±4.0 |
Human ovarian cancer cells and human ovarian epithelial cells (IOSE-386 and IOSE-397) were distributed in 96-well plates (1×103 cells/well) and treated with β-elemene at 10, 20, 40, 60, 80, 100, 120, 140, 160, 180, and 200 μg/ml for 48 h. Cell cytotoxicity was determined by the MTT assay. Values are expressed in relation to wells without β-elemene treatment (100%). The percentage cell growth was plotted against the β-elemene concentration, and the half maximal inhibitory concentration (IC50) values of β-elemene were estimated from the plots.
The IC50 value is defined as the concentration of β-elemene needed for 50% inhibition of cell growth and proliferation.
Figure 1.

Cytotoxicity of β-elemene towards human ovarian epithelial IOSE-397 cells, cisplatin-sensitive human ovarian cancer A2780 cells, and cisplatin-resistant human ovarian cancer A2780/CP cells. Exponentially growing cells were treated with β-elemene at the indicated concentrations (20–200 μg/ml) for 24, 48, 72, and 96 h. A and B: Cell survival was assessed by the MTT assay, as described in the Materials and Methods. Cell growth values are expressed in relation to growth of untreated cells (100%). C: Average half maximal inhibitory concentration (IC50) values of β-elemene were 113 μg/ml for IOSE-397 cells, 65 μg/ml for A2780 cells, and 75 μg/ml for A2780/CP cells. The results represent the means±SD of at least three independent experiments. #p<0.01 vs. IOSE-397 cells.
β-Elemene induced cell cycle arrest at the G2/M phase in ovarian cancer cells, through modulation of cell cycle regulatory molecules
Flow cytometric profiles revealed substantial increases in the fraction of A2780 and A2780/CP cells at the G2/M phase of the cell cycle, after treatment with β-elemene at 30, 50, or 70 μg/ml for 48 h (Figure 2). The percentages of A2780 cells at G2/M were 9.44, 10.89, and 12.53% after treatment with 30, 50, and 70 μg/ml of β-elemene, respectively, compared with 7.6% of untreated cells. Following β-elemene exposure, the percentage of A2780/CP cells at G2/M increased markedly and in a dose-dependent manner, from 10.63% of untreated cells to 22.83, 31.87, and 37.3% of cells treated with 30, 50, and 70 μg/ml of β-elemene, respectively. These results suggest that β-elemene suppresses ovarian cancer cell growth by inducing a specific block at the G2/M phase and that this effect of β-elemene is greater in cisplatin-resistant ovarian cancer cells than in cisplatin-sensitive cells.
Figure 2.
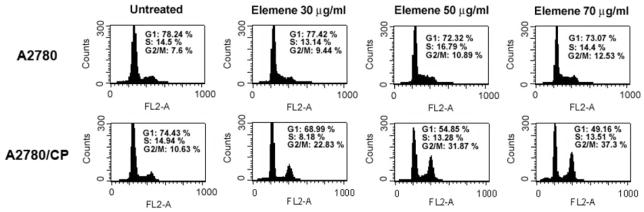
β-Elemene induced cell cycle arrest at the G2/M phase in asynchronized A2780 and A2780/CP ovarian cancer cells. Asynchronously growing cells were treated with β-elemene at the indicated concentrations for 48 h and harvested. DNA content was analyzed by flow cytometry using propidium iodide staining, as described in the Materials and Methods. The percentage of cells in each cell cycle phase was calculated using the CellQuest Pro software (Becton-Dickinson) and is indicated at the upper right of each profile. A representative profile for each treatment is shown. β-Elemene caused cell cycle arrested at G2/M, in a concentration-dependent manner.
To investigate the molecular mechanism leading to G2/M arrest in ovarian cancer cells, we analyzed the expression of several cell cycle regulatory proteins on western blots. The protein expression of CDC2, cyclin A, and cyclin B1 was down-regulated, and the protein expression of p53 and p21WAF1/CIP1 was up-regulated by β-elemene treatment in both A2780 and A2780/CP cells. In addition, β-elemene-treated cells exhibited decreased phosphorylation of CDC2 at Thr161 and increased phosphorylation of CDC2 at Tyr15. As these changes were positively correlated with the G2/M cell population and negatively correlated with cyclin B1 and CDC2 expression (Figure 3), they may be responsible for G2/M arrest in ovarian cancer cells in response to β-elemene.
