

Abstract
Brain development requires a precise balance between expansion of the neural progenitor pool and the production of postmitotic neurons and glia. Disruption of this equilibrium results in a myriad of structural abnormalities and disorders of the nervous system. The molecular mechanism that restricts neural progenitor expansion is poorly understood. Here we show that the tumor suppressor neurofibromatosis 2 (Nf2; merlin) limits the expansion of neural progenitor cells (NPCs) in the mammalian dorsal telencephalon. Nf2 is localized at the apical region of NPCs. In the absence of Nf2, NPCs of the cortical hem, hippocampal primordium and neocortical primordium overexpand, while production of Cajal-Retzius cells and hippocampal neurons decreases, resulting in severe malformation of the hippocampus in adult mice. We further show that Nf2 functions by inhibiting the Yap/Taz transcriptional coactivators, probably through a mechanism that is distinct from the canonical Hippo pathway. Overexpressing human YAP in NPCs causes a hippocampal malformation phenotype that closely resembles that of Nf2 mutants and, importantly, deleting Yap in the Nf2 mutant background largely restores hippocampal development. Our studies uncover Nf2 as an important inhibitor of neural progenitor expansion and establish Yap/Taz as key downstream effectors of Nf2 during brain development.
Keywords: Radial glial cells, Neurogenesis, Archicortex, Mouse, Yap1, Wwtr1
INTRODUCTION
Neurofibromatosis type 2 (NF2) is an autosomal dominant disorder characterized by the development of nervous system tumors (Baser et al., 2003). It is caused by inactivating mutations of neurofibromin 2 (NF2; also known as neurofibromatosis 2 or merlin), which is also mutated in a large proportion of sporadic schwannomas, meningiomas and ependymomas. The Nf2 protein is structurally similar to the ERM proteins ezrin, radixin and moesin. Functionally, however, Nf2 is unique in having tumor-suppressor activities.
The molecular function of Nf2 appears to be complex. Numerous studies have shown that Nf2 can mediate the formation of adherens junctions, regulate the distribution of cell-surface receptors, and modulate a diverse array of signaling effectors ranging from the EGFR-Ras-ERK, Rac-PAK, PI3K-Akt, mTORC1 and Hippo-YAP pathways at the cell cortex to the CRL4DCAF1 E3 ubiquitin ligase in the nucleus (reviewed by Li et al., 2012). Although Nf2 is widely expressed during mammalian embryogenesis (Akhmametyeva et al., 2006), its role during development is poorly understood. Nf2 knockout mouse embryos fail to initiate gastrulation (McClatchey et al., 1997). It has been suggested that defects in extra-embryonic tissues are to blame, but the nature and cause of the defects remain elusive. Two studies have shown that liver-specific Nf2 deletion results in liver overgrowth and tumorigenesis (Benhamouche et al., 2010; Zhang et al., 2010), yet the primary cell type that is autonomously affected by Nf2 loss is unclear and the signaling pathways responsible for the phenotypes remain controversial (Breuhahn and Schirmacher, 2010). Loss of Nf2 in the skin leads to defective tight junctions and loss of progenitor cell polarity, but has no effect on progenitor proliferation (Gladden et al., 2010). Thus, Nf2 function and its mechanism of action are highly cell type dependent.
The mammalian brain is a highly structured ensemble comprising a vast number of diverse types of cells. The development of such a complex organ requires the exquisite coordination of progenitor proliferation and differentiation. Neural stem/progenitor cells (NPCs), including neuroepithelial cells and radial glial cells, form a pseudostratified epithelial layer that lines the ventricle of the neural tube (reviewed by Götz and Huttner, 2005; Kriegstein and Alvarez-Buylla, 2009). Many of the cellular processes in which Nf2 has been implicated, including cell junction formation, polarity establishment and proliferation, significantly affect NPC behavior. Although Nf2 is highly expressed in NPCs during brain development (McLaughlin et al., 2007), its role in NPCs is poorly understood. A previous study using a conditional loss-of-function allele of Nf2 (Nf2flox2, referred to here as Nf2F) (Giovannini et al., 2000) and a Nestin-Cre line that drives mosaic recombination in NPCs as early as embryonic day (E) 8.5 found that Nf2 loss led to detachment of NPCs from the neuroepithelium and tissue fusion defects, including neural tube closure defects (McLaughlin et al., 2007). The authors suggested that Nf2 is required specifically for the assembly, but not the maintenance, of the neuroepithelial junctional complex. However, the role of Nf2 in NPC self-renewal and differentiation was not assessed.
Here, we bypassed the early structural defects to specifically address the role of Nf2 in NPC maintenance, proliferation and differentiation during brain development. We found that Nf2 loss resulted in a severe reduction in hippocampus size. Counterintuitively, NPCs in the developing hippocampus and cortical hem, which is the hippocampal organizer, of Nf2 mutants maintained their progenitor properties longer than normal and overexpanded. Nf2 loss also caused an overexpansion of the neocortical progenitor pool, suggesting that Nf2 limits the expansion of neural progenitor populations during brain development. Through molecular and genetic approaches, we demonstrate that Nf2 functions by inhibiting the transcriptional coactivators Yap (Yap1 - Mouse Genome Informatics) and Taz (Wwtr1 - Mouse Genome Informatics). This work thus reveals a novel function of Nf2 as an inhibitor of neural progenitor expansion during brain development and establishes Yap/Taz as key mediators of Nf2 function.
