

Abstract
Systemic lupus erythematosus (SLE) is a disease that disproportionately affects females. Despite significant research effort, the mechanisms underlying the female predominance in this disease are largely unknown. Previously, we showed that estrogen receptor alpha knockout (ERαKO) lupus prone female mice had significantly less pathologic renal disease and proteinuria, and significantly prolonged survival. Since autoantibody levels and number and percentage of B/T cells were not significantly impacted by ERα genotype, we hypothesized that the primary benefit of ERα deficiency in lupus nephritis was via modulation of the innate immune response. Using BMDCs and spleen cells/B cells from female wild-type or ERαKO mice, we found that ERαKO-derived cells have a significantly reduced inflammatory response after stimulation with TLR agonists. Our results indicate that the inflammatory response to TLR ligands is significantly impacted by the presence of ERα despite the absence of estradiol, and may partially explain the protective effect of ERα deficiency in lupus-prone animals.
Keywords: Estrogen receptor alpha, Systemic lupus, erythematosus, Dendritic cells, Toll-like receptors
1. Introduction
Systemic lupus erythematosus and many other autoimmune diseases disproportionately affect females, yet the mechanisms underlying the female predominance in these diseases remain a key unanswered question. The basis of the gender bias is almost certainly multifactorial, likely involving the sex chromosomes themselves, as well as the sex hormones and their receptors. Given that the gender bias is most prominent during the reproductive years, estrogen and its receptors are implicated in disease pathogenesis. Estrogen’s primary effects are mediated via estrogen receptors alpha and beta (ER α/β) that are expressed in many tissues, including most immune cells, where they have pleiotropic effects in both the innate and adaptive immune responses [1]. The classical roles of ERs as nuclear hormone receptors are, when ligand-bound, to directly bind estrogen response elements (EREs) in gene promoters to regulate transcription of target genes. ERs can also, however, tether to other transcription factors to regulate transcriptional activity, without direct DNA binding [2–4]. Additionally, via “non-classical effects”, cytoplasmic ER and membrane associated ER can impact specific kinase signaling pathways directly to regulate the cellular milieu [5–7], making the potential regulatory impact of ERs even greater. These “non-classical” effects of ERs can occur in the absence of ligand binding, i.e. estradiol-independent.
Previously, we showed that ERα−/− NZM2410 and ERα−/−MRL/lpr lupus prone mice had significantly prolonged survival compared to ERα+/+ animals [8]. ERa−/− mice manifested significantly less pathologic renal disease and proteinuria, despite similar levels of autoantibody production and immune complex deposition as wild-type mice. These findings in ERα deficient mice indicated that autoantibody production/immune complex deposition is necessary, but not sufficient, for full expression of lupus nephritis. Given that the adaptive immune response appears minimally affected by ERα deficiency, we hypothesized that one of the protective effects of ERα deficiency in lupus is by modulating the innate immune response.
Toll-like receptors (TLRs) are critical factors in the innate immune response and TLR activation by autoantigens can potentially amplify autoimmune responses [9]. Multiple lines of evidence suggest that TLRs are involved in the pathogenesis of lupus. For example, the Yaa locus, a duplication of the TLR7 gene on the Y chromosome, is responsible for lupus susceptibility in BXSB mice [10,11]. Consistent with this, TLR7- and TLR7/9-deficient lupus prone animals have diminished disease [12]. Interestingly, TLR9 deficiency alone exacerbates disease, however, disease worsening was dependent on the presence of TLR7 [13]. This recent work suggests that TLR9 acts both in parallel and in interdependent pathways with TLR7 to regulate immune responses. TLR3 is also implicated in lupus disease expression [14], but its effects overall are still unclear as renal injury in TLR3-deficient mice was unaffected [15].
TLRs are highly expressed on DCs, potent antigen presenting cells important in the innate immune response that are dysregulated in lupus [16,17]. Estradiol regulates DC differentiation, and is, in fact, required for the development of the Langerhans-like subset of DCs from myeloid progenitors (MP) [18]. A functional ERα is required for this effect. In contrast, estradiol inhibits DC differentiation when MP are supplemented with Flt3 ligand, which predominates in homeostatic DC development [19,20]. This finding suggests a role for estradiol and ERs in differential regulation of DCs depending on the cellular milieu (inflammatory vs. non-inflammatory). Since DCs can stimulate Th1, Th2 and Th17 responses, modulate NK-T cell and plasma cell function, as well as directly impact B cell proliferation, differentiation and isotype switching [21–23], they have far reaching effects on the immune response when inappropriately activated. DCs are dysregulated in SLE; DCs derived from both SLE patients and lupus-prone animals display an activated phenotype characterized by increased CD80/86 and MHCII expression as well as increased inflammatory cytokine production [24–27]. Recently, Seillet et al. showed that 17β-estradiol treatment of normal post-menopausal women enhanced TLR7/9 pDC production of IFNα, an effect that required ERα [28].
We hypothesized that ERα deficiency impacts lupus disease expression by modulating the innate immune response to TLR signaling. In this study we showed that DC development and TLR-stimulated DC function were affected by ERα absence in murine lupus. The absolute number of DCs isolated from lupus prone ERαKO mice was reduced, and these DCs produced significantly less IL-6 and MCP-1 in response to TLR9 stimulation compared with wild-type littermates. Interestingly, we also find fewer plasmacytoid-type DCs (pDCs) in ERαKO lupus prone mice. After cell-sorting and standardizing numbers, pDCs derived from ERαKO mice also produced less IFNα in the absence of estradiol. We also report for the first time that ERα deficiency impacts IL-23, IL-23R and IL-17 expression in lupus prone mice. These cytokines were reduced in DCs and ex vivo spleen cells, respectively, when cultured from ERαKO lupus prone mice treated with TLR7/9 agonists in the absence of estradiol. These experiments indicate that ERα may modulate lupus disease expression by influencing DC development and TLR signaling, and may contribute to the development of pro-inflammatory cell types such as pDCs and Th17 cells that are implicated in lupus pathogenesis. This study provides partial mechanistic understanding of how ERα deficiency may protect against development of lupus nephritis in murine lupus.
2. Materials and methods
2.1. Mice
ERα−/−mice on the C57BL/6 background (kind gift of Ken Korach, NIEHS, RTP, NC) were backcrossed for 9 generations to NZM2410 or MRL/lpr mice (Jackson Laboratory, Bar Harbor, ME) and congenic status was verified as previously described [8]. Only female mice were used for these experiments. All mice were maintained at the Ralph H. Johnson VAMC Animal Care Facility (Charleston, SC) using Institutional Animal Care and Use Committee approved protocols #421 and #498 approved August 2008 and 2011 respectively.