Figure 3.

Altered expression of cell cycle regulatory proteins in A2780 and A2780/CP cells following β-elemene treatment. A2780 and A2780/CP cells were treated with β-elemene at the indicated concentrations for 48 h, and cell proteins were extracted. Extracted proteins (60 μg) from drug-treated and untreated cells were separated by SDS-PAGE and immunoblotted with antibodies against cyclin A, cyclin B1, cyclin-dependent kinase 1 (CDK1), also known as cell division control protein 2 homolog (CDC2), phospho-CDC2 (Tyr15), phospho-CDC2 (Thr161), p53, p21WAF1/CIP1, and CDC25C, respectively. β-Actin was used as a loading control. Western blot data are representative of those obtained from three separate experiments.
β-Elemene induced apoptotic cell death in cisplatin-sensitive and -resistant ovarian cancer cells
To detect apoptosis, cells were stained with fluorescently labeled annexin V, which binds to phosphatidylserine that has been transported to the outer leaflet of the cytoplasmic membrane during apoptosis. Necrotic cells, in which the cytoplasmic membrane has been completely disrupted, were detected by staining with PI in addition to annexin V. Flow cytometry of stained cells revealed increased annexin V staining in both cisplatin-sensitive and -resistant ovarian cancer cells following β-elemene exposure. The percentages of early and late apoptotic A2780 cells were 1.67, 16.11, 51.71, and 65.33% after treatment with 0, 30, 50, and 70 μg/ml β-elemene, respectively (Figure 4A). In A2780/CP cells, the percentages of early and late apoptotic cells were 1.35, 8.72, 20.17, and 93.09% after treatment with 0, 30, 50, and 70 μg/ml β-elemene, respectively (Figure 4A). These results demonstrate that β-elemene induced apoptosis in both sensitive and resistant ovarian cancer cells.
Figure 4.

Apoptotic responses to β-elemene challenge in A2780 and A2780/CP cell lines. A: Flow cytometry of annexin V-stained cells. Cells were treated with β-elemene at the indicated concentrations for 48 h and then stained with fluorescein isothiocyanate (FITC)-labeled annexin V and propidium iodide to detect cell membrane changes, as described in the Materials and Methods. Untreated cells were used as controls. B and C: Small nucleotide fragments, produced as a result of cell apoptosis, were quantified by ELISA using a commercially available apoptosis detection kit. The enrichment factor, which correlates with the number of apoptotic cells, is shown as an apoptotic index. Cells were treated with β-elemene at the indicated concentrations for 24, 48, and 72 h. Data are representative of three independent experiments. *p<0.05 and #p<0.01, vs. untreated control groups.
During apoptosis, DNA degradation occurs several hours before the plasma membrane breaks down. The small nucleotide fragments produced during apoptosis were quantified using a Cell Death Detection ELISA kit. As an apoptotic index, an enrichment factor was calculated as the ratio of fragmented DNA in treated cells versus that in untreated cells, which was arbitrarily set at 1.0. The enrichment factors were significantly increased in A2780 and A2780/CP cells after treatment with 50 and 70 μg/ml β-elemene for 24, 48, and 72 h (Figure 4B and 4C; #p<0.01 and *p<0.05). In addition, apoptotic nuclei in ovarian cancer cells were evaluated by TUNEL staining. Compared with untreated cells, both A2780 and A2780/CP cells treated with β-elemene for 48 h, particularly at 50 and 70 μg/ml, exhibited an increased number of TUNEL-positive cell nuclei (Figure 5). These data further confirm the dose- and time-dependent induction of apoptosis by β-elemene in both sensitive and resistant ovarian cancer cells.
Figure 5.
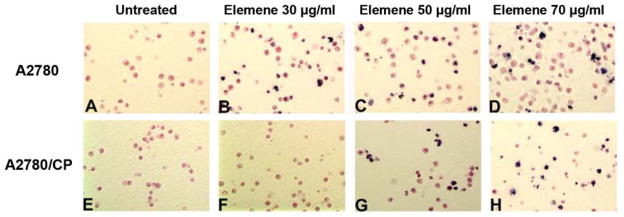
Terminal deoxynucleotidyltransferase-mediated uridine triphosphate end-labeling (TUNEL) staining revealed β-elemene-induced apoptotic nuclei in A2780 and A2780/CP cells. Cells were treated with β-elemene at the indicated concentrations for 48 h, harvested, fixed with 4% formaldehyde, and stained according to the manufacturer’s instructions. TUNEL-positive cells were counted under a light microscope. A-D: A2780 cells; E-H: A2780/CP cells.