MATERIALS AND METHODS
Animals
Animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of St Jude Children’s Research Hospital. Emx1IRES-Cre (stock no. 005628) and Nes-Cre lines (stock no. 003771) were obtained from the Jackson Laboratory. TetO-YAP1 line was provided by Thijn Brummelkamp and Fernando Camargo (Children’s Hospital, Boston, MA, USA) and Nestin-rtTA line by Steven Kernie (UT Southwestern Medical Center at Dallas, TX, USA). Doxycycline (Dox) was administered via diet (Bio-Serve; 200 mg/kg, E11.5 microarray experiment) or drinking water (100 mg/l from E9.5 to E14.5 and 200 mg/l from E14.5 to E17.5, phenocopy experiment). For BrdU labeling, timed pregnant mice were intraperitoneally injected with BrdU at 100 mg/kg body weight.
Histology, electron microscopy, immunostaining and TUNEL assay
Luxol Blue and Cresyl Violet stainings were performed using the Kluver-Barrera method (EMS #26681). For electron microscopy analysis, embryonic brains were fixed with 2.5% paraformaldehyde and 2.5% glutaraldehyde overnight, postfixed in 2% osmium tetroxide with 1.5% potassium ferrocyanide, and examined using a JEOL 1200EX transmission electron microscope. Immunostainings were preformed on 10-20 μm cryosections according to standard protocols. Primary antibodies are listed in supplementary material Table S1. Alexa 488-, Alexa 568- and Alexa 647- (Invitrogen), and DyLight 549- and DyLight 649- (Jackson ImmunoResearch) conjugated secondary antibodies were used. TUNEL assays were performed with the ApopTag Kit (Millipore). Low-magnification images were obtained with a Zeiss AxioImager M2 microscope. The remaining images were obtained with a Zeiss LSM 510 or Yokogawa CSU-X spinning disk confocal microscope. Image quantifications were performed using Imaris (Bitplane) or ImageJ (NIH) software. Statistical significance was evaluated by two-tailed, unpaired t-test.
In situ hybridization
In situ hybridization was performed as described (Schaeren-Wiemers and Gerfin-Moser, 1993). The following probes were used: Wnt3a, Wnt5a (Andrew McMahon, University of Southern California, Dornsife, CA, USA); Fzd10, SCIP, KA1 (Elizabeth Grove, University of Chicago, IL, USA); Axin2 (Juan Pedro Martinez-Barbera, University College London, UK); Lef1 (Gabriele Kardon, University of Utah, Salt Lake City, UT, USA).
Microarray and quantitative RT-PCR
Total RNA was isolated with Trizol reagent (Invitrogen). For each genotype, three embryos were used for microarray analyses. 100 ng of total RNA was processed using the Affymetrix 3′ IVT Express Kit. Biotin-labeled cRNAs were hybridized to the Affymetrix GeneChip HT MG-430 pm array and scanned on the GeneTitan system (Affymetrix). Data were summarized using Affymetrix Expression Console software v1.1 to apply the robust multi-array average algorithm. Differentially expressed transcripts were identified by ANOVA (Partek Genomics Suite v6.5) or by the local pooled error (LPE) t-test (S-Plus v6.2) (Jain et al., 2003). The false discovery rate (FDR) was estimated using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). Data files are available at the NCBI Gene Expression Omnibus (GEO) under Accession Number GSE48078. Additional sets of three embryos per genotype were used for quantitative RT-PCR analyses to confirm the microarray results. Primer sequences are listed in supplementary material Table S2.
Subcellular fractionation and quantitative western blot analysis
Total brain or cell lysates were prepared in 20 mM HEPES (pH 7.4), 150 mM NaCl, 2% SDS and 5% glycerol supplemented with Halt protease and phosphatase inhibitors (Thermo Scientific). Nuclear and cytosol/membrane fractions were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific). For quantitative western blot analyses, 20 μg protein was loaded per lane and detected by IRDye 600LT-conjugated and 800CW-conjugated secondary antibodies with the ODYSSEY infrared imaging system (LI-COR). The following primary antibodies were used: Yap (Sigma, WH0010413M1), Yap/Taz (Cell Signaling, 8418), phospho-S127-Yap (Cell Signaling, 4911), Nf2 (Sigma, HPA003097), actin (Ambion, AM4302) and PARP (Cell Signaling, 9542).
NPC culture and protein stability assay
Primary NPCs isolated from E13.5 cortices were cultured in the presence of FGF and EGF (20 ng/ml each) for 60 hours. Resulting neurospheres were treated with cycloheximide (100 μg/ml; Fisher).
RESULTS
Nf2 is localized at the apical region of NPCs but is not required to maintain the apical junctions or polarity of NPCs
To understand Nf2 function during murine brain development, we generated Nf2 conditional knockout (cKO) mice by crossing mice harboring the Nf2F allele (Giovannini et al., 2000) with the Emx1IRES-Cre line, which expresses Cre in the dorsal telencephalon by E10.5 (Gorski et al., 2002). At E12.5, Nf2F/F;Emx1IRES-Cre/+ (referred to as Nf2F/F;Emx1-Cre) embryos lacked Nf2 immunostaining signal at the ventricular surface of the dorsal, but not of the ventral, telencephalon (Fig. 1A,B, arrows). These Nf2 mutant mice were born at the expected Mendelian ratio and survived to adulthood. Histological examination of adult mutant brains revealed two major defects: dysgenesis of the corpus callosum and malformation of the hippocampus (Fig. 1C,D). We focused on the hippocampus phenotype in this study.
Fig. 1.