2.2. Generation of BMDCs and spleen cells
Bone marrow-derived DCs were generated using a modified version of the protocol originally described by Inaba et al. [29], without lymphocyte depletion. Briefly, BM was flushed from the femurs and tibias of mice and then depleted of red blood cells by incubation with RBC lysis buffer (144 mM NH4Cl and 17 mM Tris, pH 7.6). Equal numbers of BM cells from WT and knockout mice were suspended in complete RPMI and supplemented with murine GM-CSF (20 ng/ml) and murine IL-4 (20 ng/ml) and cultured in T75 flasks at 1×106 cells/ml (~20×106/flask). On day 3 of culture, fresh complete RPMI medium was added to increase the total volume by 50%. Cytokines were replenished for the final total volume of medium. On day 7, the cells were harvested, washed twice and the phenotype of cDCs (CD11c+CD11b+) was confirmed by flow cytometry. This method yielded 50–80% conventional myeloid DCs depending on the strain. For spleen cells, mice were sacrificed and spleens were harvested and kept in ice-cold RPMI. Spleens were processed and subjected to red blood cell lysis. Cells were washed twice in cold RPMI before being counted and cultured in 12-or 6-well plates at 1×106 cells/ml (2–4×106/well) for 24 h. For B cell isolation, mouse depletion dynabeads were used (Dynal Mouse B Cell Negative Isolation Kit, Invitrogen, Carlsbad, CA) with a starting leukocyte working volume of 5×107 cells. An enriched B cell population was validated by FACS analysis and found to be > 92% pure.
2.3. Treatment of DCs with TLR agonists
BMDCs were harvested on day 7 from BM cultures as described above, washed twice with PBS and then counted with a hemocytometer. Cells were seeded at 1×106 cells/ml into 6- or 12-well plates in estrogen free phenol red-free RPMI with 10% charcoal-dextran-stripped FCS and treated with vehicle or TLR agonist: loxoribine (TLR7/8 agonist, 50–200 μmol), polyI:polyC (TLR3 agonist 10–100 mmol), LPS (TLR4 agonist; 1 μg/ml) or mouse CpG DNA (TLR9 agonist; 1μg/ml) for 24 h.
2.4. Antibodies and reagents
Antibodies including anti-mouse CD16/CD32 (Fc receptor block), and fluorescent conjugated anti-CD11b, anti-CD11c, anti-CD317 (PDCA1), anti-CD3e and anti-IL-17A were purchased from eBiosciences, Inc. (San Diego, CA). Anti-human/mouse RORγt-APC was purchased from R&D Systems (Minneapolis, MN). PE conjugated-120G8.04 mAb to pDC was purchased from Imgenex/Dendritics (San Diego, CA). Murine recombinant GM-CSF and IL-4 cytokines were purchased from R&D systems (Minneapolis, MN) and reconstituted in PBS before use. TLR agonists: polyI:polyC, loxoribine, and lipopolysaccharide were all purchased from Sigma–Aldrich, (St. Louis, MO). The TLR9 agonist CpG was purchased from Hycult Biotech (Canton, MA).
2.5. Flow cytometry
Fresh single-cell suspensions were prepared and 1×106 cells were treated with anti-CD16/CD32 for 5 min on ice. Cells were then stained with the indicated conjugated mAbs, and incubated for 30 min on ice. These cells were washed twice and re-suspended in 0.3 ml of 0.5% BSA, 0.02% sodium azide in PBS solution. To determine IL-17 and RORγt expression, spleen cells were cultured in complete RPMI±GolgiStop before intracellular staining was performed. Cells were fixed and permeabilized using an intracellular staining kit from BD Biosciences (San Jose, CA), and stained for IL-17 and RORγt. FACS acquisition was done on BD FACS Calibur machine. Cells were analyzed by flow cytometry using the FlowJo software package (Tree Star, Inc., Ashland, OR).
2.6. RNA preparation
Following 24 h culture, media was removed and RNA was prepared from cells using a hybrid Trizol/RNeasy protocol (adapted from Alvydas Mikulskis and the Qiagen RNeasy protocol). Briefly, samples were suspended in an appropriate volume of trizol and left at room temperature for 30 min. Chloroform was then added at a volume of 1:5. After a 15 min centrifugation at 12,000×g, the aqueous phase was removed and 70% EtOH added. Samples were loaded into RNeasy columns and washed per protocol before elution in TE buffer.
2.7. Analysis of gene expression in DCs and spleen cells by quantitative RT-PCR
Primers (IL1β, IL23, IL23R, GAPDH, 18 S) were purchased from SABiosciences (Frederick, MD); L19 was synthesized by Integrated DNA Technologies (Coralville, Iowa). All primers were used at 5 mM. First Strand Kit (SABiosciences) was used to generate cDNA. RT-PCR was performed as follows: for each real-time qPCR, 1ug cDNA was added to a master mix containing primers, IQ SYBR-green (Bio-Rad) and nuclease-free water. PCR conditions were 10 m at 95 °C, 40 cycles of 15 s at 95 °C, 60 s at the appropriate annealing temperature (generally 60 °C), 60 s at 95 °C, and 60 s at 55 °C. Subsequent melt curve analysis commenced at 55 °C for 10 s and increased by 0.5 °C every 10 s for 80 cycles. A melt curve for each qPCR, with a single peak at the correct melting temperature, was indicative of a reliable and desired PCR product. All samples and controls were analyzed in triplicate, and a no-cDNA control was included. The relative gene expression of the genes of interest was calculated after normalization to at least 1 housekeeping gene (GAPDH, 18 s, or L19).
2.8. Cytokine release assay
Cytokine release by DCs and spleen cells was determined by culturing 1×106/ml cells with either vehicle or TLR agonist. After 24 h, culture supernatants were harvested and cytokine (IL-6, MCP-1 or IL-23) concentrations were measured by sandwich ELISA per the manufacturer’s protocol (eBioscience, Inc. San Diego, CA) using a microplate luminometer (Thermo Scientific Multiskan Ascent).
2.9. WISH cell assay
WISH cells, purchased from ATCC Cat #CCL-25 (Manassus, VA), were plated in 12-well plates at 3×105, and cultured in either control media (RPMI +10% FCS), control media plus Universal Type I Interferon 100 units (PBL Interferon Source, Piscataway, NJ) or conditioned media from PDCA1+ sorted pDCs (ERα+/+ or ERα−/−) post-treatment with CpG for 18 h. RNA was extracted from WISH cells after 6 h incubation and qRT-PCR was performed as above with 3 primer pairs (PKR, IFIT, MX1) to IFN target genes as described by Hua et al., 2006. Primer pairs were synthesized by Integrated DNA Technologies (Coralville, IA): MX1 (forw) 5′- TAC CAG GAC TAC GAG ATT G -3′; MX-1 (rev) 5′- TGC CAG GAA GGT CTA TTA G -3′; PKR (forw) 5′- CTT CCA TCT GAC TCA GGT TT -3′; PKR (rev) 5′- TGC TTC TGA CGG TAT GTA TTA -3′; IFIT (forw) 5′-CTC CTT GGG TTC GTC TAT AAA TTG -3′; IFIT (rev) 5′-AGT CAG CAG CCA GTC TCA G 3′.
2.10. Statistics
Numerical data obtained from each experiment were expressed as mean±SEM and the statistical differences between experimental and control groups were assessed using GraphPad Prism software. P values less than 0.05 were considered statistically significant. For experiments that included both mouse strain and treatment variables, a grouped analysis was done by performing a two-way ANOVA with subsequent Bonferroni’s post-test correction. A sign test was performed for paired animals (WT versus KO).