β-Elemene-induced apoptosis in ovarian cancer cells was mitochondria- and caspase-dependent
The mitochondrial transmembrane potential (ΔΨm) was assessed based on the fluorescence of BD MitoSensor™ reagent-stained cells. A higher geometric mean fluorescence of the cells indicates a lower ΔΨm and a higher incidence of apoptosis. The fluorescence of untreated cells served as a baseline. Compared with untreated cells, β-elemene-treated A2780 and A2780/CP cells had significantly higher geometric mean fluorescence values, indicating a significantly lower ΔΨm (Figure 6A). The fluorescence increased in a β-elemene concentration-dependent manner. Furthermore, the level of cytochrome c in the cytosolic fraction of A2780 and A2780/CP cells was increased after treatment with β-elemene for 48 h (Figure 6B).
Figure 6.

Detection of mitochondrial transmembrane potential (ΔΨm) and cytochrome c release into the cytosol in A2780 and A2780/CP cells. A: ΔΨm was assessed using the BD MitoSensor™ reagent staining. Cells were treated with β-elemene at the indicated concentrations for 48 h, harvested, incubated with the BD MitoSensor™ reagent (final concentration, 5 μg/ml) at 37°C in an incubator with 5% CO2 for 20 min, and then rinsed in suspension. Apoptotic cells were detected by flow cytometry as described in the Materials and Methods. The geometric mean fluorescence of the cells was calculated. A higher value reflects a lower ΔΨm and more apoptosis. The geometric means are shown in each panel. Untreated cells were used as controls. B: Release of cytochrome c from mitochondria into the cytosol in A2780 and A2780/CP ovarian cancer cells was detected by western blots.
To examine the involvement of caspase activation and cleavage in β-elemene-induced apoptosis in ovarian cancer cells, we measured the activities of caspase-3, -8 and -9 in β-elemene-treated A2780 and A2780/CP cells. The activities of all three caspases were significantly increased at 48 h after treatment with β-elemene (Figure 7A and 7B; #p<0.01 and *p<0.05). The increase in caspase activity was blocked by the general caspase inhibitor Z-VAD-FMK (data not shown). Moreover, the auto-cleavage of caspase-9 was increased following β-elemene treatment in both A2780 and A2780/CP cells (Figure 7C). We also assessed whether other apoptotic effectors such as BCL-2 family proteins participated in the apoptotic response, triggered by β-elemene. Western blot analysis of treated and untreated A2780 and A2780/CP cells showed that the BCL-2 and BCL-XL protein levels were down-regulated, whereas the BAX protein level was up-regulated after β-elemene treatment (Figure 8).
Figure 7.

β-Elemene triggered caspase activation and cleavage in A2780 and A2780/CP cells. A and B: Caspase-3, -8 and -9 enzymatic activities in A2780 and A2780/CP cells were measured using a CasPASETM apoptosis assay kit. Cells were treated with β-elemene (ELE) at 30, 50, and 70 μg/ml for 48 h, harvested, and washed twice with ice-cold PBS. Cytosolic extracts were prepared and incubated with 5 μl of 1 mM caspase substrate at 37°C for 2 h. Absorbance of the cleaved substrate was measured at 405 nm. Untreated cell lysate was used as a control. Relative caspase activity was determined by calculating the fold increase of caspase activity in β-elemene-treated cells vs. untreated cells. *p<0.05 and #p<0.01 vs. the untreated groups. C: Caspase-9 cleavage in A2780 and A2780/CP cells is seen on western blots.
Figure 8.