Loss of Nf2 results in dysgenesis of the corpus callosum and malformation of the hippocampus. (A,B) Immunostaining showing Nf2 concentrated at the apical/ventricular surface of E12.5 control brains (arrows, A). Nf2 immunoreactivity is specifically eliminated in the dorsal telencephalon of Nf2F/F;Emx1-Cre embryos (arrows, B). Magnified views of the boxed areas are shown to the right. (C,D) Luxol Blue staining for myelinated axons (blue) and Cresyl Violet for neuronal cell bodies (purple) show dysgenesis of the corpus callosum (arrows) and malformation of the hippocampus (boxed) in 2-month-old Nf2F/F;Emx1-Cre mice (D) as compared with control (C). (E,F) Whole-mount immunostaining of E12.5 neocortex, viewed from the ventricular surface (single plane views and corresponding 5 μm z-stack views at the position of the red line), showing Nf2 enrichment at NPC apical regions in the control (E). Nf2 immunoreactivity is greatly reduced in Nf2F/F;Emx1-Cre neocortices without affecting β-catenin or ZO-1 localization (F). Scale bars: 500 μm in A,B; 5 μm in E,F.
Because Nf2 is required for tight junction formation and polarity in epidermal cells (Gladden et al., 2010), we first analyzed whether Nf2 mutant dorsal telencephalons had defects in NPC apical junctions and polarity. At E12.5, Nf2 is concentrated at the apical region of wild-type NPCs, as shown by colabeling with the apical junctional components β-catenin and ZO-1 (Tjp1 - Mouse Genome Informatics) (Fig. 1E). However, loss of Nf2 did not affect the localization of β-catenin or ZO-1 at E12.5 (Fig. 1F). Apical junctions remained intact in E15.5 mutant dorsal telencephalons, as observed by electron microscopy (supplementary material Fig. S1A-B′) and ZO-1 immunostaining (supplementary material Fig. S1C,D). We did not observe any changes in the localization of the apical polarity marker aPKC either (supplementary material Fig. S1E,F).
In the vertebrate central nervous system (CNS), NPCs undergo interkinetic nuclear migration during their cell cycle and divide at the ventricular surface (Götz and Huttner, 2005). Loss of NPC polarity is often associated with ectopic divisions outside the ventricular surface. We found that, similar to those in control littermates, NPCs in Nf2 mutant dorsal telencephalons divided along the ventricular surface, as shown by immunostaining of the mitotic marker phospho-histone H3 (supplementary material Fig. S1G,H). Furthermore, in contrast to the findings of a previous study (McLaughlin et al., 2007), we never observed NPC detachment from the neuroepithelium or any neural tube defects in Nf2 mutants (n>50). Together, these results indicate that Nf2 is not required to maintain the apical junctions or polarity of cortical progenitors after E10.5.
Nf2 regulates growth and developmental progression of the cortical hem
To understand how the hippocampal phenotype arose, we tracked the developmental progression of the dorsal telencephalon in control and Nf2 mutant mice. We first examined the cortical hem, a signaling center that instructs hippocampus formation (Grove et al., 1998; Mangale et al., 2008; Subramanian et al., 2009). Using the hem marker Lmx1a, we found that the hem was significantly enlarged in mutant brains by E14.5, and by E15.5 it was about three times the size of that in control brains (Fig. 2A-E; supplementary material Fig. S2A-I). The enlarged region also expressed genes known to be specifically or preferentially expressed at the hem, including Wnt3a, Wnt5a and Fzd10 (Caronia et al., 2010; Grove et al., 1998), confirming that it was a true expansion of the hem (supplementary material Fig. S2J-O).
Fig. 2.
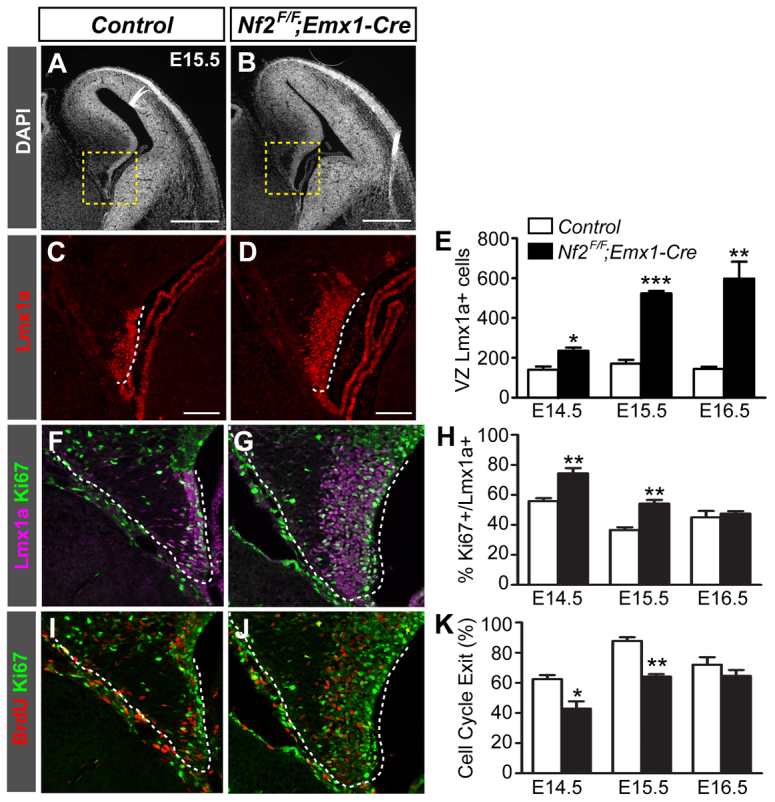
Loss of Nf2 leads to a marked expansion of the cortical hem. (A,B) DAPI staining shows the overall structure of control and Nf2F/F;Emx1-Cre dorsal telencephalons. (C,D) Lmx1a immunostaining shows enlargement of the cortical hem (dashed lines) in Nf2F/F;Emx1-Cre mouse embryos. C and D are enlargements of the boxed areas in A and B, respectively. (E) Quantification of the number of ventricular zone (VZ) Lmx1a+ hem cells per section. (F-H) Ki67 staining showing increased proliferation of VZ Lmx1a+ hem cells in Nf2F/F;Emx1-Cre embryos at E14.5 and E15.5. (I-K) After a 24-hour BrdU pulse, quantification of Ki67- cells among BrdU+ cells show decreased cell cycle exit of Nf2F/F;Emx1-Cre hem cells at E14.5 and E15.5. All images correspond to the medial level along the anterior-posterior axis of the hem. Quantifications were performed on sections at a similar level. Values are mean ± s.e.m. of three embryos. *P<0.05, **P<0.01, ***P<0.001. Scale bars: 500 μm in A,B; 100 μm in C,D.