3. Results
3.1. TLR-induced IL-6 production is significantly reduced in spleen cells and isolated B cells
To test the hypothesis that TLR-induced inflammation is downregulated in immune cells from ERα deficient lupus prone mice, we examined production of IL-6 by spleen cells and isolated B cells following treatment with TLR agonists under estrogen-free conditions. Animals were sacrificed prior to onset of disease at 8–12 weeks of age and littermates were utilized when possible. Spleens were harvested and cells were isolated for overnight culture. A subset of spleen cells was used for isolation of B cells by negative selection (>92% pure). Treatment for 24 h with LPS 1 μg/ml (TLR4), polyI:C 10 or 100 μg/ml (TLR3), or loxoribine 200 mmol (TLR7) resulted in significantly decreased levels of IL-6 production by spleen cells (Fig. 1A) and isolated B cells (Fig. 1B) from lupus prone ERαKO animals compared to wildtype littermates, as measured by ELISA. This result suggested that TLR-induced inflammatory cytokine production is blunted in the absence of ERα.
Figure 1.

TLR-induced IL-6 production is significantly reduced in isolated spleen cells (A) and B cells (B) from ERα−/− mice. Treatment with a TLR4 agonist (LPS 1ug/ml), TLR3 agonist (polyI:C 10 or 100ug/ml) or a TLR7 agonist (loxoribine 200 mmol) for 24 h resulted in IL-6 induction that was reduced by at least 50% in spleen cells or B cells from ERα−/− animals. Representative of 3 independent experiments with a total of 10 animals per group minimum. *=p<0.05, **=p<0.001.
3.2. TLR9-induced IL-6 and MCP-1 production by dendritic cells is reduced in both B6 and lupus prone ERαKO mice
Dendritic cells (DCs) express high levels of TLRs, are key mediators of the innate immune response, and are implicated in the pathogenesis of lupus. We isolated bone marrow hematopoietic cells from WT or ERαKO mice (B6, MRL/lpr and NZM2410) and derived DCs with selective/supplemented media. Following harvest on d7, a subset of DCs was analyzed by FACS (see Supplemental Fig. 1) and the remaining DCs were divided into 6- or 12-well plates. As DC development requires estrogen, the cells were derived in usual complete media (which contains the equivalent of micromolar concentrations of estradiol). Post derivation, stimulation of the DCs was performed under estrogen-free conditions. Following treatment with vehicle (PBS), loxoribine (50 nmol), or CpG DNA (1ug/ml) for 24 h, media was removed and the pro-inflammatory cytokines, IL-6 and MCP-1, were measured. In DC cultures derived from wild-type B6 mice, the TLR9 agonist CpG stimulated robust IL-6 production, however, ERαKO DC IL-6 production was significantly decreased. CpG stimulation increased IL-6 levels more than 20-fold in media from B6 WT DCs, with the stimulation index reduced by ~50% in ERαKO animals (Fig. 2A). MCP-1 levels increased 3-fold in response to CpG, and again there was a significantly reduced response by ERαKO DCs. Cytokine levels were in the range of 120 pg/ml–47 ng/ml, indicating a robust response. These data indicate that ERα significantly modulates TLR9 responses by DCs.
Figure 2.
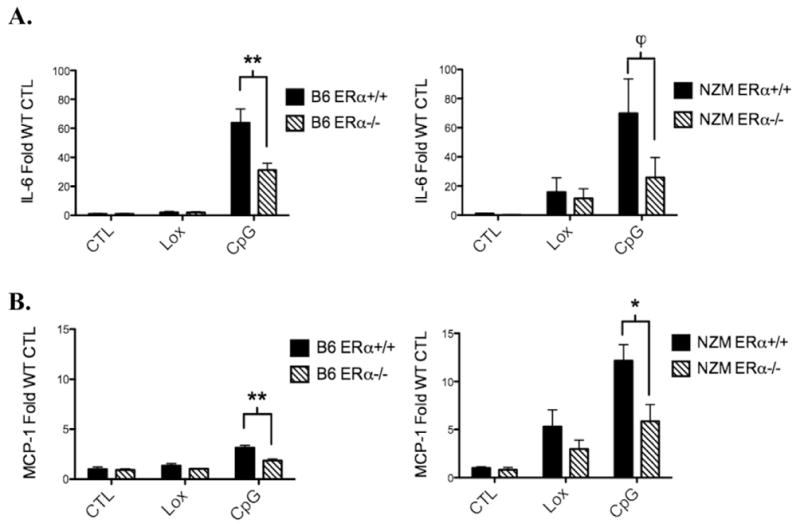
TLR9-induced IL-6 and MCP-1 production by bone-marrow derived dendritic cells cultured from ERα−/− mice is reduced in both B6 and NZM2410 strains. BMDCs from ERα+/+ and ERα−/− mice were cultured for 24 h with vehicle (CTL), loxoribine (Lox) 50 nmol, or CpG 1ug/ml under estradiol-free conditions. Media was removed and IL-6 (A) or MCP-1 (B) was measured by ELISA. Cytokine levels were in the range of 120 pg/ml–47 ng/ml. Pooled data from n=4 separate experiments with total number of 12 animals per group minimum. ϕ=p<.05, *=p<.01, **=p<0.0001.
Consistent with our findings in B6 ERαKO mice, DCs from ERα−/− lupus prone MRL/lpr and NZM2410 mice also exhibited a significantly blunted response to TLR agonists. In NZM2410 derived DCs, CpG treatment resulted in 60-fold increase in IL-6, with a significant reduction in IL-6 production in the absence of ERα (Fig. 2B). Similarly, MCP-1 production was significantly increased in CpG-treated DCs isolated from NZM mice, and this effect was reduced by ~50% in the setting of ERα deficiency, again indicating a role for ERα in modulating the effect of the TLR stimulated inflammatory response independent of ligand. Similar trends were observed in MRL/lpr WT and ERαKO animals (data not shown). TLR7 stimulation with loxoribine (50 nmol) also variably increased IL-6 and MCP-1 levels, with a trend towards decreased levels in ERα-deficient mice, however, the effects were not significant due to a low stimulatory index in both WT and ERαKO mice.
3.3. IL-1β and IL-23 expression is induced by a TLR9 agonist in wild-type but not ERαKO mice
CD4+ T-cells differentiate to become Th1, Th2, Tregs, or Th17 cells (among other subsets), based on intercellular interactions with APCs and the cytokine environment. IL-6 is an essential cytokine for driving the development of Th17 cells, which are implicated in the pathogenesis of SLE. Because TLR-induced levels of IL-6 were modulated by the presence or absence of ERα, we investigated the expression of IL-1β and IL-23, two TLR-induced cytokines that are produced by APCs and also promote the expansion and survival of Th17 cells.