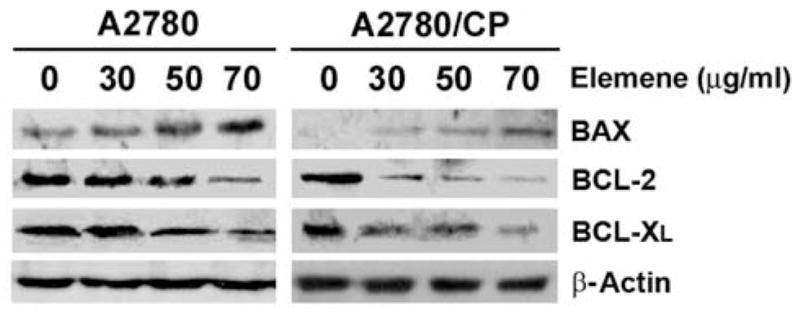
BCL-2 family proteins were involved in β-elemene-induced apoptosis in A2780 and A2780/CP cells. Each cell line was treated with β-elemene at the indicated concentrations for 48 h. The expression of BCL-2 family proteins was determined by western blot analysis as described in the Materials and Methods. Equal sample loading was verified by using anti-β-actin antibody.
Discussion
We previously showed that β-elemene significantly suppresses cell growth and proliferation of several types of human cancer cells, with no significant toxicity towards normal lung fibroblasts (11–15). In the current study, we demonstrated that β-elemene dramatically inhibited cell survival and growth of both cisplatin-sensitive and cisplatin-resistant human ovarian cancer cells, while it had only a moderate inhibitory effect on the proliferation of human ovary epithelial cells. The IC50 values for normal human ovary epithelial cell lines were nearly twice those for both sensitive and resistant ovarian cancer cell lines. Our study further revealed that the antitumor activity of β-elemene in ovarian cancer cells was mediated mainly through cell cycle arrest at the G2/M phase and the induction of apoptosis. These results strongly indicate that cisplatin-resistant ovarian tumor cells are sensitive to β-elemene in vitro, suggesting that β-elemene has a potential for replacing cisplatin in ovarian cancer chemotherapy when cisplatin fails.
CDKs regulate eukaryotic cell cycle progression. Their activities are positively regulated by cyclins and negatively regulated by CDK inhibitors (32). Changes in the expression of specific CDKs or their regulatory proteins can lead to uncontrolled cell proliferation and eventually to carcinogenesis (23, 33). Additionally, cell cycle checkpoint proteins play crucial roles in controlling cell cycle progression, eventually determining the fate of cells. Targeting the G1 and G2 checkpoint proteins has been the major focus for anticancer drug manipulation (22). To uncover the molecular mechanisms underlying the antiproliferative effect of β-elemene in human ovarian cancer cells, we analyzed β-elemene-treated cells by flow cytometry, which revealed that β-elemene induced cell cycle arrest at the G2/M phase in both cisplatin-sensitive and cisplatin-resistant ovarian cancer cells, with a greater effect in cisplatin-resistant cells. Western blot analysis showed that β-elemene down-regulated the protein expression of CDC2, cyclin A, and cyclin B1, and up-regulated the expression of p21WAF1/CIP1 and p53 in both A2780 and A2780/CP cells. Furthermore, β-elemene reduced the phosphorylation of CDC2 at Thr161 and increased that at Tyr15 in both cell lines. These results suggest that β-elemene suppresses ovarian cancer cell proliferation through cell cycle arrest at the G2/M phase, and that this occurs via β-elemene activation of p53, which in turn activates p21WAF1/CIP1. Activated p21WAF1/CIP1 then inhibits the activities of CDC2-cyclin B1 complex and other proteins controlling the G2 checkpoint, leading to cell cycle arrest and cell growth suppression in ovarian cancer cells.
Cells arrested at the G2/M phase very often enter apoptosis. Our previous studies have shown that β-elemene induces apoptotic responses in multiple types of cancer cell lines (11–15). In the present study, the results of several analytical methods, including ELISAs of fragmented DNA, annexin V and PI binding assays, and in situ TUNEL staining, demonstrated that β-elemene also induced apoptosis in both cisplatin-sensitive and cisplatin-resistant human ovarian cancer cells.