The hem expansion in Nf2 mutants is unlikely to be caused by cortex-to-hem fate conversion, as seen in Lhx2-/- and Foxg1-/- embryos (Mangale et al., 2008; Muzio and Mallamaci, 2005), because the Emx1IRES-Cre line only becomes active after the cortex and hem fates have been specified. In fact, even conditional deletion of Lhx2 with the same Cre line does not cause hem expansion (Mangale et al., 2008). Moreover, fate conversion is typically associated with an elongation of the hem domain by E12.5, which was not observed in Nf2 mutants as late as E14.5 (supplementary material Fig. S2A′,B′). To exclude the possibility that Nf2 mutant hem cells had adopted a hem/cortex hybrid fate, we examined the expression of the cortex markers Pax6 and Lhx2. Similar to control hem cells, Nf2 mutant hem cells expressed low to undetectable levels of these markers at E14.5 and E16.5 (supplementary material Fig. S3), demonstrating that the cortex and hem fates were properly specified and maintained in Nf2 mutants.
We next tested whether changes in cell proliferation might have caused hem expansion in Nf2 mutants. We observed a higher percentage of Ki67+ hem cells in Nf2 mutants at E14.5 and E15.5 (Fig. 2F-H). Furthermore, 24 hours after a BrdU pulse, the fraction of BrdU+ hem cells that were Ki67- (i.e. cells that exited the cell cycle within the 24-hour period) was decreased in mutants at E14.5 and E15.5 (Fig. 2I-K). Together, these results demonstrate that Nf2 limits the proliferation capacity of hem cells.
To determine whether the increased proliferation of Nf2 mutant hem cells is accompanied by the maintenance of progenitor characteristics, we analyzed the expression of RC2, a radial glia-specific marker, and Blbp (Fabp7 - Mouse Genome Informatics), a marker of some radial glia and glia populations (Hartfuss et al., 2001; Kurtz et al., 1994; Misson et al., 1988). RC2 immunoreactivity at the hem of control embryos was low at E14.5 and was further diminished by E16.5 (Fig. 3A,B), especially when compared with the nearby dentate neuroepithelium (DNE) (Fig. 3, arrows). However, RC2 levels at the hem of Nf2 mutants were much higher at E14.5 and E16.5, and only became diminished by E18.5 (Fig. 3D-F). Blbp expression exhibited the opposite dynamics. In control brains, Blbp was induced at the dorsal hem at E14.5 and reached high expression levels at E15.5. In Nf2 mutants, the induction of Blbp was normal, but its expression level increased only moderately thereafter and never reached the intensity of that seen in control brains, except for a few cells at the dorsal edge of the hem (Fig. 3G-L′; supplementary material Fig. S4). These results suggest that the developmental progression of hem cells, manifested by the downregulation of RC2 and upregulation of Blbp, is incomplete upon Nf2 loss; Nf2 mutant hem cells maintain their early progenitor properties longer, which probably explains why they have increased proliferative capacity. However, as the lineage relationship between RC2+ and Bblp+ hem cells has not been formally established, it is also possible that these are two separate pools of hem cells and that RC2+ cells are preferentially expanded in Nf2 mutants. Nevertheless, together with the finding that Cajal-Retzius cells produced by the hem are reduced in Nf2 mutants (see below; Fig. 5C), our results indicate that Nf2 promotes hem cell maturation/differentiation.
Fig. 3.
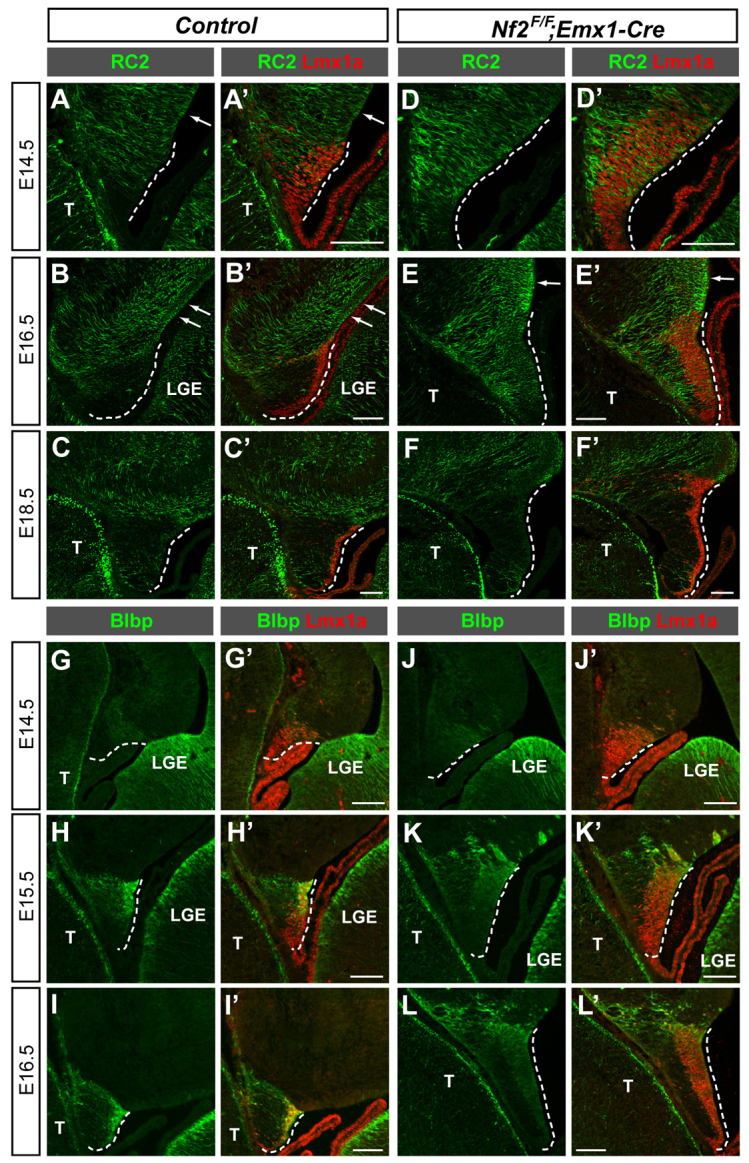
Loss of Nf2 impairs the developmental progression of cortical hem cells. (A-C′) RC2 immunoreactivity is detected at the E14.5 cortical hem (dashed line) and is diminished by E16.5 in control brains. (D-F′) RC2 immunoreactivity is notably stronger at the E14.5 Nf2F/F;Emx1-Cre hem, which persists past E16.5, and becomes diminished by E18.5. (G-I′) In control brains, weak Blbp immunoreactivity is detected at the dorsal hem at E14.5 and the immunoreactivity reaches high levels by E15.5. (J-L′) Although Blbp expression is induced normally at E14.5 Nf2F/F;Emx1-Cre hem, it remains expressed at low levels at E15.5 and E16.5, except for a few cells at the dorsal end of the hem. Arrows point to the dentate neuroepithelium. T, thalamus; LGE, lateral ganglionic eminence. Scale bars: 100 μm.