In DCs isolated from B6 WT mice, IL-1β was induced >90-fold by CpG treatment. This effect was reduced by 80% in the ERα−/− animal (Fig. 3A). Loxoribine induced IL-1β mRNA expression by 10-fold and 3-fold in B6 WT and ERα KO mice respectively. CpG also stimulated IL-23 mRNA expression by B6 WT DCs, but did not induce IL-23 in ERα KO animals, suggesting that TLR9 stimulation of IL-23 requires ERα. IL-23 expression was also determined at the protein level by ELISA. Low concentrations of IL-23 were detected in media removed from cultured B6 WT DCs, but only in CpG-treated cultures. This effect was abrogated in B6 ERα−/− DC cultures (Fig. 3B). Similar results were found in NZM2410 mice, with the exception that higher levels of IL-23 were produced. In summary, expression of the pro-inflammatory cytokines IL-1β and IL-23 by B6 or NZM2410 DCs, following TLR9 stimulation, was significantly reduced in the setting of ERα deficiency, further implying an important role for ERα in modulating TLR9 signaling independent of estradiol.
Figure 3.
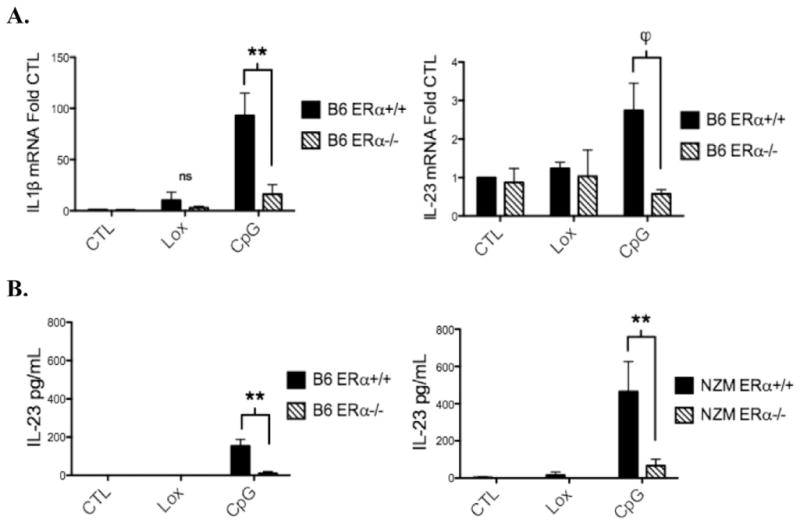
TLR9-induced mRNA expression of the inflammatory cytokines IL-1β and IL-23 by dendritic cells is reduced in B6 ERα−/−mice. BMDCs from ERα+/+ and ERα−/− mice were cultured for 24 h with vehicle (CTL), loxoribine (Lox) 50 nmol, or CpG 1ug/ml under estradiol-free conditions. A, RNA was isolated and IL-1β and IL-23 mRNA were measured by qRT-PCR. B, IL-23 protein levels were measured by ELISA (p19-specific capture antibody and a p40 specific detection antibody). Cytokine levels were in the range of 60 pg/ml–700 pg/ml. CpG-induced IL-23 production was reduced in ERα−/− animals in both B6 and NZM strains, n=3 or 4 separate experiments, **=p<0.001, ϕ=p<.05.
3.4. TLR9 stimulation induces IL-23R expression in wild-type but not ERαKO mice
To further define ERα effects on immune responses, we evaluated IL-23R expression. In addition to IL-23R upregulation on Th17 cells, a subset of dendritic cells (and macrophages) expressing IL-23R can be directly activated by IL-23, resulting in the production of inflammatory mediators [30,31]. We assessed the level of IL-23R mRNA expression in both WT and lupus prone animals to determine if its expression was affected by the presence or absence of ERα. As with cytokine levels of IL-23, IL23 receptor levels were also significantly reduced in the setting of ERα deficiency. In B6 WT DCs, IL-23R mRNA expression was stimulated 2–3-fold by both TLR7 and 9 ligands, however, TLRs did not induce IL-23R expression in DCs isolated from ERαKO mice (Fig. 4). TLR9 stimulation of DCs and spleen cells from MRL/lpr mice resulted in a robust increase in IL-23R expression, which was decreased by >50% in ERαKO mice (data not shown). A similar trend was noted in NZM2410 mice, but did not reach statistical significance. Combined, these data suggest that ERα plays a role in modulating TLR stimulation of the IL-23/IL-17 pathway that contributes to the pro-inflammatory phenotype in these animals.
Figure 4.
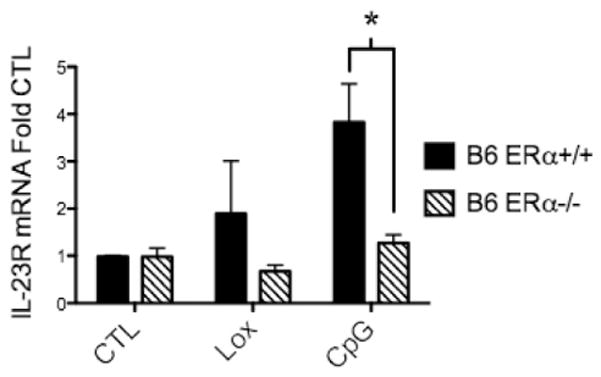
TLR9-induced mRNA expression of IL-23R by dendritic cells was reduced in B6 ERα−/− mice. BMDCs from ERα+/+ and ERα−/− mice were cultured for 24 h with vehicle (CTL), loxoribine (Lox) 50 nmol, or CpG 1ug/ml under estradiol-free conditions. RNA was isolated from DCs and IL-23R was measured by qRT-PCR. A similar trend was observed in NZM and MRL/lpr mice that reached statistical significance in MRL DCs and spleen cells (data not shown), n=3 separate experiments, min. 5 animals per group, *=p<0.01.
3.5. Percent of IL-17+/RORγt+spleen cells are reduced in lupus prone ERαKO mice
RORγt is a key transcription factor for Th17 differentiation and expansion, including upregulation of IL-23R to stabilize the Th17 phenotype. Evaluation of IL-17+/RORγt+spleen cells by flow cytometry was performed to determine whether the presence or absence of ERα in lupus prone animals impacted the development of IL-17 producing cells. Spleen cells were harvested and cultured for 24 h prior to being fixed and permeabilized for intracellular staining. In each experiment, spleen cells isolated from NZM ERα−/−animals had significantly fewer IL-17+/RORγt+cells than NZM WT animals (Fig. 5), again supporting the hypothesis that ERα is necessary for a robust inflammatory response to TLR stimulation, with one endpoint being production of IL-17-secreting cells.
Figure 5.