Apoptosis is executed by two pathways, an extrinsic pathway and an intrinsic pathway. Although the pathways differ, they partially overlap, allowing downstream crosstalk between the two pathways (34). The extrinsic pathway is initiated by the binding of extracellular death ligands to their cell surface receptors, leading to the activation of caspase-8/caspase-10 (35). A caspase-8-mediated process can subsequently trigger the release of mitochondrial proteins, activating the intrinsic pathway. In addition, a variety of stress signals generated within the cell can initiate the intrinsic pathway, inducing the permeabilization of the outer mitochondrial membrane and the release of cytochrome c from mitochondria into the cytosol. This leads to the activation of caspase cascades (36–38), which are tightly regulated by BCL-2 family proteins, including proapoptotic mediators (e.g., BAX and BAK) and antiapoptotic effectors (e.g., BCL-2 and BCL-XL) (34, 35, 39, 40, 41 ).
To further elucidate the molecular mechanisms through which β-elemene triggers apoptosis in ovarian cancer cells, we examined caspase activation and cleavage, and showed significantly increased activities of caspase-3, -8 and -9 in both cell lines following β-elemene treatment. These caspase activities were blocked by the caspase inhibitor Z-VAD-FMK. Moreover, the cleavage of caspase-9 was increased in β-elemene-treated cells. Thus, β-elemene appears to induce apoptotic cell death via a caspase-dependent apoptosis pathway in ovarian cancer cells. In addition, β-elemene enhanced BAX expression, increased phosphorylation of BCL-2 at Ser70 (data not shown), and reduced BCL-2 and BCL-XL protein levels in both A2780 and A2780/CP cells. Taken together, these data strongly support the involvement of BCL-2 family proteins in β-elemene-induced apoptosis in human ovarian cancer cells.
Cytochrome c is a component of the mitochondrial electron-transfer chain present in the inter-membrane space. The release of cytochrome c into the cytosol is an early event of apoptosis (42, 43). In the present study, cytosolic cytochrome c was increased in ovarian cancer cells after treatment with β-elemene. Cytochrome c in the cytosol is thought to activate downstream caspase-9 and caspase-3, leading to apoptosis. Loss of the mitochondrial transmembrane potential has been shown to result in a mitochondrial permeability transition, which causes the release of mitochondrial apoptogenic proteins (44, 45). In the present study, the mitochondrial transmembrane potential was reduced in ovarian cancer cells after treatment with β-elemene. These findings support the notion that β-elemene-induced apoptosis in ovarian cancer cells is mitochondria-dependent.
In conclusion, we provide evidence that β-elemene inhibits cell growth and survival in both cisplatin-sensitive and cisplatin-resistant human ovarian cancer cells. This effect of β-elemene in ovarian cancer cells is mediated through cell cycle arrest at the G2/M phase and the induction of apoptosis, suggesting that β-elemene may have multiple therapeutic targets. Our results support β-elemene as a novel chemotherapeutic candidate for cisplatin-resistant ovarian carcinoma.
Acknowledgments
This study was supported by grants from the National Institutes of Health (Nos. P20RR16440-010003, P20RR16440-020003, P20RR16440-030003, P20RR16440-040003 to QQL,) and West Virginia University School of Medicine (to QQL).
References
- 1.Sfakianos GP, Havrilesky LJ. A review of cost-effectiveness studies in ovarian cancer. Cancer Control. 2010;18:59–64. doi: 10.1177/107327481101800109. [DOI] [PubMed] [Google Scholar]
- 2.Ozols RF, Young RC. Chemotherapy of ovarian cancer. Semin Oncol. 1984;11:251–263. [PubMed] [Google Scholar]
- 3.McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, Clarke-Pearson DL, Davidson M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage iii and iv ovarian cancer. N Engl J Med. 1997;334:1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 4.Sood A, Buller R. Drug resistance in ovarian cancer: From the laboratory to the clinic. Obstet Gynecol. 1998;92:312–319. doi: 10.1016/s0029-7844(98)00184-7. [DOI] [PubMed] [Google Scholar]