Fig. 5.

Loss of Nf2 results in fewer Cajal-Retzius cells at the hippocampal region, a smaller hippocampal fissure and malformation of the radial glial scaffold. (A-H) Immunostaining for the Cajal-Retzius (CR) cell markers p73 (arrowheads in A,B), reelin and calretinin shows fewer CR cells at the E14.5 Nf2F/F;Emx1-Cre hippocampal primordium. Tbr2+ cells at the hem are also reduced (D). (C,D) Mean number of positive cells per section ± s.d. (n=3 embryos). *P<0.05, ***P<0.001. (I-N) The hippocampal fissures (arrows) are much smaller in E17.5 Nf2F/F;Emx1-Cre brains. (O,P) Gfap immunostaining showing abnormal dentate glial scaffolds in E17.5 Nf2F/F;Emx1-Cre brains, manifested by the presence of ectopic Gfap+ fibers at the hippocampal and hem regions (yellow arrows), lack of a tightly bundled radial glial shaft (white arrows), and absence of glial fibers below the reelin+ hippocampal fissure (arrowheads). Blue signals are DAPI staining. Scale bars: 100 μm in A-H; 200 μm in I-P.
Nf2 is required for hippocampal morphogenesis
Next, we characterized hippocampus development in Nf2F/F;Emx1-Cre embryos. The hippocampal primordium develops at the caudal dorsomedial telencephalon, flanked by the cortical hem and the neocortex primordium. It gives rise to the mature hippocampus consisting of Ammon’s horn and the dentate gyrus. Pyramidal neurons in Ammon’s horn arise from the hippocampal ventricular zone (VZ) and arrive at their final location through radial migration. Dentate granule neurons originate from a narrow VZ area dorsal to the hem, the DNE, and migrate tangentially through the subpial region to form the dentate gyrus structure (Altman and Bayer, 1990; Nakahira and Yuasa, 2005).
We first examined the expression of a series of markers at E14.5, including: Axin2, which is normally expressed at the hem and ventral hippocampal primordium (Diep et al., 2004); Lef1, which is expressed at the ventral hippocampal primordium (Galceran et al., 2000); the DNE marker Prox1 (Lavado and Oliver, 2007); and the hippocampus marker Zbtb20 (Xie et al., 2010). These markers all showed normal expression patterns in Nf2 mutants (Fig. 4A-H). Furthermore, in both control and Nf2 mutant brains, Tbr2+ (Eomes - Mouse Genome Informatics) intermediate progenitors of pyramidal neurons occupied the subventricular zone (SVZ) of the hippocampal primordium, and Tbr2+ progenitors of the dentate granule neurons migrated as a stream from the SVZ towards the pia and into the dentate anlage (Fig. 4I,J, arrowheads) (Li et al., 2009). Thus, despite the enlargement of the hem, hippocampal patterning and initial morphogenesis are largely unaffected by Nf2 deletion.
Fig. 4.

Loss of Nf2 leads to defective hippocampal development. (A-H) Similar expression patterns of Axin2, Lef1, Prox1 and Zbtb20 in control and Nf2F/F;Emx1-Cre E14.5 hippocampal primordium. (I,J) In both control and Nf2F/F;Emx1-Cre E14.5 brains, Tbr2+ cells are found in the subventricular zone (SVZ) of the hippocampal primordium and in the migratory stream leaving the dentate SVZ (arrowheads). (K,L) Immunostaining for the NPC marker Sox2 shows the increased width of the hippocampal VZ of E15.5 Nf2F/F;Emx1-Cre brains. (M-T) At E17.5, the dentate gyrus (DG) labeled by Prox1 (M,N, arrowheads) and the pyramidal cell layer (CA1 and CA3) labeled by SCIP (Pou3f1), KA1 (Grik4) and Zbtb20/Brn1a, are much smaller in Nf2F/F;Emx1-Cre brains. At this stage, KA1 is normally expressed in both CA3 and DG (Q), but these two fields are morphologically not well-separated in Nf2 mutants (R, arrowhead). Blue signals in M,N,S,T are DAPI staining. (U,V) The nestin expression level is considerably stronger in hippocampal (arrowheads) and neocortical (asterisks) neuroepithelia of E17.5 Nf2F/F;Emx1-Cre mouse embryos. Scale bars: 100 μm in A-J; 200 μm in K-V.