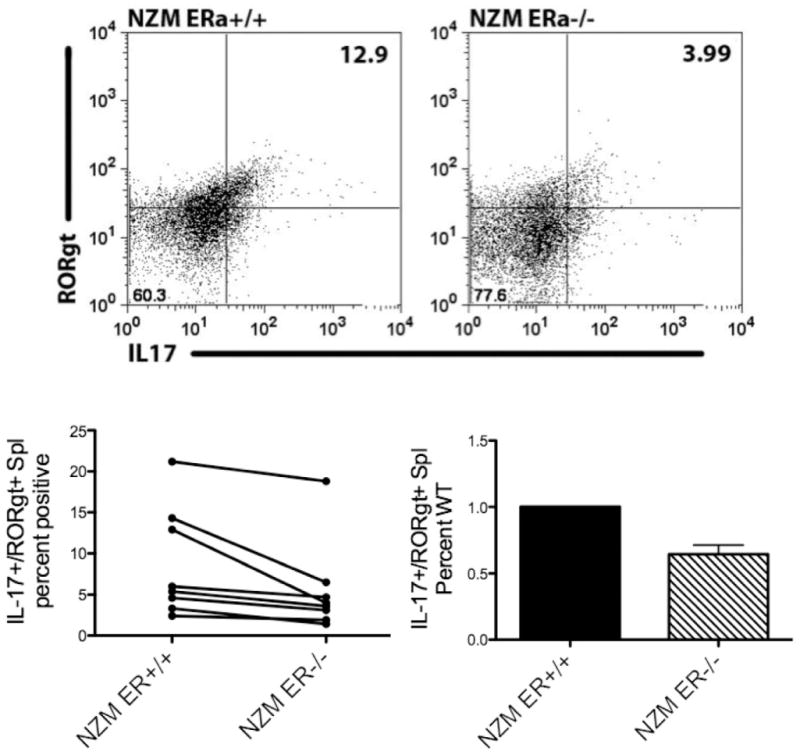
Unstimulated spleen cells derived from NZM ERα−/− animals displayed reduced numbers of IL-17+/RORγt+cells. Spleen cells were isolated from NZM ERα+/+ and NZM ERα−/− mice and cultured for 24 h +/− GolgiStop. Cells were fixed, permeabilized and stained with IL-17-PE and RORγt-APC. Isotype controls: rat IgG2a for IL-17 and anti-human/anti-mouse IgG mix for RORγt were used to set gates. Results are compiled from 8 sets of animals, n=5 separate experiments. For the difference between WT and ERαKO, both a paired T test and a sign test indicate a significantly larger percent of IL17+/RORγt+spleen cells in WT relative to KO animals (p=0.025 paired t-test, p=0.008 sign test).
3.6. BMDCs from ERαKO mice display a decreased plasmacytoid phenotype
In addition to finding fewer IL-17 producing cells in the absence of ERα, we tested the hypothesis that IFNα production is also impacted by ERα. The plasmacytoid subset of DCs is the major percentages were decreased significantly compared to their wild-type littermates, indicating that ERα impacts the development of pDCs under these culture conditions. producer of IFNα and expresses high levels of TLR7 and TLR9. Of note, in this report, DCs were derived from BM cells cultured for 7 d in GM-CSF and IL-4 supplemented media, an established culture method for deriving conventional, rather than plasmacytoid DCs (pDCs), while selecting against macrophages. With this method, approximately 50–80% of cells cultured expressed both CD11c and CD11b (Supplemental Fig. 1). Only a few cells (2–10%) were stained with the pan-macrophage marker F4-80 (data not shown) suggesting very little macrophage contamination. Thus we hypothesized that the remaining cells were pDCs. Mouse pDCs express low levels of CD11c or lose CD11c expression, but have other specific markers. To determine whether pDCs were present in our DC cultures, we used 2 different murine pDC markers, PDCA1 and 120 G8. By both markers, pDC percentage and number were significantly increased in NZM2410 (ERα+/+ approximately 42%) compared to B6 mice (5–10%) (Fig. 6). Most notably, we also found that in ERαKO strains, pDC numbers and
Figure 6.
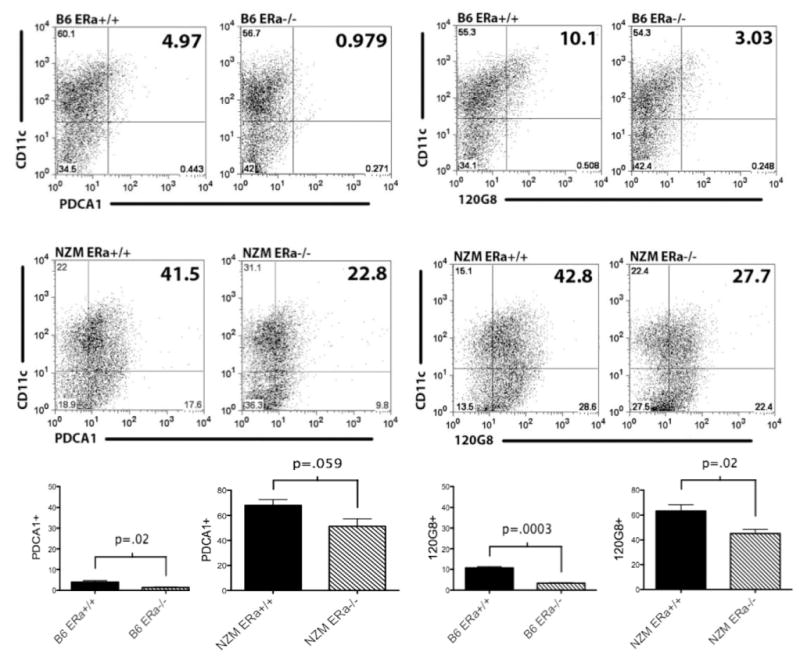
BMDCs from lupus-prone mice (middle panel) display increased plasmacytoid DC markers compared with wild-type B6 mice (top panel), despite culturing under traditional cDC conditions, using 2 different pDC-specific markers (PDCA or 120 G8). There is a clear trend toward reduced numbers of PDCA+or 120 G8+ cells in ERα−/− BMDCs which is statistically significant in B6 animals and in NZM mice staining with 120 G8. Flow cytometry plots represent 3 separate experiments, n=6 B6 and n=15 NZM animals (compiled in bottom panel).
3.7. IFNα and IL-6 production by plasmacytoid dendritic cells derived from ERαKO lupus-prone mice is significantly reduced
pDCs produce both IFNα and IL-6. Since we previously found decreased levels of IL-6 production by BMDCs, we first wanted to determine whether the overall reduced number of pDCs in the ERαKO animals was responsible for the cytokine decrease rather than being a functional difference. BMDCs were harvested on d7 and pDCs were stained with PDCA1. PDCA1+ cells were then sorted by MoFlo to separate pDCs from cDCs. Similar to the prior experiment, approximately 40–50% of NZM2410 ERαWT-derived DCs were PDCA1+ whereas only 20–30% of the DCs derived from ERαKO mice were PDCA1+. Once sorted, the pDC numbers were standardized to 1×106 cells/well and CpG added for 18 h. Media were removed and IL-6 levels were measured by ELISA. Consistent with our whole BMDC cultures, IL-6 production by ERαKO pDCs was significantly reduced compared with NZM2410 WT (Fig. 7A).
Figure 7.