- 5.ReConte P, Bruzzone M, Chiara S, Sertoli M, Daga M, Rubagotti A, Conio A, Ruvolo M, Rosso R, Santi L. A randomized trial comparing cisplatin plus cyclophosphamide versus cisplatin, doxorubicin, and cyclophosphamide in advanced ovarian cancer. J Clin Oncol. 1986;4:965–971. doi: 10.1200/JCO.1986.4.6.965. [DOI] [PubMed] [Google Scholar]
- 6.Kita T, Kikuchi Y, Hirata J, Nagata I. Prognosis of ovarian cancer today. Cancer J. 1998;11:201–207. [Google Scholar]
- 7.Glaysher S, Yiannakis D, Gabriel FG, Johnson P, Polak ME, Knight LA, Goldthorpe Z, Peregrin K, Gyi M, Modi P, Rahamim J, Smith ME, Amer K, Addis B, Poole M, Narayanan A, Gulliford TJ, Andreotti PE, Cree IA. Resistance gene expression determines the in vitro chemosensitivity of non-small cell lung cancer (nsclc) BMC Cancer. 2009;9:300. doi: 10.1186/1471-2407-9-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kudoh KRM, Ravatn R, Elkahloun AG, Bittner ML, Meltzer PS, Trent JM, Dalton WS, Chin KV. Monitoring the expression profiles of doxorubicin-induced and doxorubicin-resistant cancer cells by cdna microarray. Cancer Res. 2000;60:4161–4166. [PubMed] [Google Scholar]
- 9.Ozols R. Future directions in the treatment of ovarian cancer. Semin Oncol. 2002;29:32–42. doi: 10.1053/sonc.2002.31594. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Zhang H, Sun Y. Phase iii clinical trial of elemenum emulsion in the management of malignant pleural and peritoneal effusions. Zhonghua Zhong Liu Za Zhi. 1996;18:464–467. [PubMed] [Google Scholar]
- 11.Wang G, Li X, Huang F, Zhao J, Ding H, Cunningham C, Coad JE, Flynn DC, Reed E, Li QQ. Antitumor effect of beta-elemene in non-small-cell lung cancer cells is mediated via induction of cell cycle arrest and apoptotic cell death. Cell Mol Life Sci. 2005;62:881–893. doi: 10.1007/s00018-005-5017-3. [DOI] [PubMed] [Google Scholar]
- 12.Zhao J, Li QQ, Zou B, Wang G, Li X, Kim JE, Cuff CF, Huang L, Reed E, Gardner K. In vitro combination characterization of the new anticancer plant drug beta-elemene with taxanes against human lung carcinoma. Int J Oncol. 2007;31:241–252. [PubMed] [Google Scholar]
- 13.Li QQ, Wang G, Zhang M, Cuff CF, Huang L, Reed E. Beta-elemene, a novel plant-derived antineoplastic agent, increases cisplatin chemosensitivity of lung tumor cells by triggering apoptosis. Oncol Rep. 2009;22:161–170. doi: 10.3892/or_00000420. [DOI] [PubMed] [Google Scholar]
- 14.Li QQ, Wang G, Huang F, Banda M, Reed E. Antineoplastic effect of beta-elemene on prostate cancer cells and other types of solid tumour cells. J Pharm Pharmacol. 2010;62:1018–1027. doi: 10.1111/j.2042-7158.2010.01135.x. [DOI] [PubMed] [Google Scholar]
- 15.Li QQ, Wang G, Reed E, Huang L, Cuff CF. Evaluation of cisplatin in combination with beta-elemene as a regimen for prostate cancer chemotherapy. Basic Clin Pharmacol Toxicol. 2010;107:868–876. doi: 10.1111/j.1742-7843.2010.00592.x. [DOI] [PubMed] [Google Scholar]
- 16.Zheng S, Yang H, Zhang S, Wang X, Yu L, Lu J, Li J. Initial study on naturally occurring products from traditional chinese herbs and vegetables for chemoprevention. J Cell Biochem Suppl. 1997;27 :106–112. [PubMed] [Google Scholar]
- 17.Zhou H, Shen J, Hou J, Qiu Y, Luo Q. Experimental study on apoptosis induced by elemene in glioma cells. Ai Zheng. 2003;22:959–963. [PubMed] [Google Scholar]
- 18.Zou L, Liu W, Yu L. Beta-elemene induces apoptosis of k562 leukemia cells. Zhonghua Zhong Liu Za Zhi. 2001;23:196–198. [PubMed] [Google Scholar]
- 19.Hartwell L, Weinert T. Checkpoints: Controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 20.Eastman A. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem. 2004;91:223–231. doi: 10.1002/jcb.10699. [DOI] [PubMed] [Google Scholar]
- 21.Carr AM. Cell cycle. Piecing together the p53 puzzle. Science. 2000;287:1765–1766. doi: 10.1126/science.287.5459.1765. [DOI] [PubMed] [Google Scholar]