At E15.5, however, the Sox2+ hippocampal progenitor zone was notably wider in Nf2 mutants (Fig. 4K,L). Moreover, in striking contrast to the apparently normal early patterning, the Nf2 mutant hippocampus was severely malformed by late embryogenesis. At E17.5, all the hippocampal regions that are occupied by postmitotic neurons, including the dentate gyrus region labeled by Prox1 and the Ammon’s horn pyramidal cell layer labeled by SCIP, KA1, Brn1a (Pou3f1, Grik4, Pou3f3, respectively - Mouse Genome Informatics) and Zbtb20 (KA1 and Zbtb20 are also expressed in the dentate gyrus at this stage) (Grove and Tole, 1999), were much smaller in Nf2 mutants (Fig. 4M-T). Furthermore, although control hippocampal progenitors expressed only low levels of the NPC marker nestin at this stage, nestin immunoreactivity was markedly stronger in the mutant hippocampus (Fig. 4U,V), suggesting that, similar to hem NPCs, hippocampal NPCs in Nf2 mutants maintained their early progenitor properties longer. These results indicate that Nf2 promotes the differentiation of hippocampal NPCs.
Next, we sought to obtain a better understanding of the morphogenetic defects in the Nf2 mutant hippocampus. Cajal-Retzius (CR) cells born from the hem play crucial roles during hippocampal morphogenesis by promoting the formation of the hippocampal fissure and dentate glial scaffold (Meyer et al., 2004). We therefore investigated whether the reduced cell cycle exit of Nf2 mutant hem cells affected CR cell production. Indeed, immunostaining for p73 (Trp73 - Mouse Genome Informatics), a specific marker of CR cells (Meyer et al., 2004), showed significantly fewer CR cells at the hippocampal subpial area of E14.5 Nf2 mutants (Fig. 5A-C), which was also apparent when examined with other CR cell markers reelin and calretinin (calbindin 2 - Mouse Genome Informatics) (Fig. 5E-H) (del Río et al., 1995; Ogawa et al., 1995). Tbr2+ cells at the hem region were also decreased (Fig. 5D). By E17.5, the hippocampal fissure, marked by a collection of CR cells that separates CA1 and the dentate gyrus, was much smaller in Nf2 mutants as revealed by p73, reelin and calretinin immunostaining (Fig. 5I-N, arrows). The dentate glial scaffold, which is thought to support dentate cell migration, appeared in control brains as a shaft of Gfap+ fibers extending along the hem-DNE boundary, into the subpial space, and underneath reelin+ CR cells at the hippocampal fissure (Fig. 5O). Surprisingly, in contrast to the marked reduction of glial fibers associated with CR cell loss (Meyer et al., 2004), Gfap+ fibers became overabundant but severely disorganized upon Nf2 deletion; they were no longer restricted to the hem-DNE boundary and the subpial space but were present in a more extended region encompassing much of the hippocampal primordium and the hem (Fig. 5P). These results demonstrate that Nf2 is necessary for CR cell production and proper formation of the hippocampal fissure and dentate glial scaffold.
In addition to the morphogenetic defects, the Nf2 mutant hippocampal region showed increased cell death from E14.5 to postnatal day (P) 3 (supplementary material Fig. S5), which probably also contributed to the reduction in hippocampus size.
Nf2 restricts the expansion of neocortical NPCs
During our analyses, we noticed that the neocortex of Nf2 mutants was consistently thicker than that of controls by E15.5 (supplementary material Fig. S2, C versus D and E versus F). Indeed, quantifications revealed a ∼50% increase in VZ Sox2+ Tbr2- NPCs at E15.5 (supplementary material Fig. S6). Moreover, nestin immunoreactivity was markedly stronger in E17.5 mutant neocortex (Fig. 4U,V, asterisks), suggesting that, similar to hem and hippocampal NPCs, neocortical NPCs in Nf2 mutants maintained their early progenitor properties longer. Thus, throughout the dorsal telencephalon, one major function of Nf2 is to limit NPC expansion.
Nf2 regulates Yap/Taz nuclear localization
We next investigated the molecular basis for the effects caused by Nf2 deletion. Because previous studies indicate that the Nf2 downstream effectors are highly cell type dependent, we decided to take an unbiased approach. We compared the transcript profiles of E13.5 control (Nf2F/+;Emx1-Cre) and mutant (Nf2F/F;Emx1-Cre) dorsal telencephalons by microarray analysis and found 53 annotated genes that were upregulated ≥1.5-fold in the mutant (supplementary material Table S3). In a previous experiment, we monitored the gene expression changes in E11.5 brains overexpressing human YAP (supplementary material Table S3), the key effector of the Hippo pathway, following our finding that YAP controls NPC numbers in the chick neural tube (Cao et al., 2008). In that experiment, YAP overexpression was achieved by crossing mice that carried a doxycycline (Dox)-dependent allele of human YAP1 (TetO-YAP1) with those expressing the tetracycline-dependent transactivator (rtTA) under the control of the NPC-specific nestin promoter (Nes-rtTA) (Camargo et al., 2007; Yu et al., 2005) and feeding dams with Dox-containing food from E7.5. A total of 106 annotated genes were upregulated ≥1.5-fold in TetO-YAP1;Nes-rtTA double-transgenic (dTG) brains. Interestingly, 22 of the 53 genes upregulated in Nf2 mutant dorsal telencephalons were also upregulated in E11.5 YAP-overexpression brains (supplementary material Table S3). We picked eight of the 22 genes upregulated in both experiments for quantitative RT-PCR analyses and confirmed that six genes, including the known Yap targets Ctgf and Cyr61 (Zhang et al., 2011; Zhao et al., 2008), were upregulated in both Nf2 mutant and YAP-overexpressing brains (Fig. 6A). This significant overlap between the results of the two microarray experiments, which is unlikely to occur by chance (P<10-37), suggested that Yap activity was increased in Nf2F/F;Emx1-Cre dorsal telencephalons.