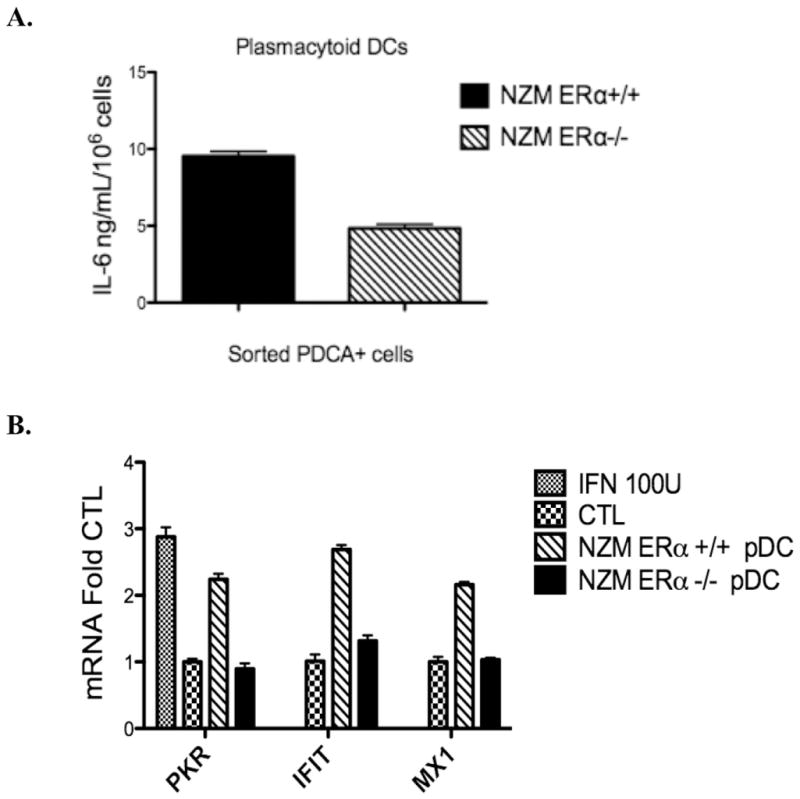
IL-6 (A) and IFNα (B) production by plasmacytoid dendritic cells derived from ERα−/− lupus-prone mice is significantly reduced. The number of PDCA+cells after cell-sorting was standardized to 1.0×106 cells/ml and cells were treated for 24 h with CpG 1ug/ml. A, IL-6 production was measured by ELISA and was significantly diminished in ERα−/− pDCs taken from NZM2410 mice, p<.001, representative of 2 independent experiments. B, IFNα activity was measured via WISH assay using conditioned media taken from CpG-treated pDCs. WISH cells were incubated with control media +/− Type I Interferon 100 units or with conditioned media from ERα+/+ or ERα−/− pDCs for 6 h. qRT-PCR was done using IFNα-responsive gene primer sets. ERα+/+ vs. ERα−/− are significantly different, p=.003, representative of 2 independent experiments.
We next determined IFNα activity via the WISH cell assay. Again, pDCs (PDCA1+ cells) were sorted and numbers standardized as above. Cells were treated with CpG for 18 h and media were removed for use in the WISH assay. WISH cells were incubated in either control or conditioned media (from pDCs treated with CpG) for 6 h prior to RNA isolation and qRT-PCR. A 2–3 fold induction of IFNα activity was observed in cells incubated with media from NZM2410-WT pDCs which was reduced to near baseline when media from NZM2410 ERαKO pDCs were tested (Fig. 7B).
4. Discussion
We previously reported that ERαKO NZM2410 and ERαKO MRL/lpr lupus prone mice had significantly reduced renal disease and significantly prolonged survival [8]. The mechanism of the renal protection afforded to ERαKO animals is currently unknown, which is the major focus of the current report. In this study, we demonstrated that ERα modulates TLR signaling. In the absence of ERα, the inflammatory response to TLR9 stimulation was significantly blunted. We also demonstrated, for the first time, that ERα is required for TLR-induced stimulation of IL-23R expression, which may have paracrine and autocrine effects on T cells and DCs involved in the IL-23/IL-17 inflammatory pathway. This may ultimately impact the numbers of Th17 cells that are expanded/stabilized, as evidenced by data in this report that fewer IL-17/RORγt-expressing spleen cells were found in ERαKO mice.
Previously published studies revealed a critical role for both estradiol and ERα in murine DC differentiation in normal animals [18–20,32,33]. Since dysregulated DC maturation and function are implicated in the pathophysiology of SLE (Reviewed in [34,35]), we investigated the role of ERα in two lupus-prone mouse strains. Our results demonstrate that ERα is an important factor in the regulation of DC development in lupus-prone mice (Supplemental Fig. 1), and that there are strain differences in the differentiation of conventional DCs, which are reduced in NZM2410 compared with MRL/lpr and B6 mice. It is possible that this difference is a reflection of the fact that BMDCs derived from NZM2410 mice have a propensity to develop into plasmacytoid DCs, despite culture conditions that favor cDC development. This is consistent with reports that a large subset of active SLE patients has increased numbers of pDCs and elevated IFNα levels [36,37]. Data by Pascual and others led to the discovery of the now well-known interferon α-signature in lupus patients and the hypothesis that SLE may be driven by unabated DC activation. Interestingly, we show in this study that pDC numbers are diminished in the absence of ERα in both B6 and NZM2410 animals, suggesting that ERα partly mediates the development of this subset of interferon-producing DCs. ERα also appears to have a functional impact on these cells; purified pDCs from ERαKO mice produced less IL-6 and IFNα than equivalent numbers of pDCs from ERα+/+ animals. Further work is necessary to characterize these cells and determine the effect on Type I interferon production and other functional parameters affected by the presence or absence of ERα.
As noted above, changes in DC maturation, as well as migration, are observed in both human and murine lupus [38–41]. DCs derived from both SLE patients and lupus-prone mice have an altered costimulatory profile, with an increase in the expression of maturation markers [25,42]. For example, in mice carrying Sle3, a lupus susceptibility locus in the NZM2410 strain, DCs are not only significantly more mature/activated, they are also less apoptotic, more proinflammatory and better at costimulating T cells in vitro [25]. DCs also express abundant TLRs and multiple lines of evidence have implicated these receptors in the development of SLE, particularly the subgroup of nucleic-acid specific TLRs.
Since ERα deficiency is renal protective in lupus-prone animals, we investigated the mechanism of ERα protection by assessing TLR-induced inflammation in the presence or absence of ERα. Perhaps not surprisingly, TLR9 stimulation caused a significant increase in pro-inflammatory mediator production by lupus-derived DCs vs. B6-derived, consistent with the notion that DCs have a more activated and/or pro-inflammatory phenotype in lupus. Importantly, we found a significant decrease in TLR-induced inflammation by multiple endpoints in ERαKO mice, indicating that ERα modulates TLR signaling. Our laboratory has generated similar data in ERαKO splenocytes and B cells in this report, as well as kidney mesangial cells (Svenson, Gilkeson, Cunningham, unpublished data) with multiple TLR agonists (TLRs 3,4,7,9); thus the effect does not appear to be TLR specific. We concentrated efforts in studying TLR7 and 9 due to their clear role in lupus pathogenesis. We further focused on TLR9 because we found the greatest stimulatory effect with CpG DNA and this was markedly different in ERαKO cells. This is the first report of which we are aware, to demonstrate that ERα is required, independent of estrogen, for robust TLR signaling. The mechanism of this effect, however, is still unclear, and our next task will be to investigate the role or ERα in TLR signaling on a molecular level.