- 22.Ree A. Cell cycle-targeted therapy. Tidsskr Nor Laegeforen. 2004;124:2479–2482. [PubMed] [Google Scholar]
- 23.Sherr C. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 24.Sherr C. The pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- 25.Sen S, D’Incalci M. Apoptosis. Biochemical events and relevance to cancer chemotherapy. FEBS Lett. 1992;307:122–127. doi: 10.1016/0014-5793(92)80914-3. [DOI] [PubMed] [Google Scholar]
- 26.Kerr J, Winterford C, Harmon B. Apoptosis: Its clinical significance in cancer and cancer therapy. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 27.Makin G, Hickman J. Apoptosis and cancer chemotherapy. Cell Tissue Res. 2000;301:143–152. doi: 10.1007/s004419900160. [DOI] [PubMed] [Google Scholar]
- 28.Segal-Bendirdjian E, Dudognon C, Mathieu J, Hillion J, Besancon F. Cell death signalling: Recent advances and therapeutic application. Bull Cancer. 2005;92:23–35. [PubMed] [Google Scholar]
- 29.Parker RJ, Eastman A, Bostick-Bruton F, Reed E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced DNA repair of cisplatin-DNA lesions and reduced drug accumulation. J Clin Invest. 1991;87:773–777. doi: 10.1172/JCI115080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Behrens BC, Hamilton TC, Masuda H, Grotzinger KR, Whang-Peng J, Louie KG, Knutsen T, McKoy WM, Young RC, Ozols RF. Characterization of a cis-diamminedichloroplatinum (ii)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987;47:414–418. [PubMed] [Google Scholar]
- 31.Maines-Bandiera S, Kruk P, Auersperg N. Simian virus 40-transformed human ovarian surface epithelial cells escape normal growth controls but retain morphogenetic responses to extracellular matrix. Am J Obstet Gynecol. 1992;167:729–735. doi: 10.1016/s0002-9378(11)91579-8. [DOI] [PubMed] [Google Scholar]
- 32.Porter L, Donoghue D. Cyclin b1 and cdk1: Nuclear localization and upstream regulators. Prog Cell Cycle Res. 2003;5:335–347. [PubMed] [Google Scholar]
- 33.Wesierska-Gadek J, Gueorguieva M, Horky M. Dual action of cyclin-dependent kinase inhibitors: Induction of cell cycle arrest and apoptosis. A comparison of the effects exerted by roscovitine and cisplatin. Pol J Pharmacol. 2003;55:895–902. [PubMed] [Google Scholar]
- 34.Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 35.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: The calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 36.Jourdain A, Martinou JC. Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol. 2009;41:1884–1889. doi: 10.1016/j.biocel.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Kagan VE, Bayir A, Bayir H, Stoyanovsky D, Borisenko GG, Tyurina YY, Wipf P, Atkinson J, Greenberger JS, Chapkin RS, Belikova NA. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: A new strategy in anti-apoptotic drug discovery. Mol Nutr Food Res. 2009;53:104–114. doi: 10.1002/mnfr.200700402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danial NN, Gimenez-Cassina A, Tondera D. Homeostatic functions of bcl-2 proteins beyond apoptosis. Adv Exp Med Biol. 2010;687:1–32. doi: 10.1007/978-1-4419-6706-0_1. [DOI] [PubMed] [Google Scholar]
- 40.Danial NN. Bcl-2 family proteins: Critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254–7263. doi: 10.1158/1078-0432.CCR-07-1598. [DOI] [PubMed] [Google Scholar]
- 41.Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los M. Apoptosis and cancer: Mutations within caspase genes. J Med Genet. 2009;46:497–510. doi: 10.1136/jmg.2009.066944. [DOI] [PubMed] [Google Scholar]
- 42.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial ca2+ uptake in apoptosis. Cell Calcium. 2006;40:553–560. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta S, Kass GE, Szegezdi E, Joseph B. The mitochondrial death pathway: A promising therapeutic target in diseases. J Cell Mol Med. 2009;13:1004–1033. doi: 10.1111/j.1582-4934.2009.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gross AMJaKS. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 45.Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann NY Acad Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]