Fig. 6.

Loss of Nf2 results in increased Yap/Taz transcriptional activity. (A) Quantitative RT-PCR analysis of eight genes found by microarray to be upregulated in both E13.5 Nf2F/F;Emx1-Cre dorsal telencephalon and E11.5 TetO-YAP1;Nes-rtTA brain confirms significant upregulation of six genes in both genotypes compared with their respective controls. (B,C) No noticeable difference in phospho-S112-Yap (pYap) immunoreactivity between E14.5 control and Nf2F/F;Emx1-Cre dorsal telencephalon. (D) Quantitative RT-PCR shows upregulation of Ctgf, Cyr61 and clusterin (Clu) transcripts in E13.5 Nf2F/F;Nes-Cre brain. (E) Quantitative western blot analysis shows increased Yap/Taz levels in E13.5 Nf2F/F;Nes-Cre brain, but the amount of pYap is unchanged. Two Yap antibodies were used; that from Cell Signaling Technology (CST) recognizes both Yap and Taz. (F) Subcellular fractionation followed by quantitative western blots shows increased Yap/Taz proteins in the nuclear and, to a lesser extent, the cytosol/membrane fractions of E13.5 Nf2F/F;Nes-Cre brains compared with those of control brains. Values are mean ± s.e.m. of three (A,D) or four (E,F) embryos per group. (G) Quantitative western blot analysis of NPC cultures treated with cycloheximide (CHX) for the indicated time showing increased stability of Yap/Taz proteins in Nf2F/F;Emx1-Cre NPCs. 20 μg protein was loaded per lane. The protein level at the zero time point is set at 1. Graphs show the mean ± s.e.m. of two control and three Nf2F/F;Emx1-Cre NPC lines. (H) The steady-state levels of pYap do not differ between control and Nf2F/F;Emx1-Cre NPCs, and those of Yap/Taz show a trend of increase. Values are mean±s.d. of two control and three Nf2F/F;Emx1-Cre NPC lines. *P<0.05, **P<0.01, ***P<0.001.
We next sought to understand how Nf2 loss led to increased Yap activity. The best-studied signaling pathway that controls the activity of Yap and its paralog Taz is the Hippo pathway, which phosphorylates Yap/Taz and causes their sequestration in the cytoplasm (reviewed by Halder and Johnson, 2011; Yu and Guan, 2013). Mutations in the Hippo pathway reduce Yap/Taz phosphorylation, allowing unphosphorylated Yap/Taz to enter the nucleus to activate transcription. We therefore examined whether Nf2 loss reduced Yap phosphorylation. Immunostaining with an antibody against phospho-S112-Yap (pYap) did not reveal a noticeable reduction in Yap phosphorylation levels in E14.5 Nf2F/F;Emx1-Cre dorsal telencephalon (Fig. 6B,C).
To better quantify Yap phosphorylation levels, we turned to biochemical methods. Because only limited amounts of tissue could be obtained from mid-gestation stage dorsal telencephalons, we generated another Nf2 cKO line using a Nestin-Cre (Nes-Cre) line that expresses Cre in NPCs throughout the CNS starting from ∼E11 (Tronche et al., 1999). The dorsal telencephalon of Nf2F/F;Nes-Cre mice exhibited phenotypes similar to those of Nf2F/F;Emx1-Cre mice (supplementary material Fig. S7A,B). Moreover, quantitative RT-PCR analysis showed similar upregulation of Yap target genes in E13.5 Nf2F/F;Nes-Cre whole brains (Fig. 6D). We therefore used whole-brain tissues from E13.5 Nf2F/F;Nes-Cre and control embryos for quantitative western blots. Again, we did not detect a significant change in Yap phosphorylation levels. However, we did find a significant increase in the amount of Yap/Taz proteins (Fig. 6E). Furthermore, subcellular fractionation experiments showed that the amount of nuclear Yap/Taz proteins increased more than 2-fold in E13.5 Nf2F/F;Nes-Cre brains, together with a small increase in these proteins in the cytosol/membrane fraction (Fig. 6F). The abundances of Yap/Taz transcripts in Nf2F/F;Emx1-Cre dorsal telencephalons and Nf2F/F;Nes-Cre brains were similar to those of controls (supplementary material Fig. S7C). Together, our results demonstrate that Nf2 inhibits Yap/Taz activity during brain development by controlling their subcellular localization and protein levels, probably through a mechanism that is distinct from the canonical Hippo pathway.
To gain a deeper understanding of how Nf2 regulates Yap/Taz, we generated NPC lines from E13.5 control and Nf2 cKO cortices and examined Yap/Taz protein levels after blocking protein synthesis with cycloheximide. Both proteins were significantly more stable in Nf2-/- NPCs than in control NPCs (Fig. 6G). The steady-state levels of Yap/Taz proteins in Nf2-/- NPCs showed a trend of increase compared with those in control NPCs, whereas pYap levels did not differ significantly (Fig. 6H). These results suggest that, at least in vitro, Yap/Taz proteins become more stable upon Nf2 loss.