On a more systemic level, our results indicate a role for ERα in the TLR induced IL-23/IL-17 inflammatory pathway. TLR7 and TLR9 agonists induced significant increases in IL-1β and IL-23 expression in ERα+/+, but not ERαKO mice. Furthermore, TLR7 and TLR9-induced upregulation of IL-23R, which is important for the stabilization of the Th17 cell phenotype, could not be demonstrated in the absence of ERα. These results led us to hypothesize that the capacity to expand Th17 cells is partially regulated by ERα (Fig. 8). In this report we provide evidence to support this hypothesis; ex vivo spleen cells taken from NZM ERαKO mice expressed significantly less IL-17 and RORγt. A reduction in IL-17 producing cells may partially explain the decrease in inflammatory renal damage and increased survival in these animals. Since numerous autoimmune diseases have a female predominance, and Th17 cells are implicated in the pathogenesis of many of these diseases, it is tempting to theorize that ERα modulation of Th17 cells is a common feature among affected females. Although more studies are required to fully understand and integrate the effects that ERα has on the immune system in SLE, these findings are an important step forward in determining more specifically the role of ERα in inflammation.
Figure 8.
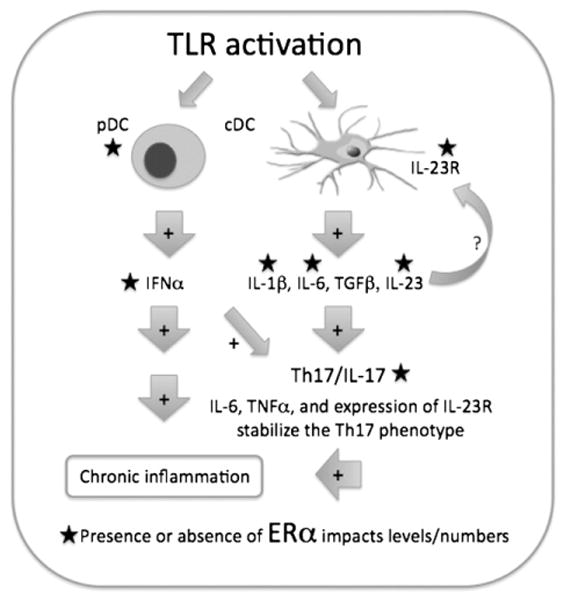
Working model for how ERα may modulate signaling by TLR-activated DCs. Activated DCs produce pro-inflammatory cytokines (ex. IL-23, IL-1β, IFNα) that promote ongoing inflammation via multiple mechanisms. As demonstrated, the expansion and stabilization of the Th17 subset of T cells via IL-6 and IL-23 and the upregulation of IL23R are all impacted by the presence of ERα. In the absence of ERα, TLR stimulation cannot fully induce IL-6, IL-1β, IL-23 or IL-23R upregulation on spleen cells (or DCs), leading to fewer IL-17-producing cells. Additionally, lupus-prone ERα−/−mice have decreased numbers of plasmacytoid DCs that feed into this pathway via production of IFNα.
There is growing evidence that the molecular mechanisms by which ERα exerts its effects are more complicated than the classic pathway of ligand-activated transcriptional activation via ERE binding. ERα also acts via multiple non-classical signaling pathways to regulate cellular responses. This study provides additional evidence for ligand-independent actions of ERα since the experiments reported herein were done under estradiol-free conditions. The full stimulatory effect of CpG and loxoribine on DC cytokine production, for example, required the presence of ERα but did not require the presence of estradiol. In addition to utilizing estradiol-free conditions, we also performed a set of experiments in the presence of ICI 182780, an estradiol antagonist, to ensure that there was no effect of any residual estradiol present in our media (phenol red-free, charcoal-dextran stripped). ICI had no effect (data not shown). Other studies have also demonstrated ligand-independent effects of ERα, and that DNA binding is not necessary for all ERα actions. For example, ERα may bind to other transcription factors such as AP-1, Sp1, and NFκB to regulate transcription [2–4]. It is also possible that ERα exerts some of its effects by recruiting co-activators or co-repressors, such as p300 [43,44], to the transcriptional complex to affect gene expression depending on the cellular milieu. This study provides evidence for a ligand-independent effect of ERα on TLR induced gene expression in dendritic cells, but does not identify definitively the molecular mechanism(s) for ERα effects on TLR signaling. Further work is needed to better understand the specific binding partners of ERα, whether DNA binding is necessary, and whether and which sites require ERα phosphorylation for these effects.
4.1. Conclusions
In summary, ERα plays an important role in TLR-induced inflammation, and appears to support the generation of both plasmacytoid dendritic cells and IL-17 producing cells, both of which are implicated in SLE (and other female predominant autoimmune diseases). Despite significant research effort and abundant evidence that sex hormones and their receptors influence disease expression, it has been difficult to disentangle the timing and specifics of their seemingly ubiquitous immune effects. Isolating the role of ERα, which clearly impacts both DC development and inflammation, suggests that modulation of lupus disease activity may be possible by targeting ERα.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.clim.2012.04.001.
References
- 1.Cunningham M, Gilkeson G. Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol. 2011;40:66–73. doi: 10.1007/s12016-010-8203-5. [DOI] [PubMed] [Google Scholar]
- 2.Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD. Crossroads of estrogen receptor and NF-kappaB signaling. Sci STKE. 2005;2005:pe27. doi: 10.1126/stke.2882005pe27. [DOI] [PubMed] [Google Scholar]
- 3.Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. J Biol Chem. 2001;276:13615–13621. doi: 10.1074/jbc.M008384200. [DOI] [PubMed] [Google Scholar]
- 4.Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- 5.Lambert KC, Curran EM, Judy BM, Lubahn DB, Estes DM. Estrogen receptor-alpha deficiency promotes increased TNF-alpha secretion and bacterial killing by murine macrophages in response to microbial stimuli in vitro. J Leukoc Biol. 2004;75:1166–1172. doi: 10.1189/jlb.1103589. [DOI] [PubMed] [Google Scholar]
- 6.Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 7.Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003;68:1–9. doi: 10.1016/s0039-128x(02)00110-1. [DOI] [PubMed] [Google Scholar]
- 8.Svenson JL, EuDaly J, Ruiz P, Korach KS, Gilkeson GS. Impact of estrogen receptor deficiency on disease expression in the NZM2410 lupus prone mouse. Clin Immunol. 2008;128:259–268. doi: 10.1016/j.clim.2008.03.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lafyatis R, Marshak-Rothstein A. Toll-like receptors and innate immune responses in systemic lupus erythematosus. Arthritis Res Ther. 2007;9:222. doi: 10.1186/ar2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisenberg RA, Izui S, McConahey PJ, Hang L, Peters CJ, Theofilopoulos AN, et al. Male determined accelerated autoimmune disease in BXSB mice: transfer by bone marrow and spleen cells. J Immunol. 1980;125:1032–1036. [PubMed] [Google Scholar]
- 11.Izui S, Iwamoto M, Fossati L, Merino R, Takahashi S, Ibnou-Zekri N. The Yaa gene model of systemic lupus erythema-tosus. Immunol Rev. 1995;144:137–156. doi: 10.1111/j.1600-065x.1995.tb00068.x. [DOI] [PubMed] [Google Scholar]
- 12.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 13.Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patole PS, Grone HJ, Segerer S, Ciubar R, Belemezova E, Henger A, et al. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol. 2005;16:1326–1338. doi: 10.1681/ASN.2004100820. [DOI] [PubMed] [Google Scholar]
- 15.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 17.Pascual V, Banchereau J, Palucka AK. The central role of dendritic cells and interferon-alpha in SLE. Curr Opin Rheumatol. 2003;15:548–556. doi: 10.1097/00002281-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Mao A, Paharkova-Vatchkova V, Hardy J, Miller MM, Kovats S. Estrogen selectively promotes the differentiation of dendritic cells with characteristics of Langerhans cells. J Immunol. 2005;175:5146–5151. doi: 10.4049/jimmunol.175.8.5146. [DOI] [PubMed] [Google Scholar]
- 19.Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol. 2004;172:1426–1436. doi: 10.4049/jimmunol.172.3.1426. [DOI] [PubMed] [Google Scholar]
- 20.Carreras E, Turner S, Paharkova-Vatchkova V, Mao A, Dascher C, Kovats S. Estradiol acts directly on bone marrow myeloid progenitors to differentially regulate GM-CSF or Flt3 ligand-mediated dendritic cell differentiation. J Immunol. 2008;180:727–738. doi: 10.4049/jimmunol.180.2.727. [DOI] [PubMed] [Google Scholar]
- 21.Jego G, Pascual V, Palucka AK, Banchereau J. Dendritic cells control B cell growth and differentiation. Curr Dir Autoimmun. 2005;8:124–139. doi: 10.1159/000082101. [DOI] [PubMed] [Google Scholar]
- 22.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 23.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 24.Decker P, Kotter I, Klein R, Berner B, Rammensee HG. Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2006;45:1087–1095. doi: 10.1093/rheumatology/kel061. [DOI] [PubMed] [Google Scholar]
- 25.Zhu J, Liu X, Xie C, Yan M, Yu Y, Sobel ES, et al. T cell hyperactivity in lupus as a consequence of hyperstimulatory antigen-presenting cells. J Clin Invest. 2005;115:1869–1878. doi: 10.1172/JCI23049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding D, Mehta H, McCune WJ, Kaplan MJ. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J Immunol. 2006;177:5878–5889. doi: 10.4049/jimmunol.177.9.5878. [DOI] [PubMed] [Google Scholar]
- 27.Kis-Toth K, Tsokos GC. Dendritic cell function in lupus: Independent contributors or victims of aberrant immune regulation. Autoimmunity. 2010;43:121–130. doi: 10.3109/08916930903214041. [DOI] [PubMed] [Google Scholar]
- 28.Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood. 2012;119:454–464. doi: 10.1182/blood-2011-08-371831. [DOI] [PubMed] [Google Scholar]
- 29.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 31.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 32.Carreras E, Turner S, Frank MB, Knowlton N, Osban J, Centola M, et al. Estrogen receptor signaling promotes dendritic cell differentiation by increasing expression of the transcription factor IRF4. Blood. 2010;115:238–246. doi: 10.1182/blood-2009-08-236935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovats S, Carreras E. Regulation of dendritic cell differentiation and function by estrogen receptor ligands. Cell Immu-nol. 2008;252:81–90. doi: 10.1016/j.cellimm.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fragoso-Loyo H, Cabiedes J, Richaud-Patin Y, Orozco-Narvaez A, Diamond B, Llorente L, et al. Inflammatory profile in the cerebrospinal fluid of patients with central neuropsychiatric lupus, with and without associated factors. Rheumatology (Oxford) 2009;48:1615–1616. doi: 10.1093/rheumatology/kep297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fragoso-Loyo H, Cabiedes J, Orozco-Narvaez A, Davila-Maldonado L, Atisha-Fregoso Y, Diamond B, et al. Serum and cerebrospinal fluid autoantibodies in patients with neuropsychiatric lupus erythematosus. Implications for diagnosis and pathogenesis. PLoS One. 2008;3:e3347. doi: 10.1371/journal.pone.0003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin O, Kavikondala S, Sun L, Fu R, Mok MY, Chan A, et al. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus. 2008;17:654–662. doi: 10.1177/0961203308089410. [DOI] [PubMed] [Google Scholar]
- 37.Louthrenoo W, Kasitanon N, Wichainun R, Wangkaew S, Sukitawut W, Kuwata S, et al. Antiagalactosyl IgG antibodies in Thai patients with rheumatoid arthritis, systemic lupus erythematosus, and systemic sclerosis. Clin Rheumatol. 2010;29:241–246. doi: 10.1007/s10067-009-1284-y. [DOI] [PubMed] [Google Scholar]
- 38.Fragoso-Loyo H, Richaud-Patin Y, Orozco-Narvaez A, Davila-Maldonado L, Atisha-Fregoso Y, Llorente L, et al. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 2007;56:1242–1250. doi: 10.1002/art.22451. [DOI] [PubMed] [Google Scholar]
- 39.Patole PS, Pawar RD, Lech M, Zecher D, Schmidt H, Segerer S, et al. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol Dial Transplant. 2006;21:3062–3073. doi: 10.1093/ndt/gfl336. [DOI] [PubMed] [Google Scholar]
- 40.Papadimitraki ED, Choulaki C, Koutala E, Bertsias G, Tsatsanis C, Gergianaki I, et al. Expansion of toll-like receptor 9-expressing B cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum. 2006;54:3601–3611. doi: 10.1002/art.22197. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez V, Fragoso MA, Billotte C, Lamar P, Orozco MA, Dubovy S, et al. Efficacy of various drugs in the prevention of posterior capsule opacification: experimental study of rabbit eyes. J Cataract Refract Surg. 2004;30:2598–2605. doi: 10.1016/j.jcrs.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 42.Theis KA, Murphy L, Hootman JM, Helmick CG, Sacks JJ. Arthritis restricts volunteer participation: prevalence and correlates of volunteer status among adults with arthritis. Arthritis Care Res (Hoboken) 2010;62:907–916. doi: 10.1002/acr.20141. [DOI] [PubMed] [Google Scholar]
- 43.Louthrenoo W, Kasitanon N, Sukitawut W, Wichainun R. A clinical study of crystal-proven gouty arthritis in a university hospital. J Med Assoc Thai. 2003;86:868–875. [PubMed] [Google Scholar]
- 44.Louthrenoo W, Kasitanon N, Mahanuphab P, Bhoopat L, Thongprasert S. Kaposi’s sarcoma in rheumatic diseases. Semin Arthritis Rheum. 2003;32:326–333. doi: 10.1053/sarh.2002.50000. [DOI] [PubMed] [Google Scholar]