YAP gain-of-function phenocopies Nf2 loss-of-function
If Nf2 inhibits Yap/Taz, then increasing Yap/Taz activity should phenocopy the hippocampal abnormalities caused by Nf2 loss. To test this, we used the aforementioned TetO-YAP1;Nes-rtTA system that enables overexpression of a mutant version of human YAP1 (S127A) that has enhanced nuclear localization compared with the wild-type protein (Camargo et al., 2007). Subcellular fractionation experiments showed that when dams were given Dox-containing drinking water (100 mg/l) starting from E9.5, at E13.5 the amount of nuclear YAP in dTG brains was ∼8-fold that in control brains (Fig. 7A). As predicted, dTG embryos developed phenotypes very similar to those of Nf2F/F;Emx1-Cre embryos: the number of CR cells decreased significantly at E14.5, the hem became markedly enlarged by E15.5, and by E17.5 the hippocampal fissure, dentate gyrus and pyramidal cell layer were all much smaller (Fig. 7B-M). Furthermore, nestin immunoreactivity was stronger in dTG brains and the Gfap+ fibers were more abundant and disorganized, almost indistinguishable from those in Nf2F/F;Emx1-Cre brains (Fig. 7N-Q). These results demonstrate that hyperactivating Yap and inactivating Nf2 have similar effects on hippocampal development.
Fig. 7.

YAP overexpression causes similar hippocampal defects to Nf2 loss. (A) Quantitative western blots showing increased YAP in the cytosol/membrane and nuclear fractions of E13.5 TetO-YAP1;Nes-rtTA double-transgenic (dTG) brains. (B-D) p73+ CR cells (arrowheads in B,C) are reduced at the hippocampal primordium of E14.5 dTG mouse embryos. (E-G) Lmx1a immunostaining shows enlargement of the hem (dashed lines) in E15.5 dTG embryos. (H-M) At E17.5, the size of the hippocampal fissure (HF, arrows), the dentate gyrus (DG, arrowheads), and the pyramidal cell layer (CA) are all reduced in dTG brains. CH, cortical hem. (N,O) The Gfap+ radial glial scaffold is defective in E17.5 dTG embryos, as shown by the presence of ectopic Gfap+ fibers at the hippocampal and hem regions (yellow arrows), lack of a tightly bundled shaft (white arrows) and absence of Gfap+ fibers below the reelin+ HF (arrowheads). (P,Q) Nestin immunoreactivity is stronger at the E17.5 dTG hippocampus (arrowheads). Blue signals are DAPI staining. Values in A are the mean ± s.e.m. of five control and four dTG embryos; values in D,G are the mean number of positive cells per section ± s.d. (n=3 embryos). *P<0.05, ***P<0.001. Scale bars: 100 μm in B,C; 200 μm in H-Q.
Loss of Yap suppresses Nf2 mutant hippocampal phenotypes
To further confirm that Nf2 regulates hippocampal development by inhibiting Yap/Taz, we generated Nf2 and Yap double mutants (Nf2F/F;YapF/F;Emx1-Cre) (Xin et al., 2011). Although Yap deletion alone had no noticeable effect on hem or hippocampal development, it significantly suppressed the overgrowth of Nf2 mutant hem at E15.5 (Fig. 8A-C). Importantly, loss of Yap strongly suppressed the hippocampal defects of Nf2 mutants: at P0, in six out of ten Nf2F/F;YapF/F;Emx1-Cre hippocampi analyzed, the dentate gyrus, Ammon’s horn, hippocampal fissure, radial glial scaffold and the amount of cell death, all appeared identical to those in control (Nf2F/+;Emx1-Cre) brains (Fig. 8D-W; supplementary material Fig. S8), and four of the ten hippocampi analyzed showed partial rescue (supplementary material Fig. S9). These results demonstrate that Nf2 functions in NPCs by inhibiting Yap/Taz and that Yap is a major downstream effector of Nf2 during hippocampal development.
Fig. 8.

Loss of Yap suppresses the hippocampal defects associated with Nf2 deletion. (A-C) Lmx1a immunostaining shows that deleting both alleles of Yap significantly suppresses hem enlargement in E15.5 Nf2 mutants. Values are mean ± s.e.m. of a series of Nf2 and Yap single and double mutants, the genotypes of which are shown below the x-axis. **P<0.01, ***P<0.001; n.s., not significant (P>0.05). CP, choroid plexus. (D-R) Prox1, Zbtb20/Brn1a and p73 immunostaining showing the dentate gyrus (DG; D-H, arrowheads), the pyramidal cell layer (CA, I-M) and the hippocampal fissure (HF; N-R, arrows), respectively, of P0 mice of the indicated genotypes. Note the much smaller DG, CA and HF in Nf2F/F;Emx1-Cre mice (E,J,O versus D,I,N) and the normal sized DG, CA and HF in Nf2F/F;YapF/F;Emx1-Cre mice (H,M,R). Deleting one allele of Yap does not suppress the hippocampal phenotype of Nf2 mutants (G,L,Q). The hippocampus of Nf2F/+;YapF/F;Emx1-Cre mice appears normal (F,K,P). (S-W) Gfap and reelin immunostaining showing the dentate glial scaffold and HF, respectively. Note the defects associated with the dentate glial scaffold of Nf2F/F;Emx1-Cre mice (T versus S), including the excessive Gfap+ fibers at the hem (yellow arrows), the absence of the subpial glial shaft (white arrows) and the absence of glial fibers below the reelin+ HF (arrowheads). These defects are suppressed by deleting Yap (W). Blue signals are DAPI staining. Scale bars: 100 μm in A,B; 200 μm in D-W.
DISCUSSION
Since its discovery as the causal mutation for neurofibromatosis type 2, NF2 has been recognized as having tumor-suppressor activities in many tumors, including ependymomas that arise from radial glial cells, a type of NPC (Baser et al., 2003; Taylor et al., 2005). Nf2 function in NPCs is, however, poorly understood. Although extensive studies have implicated a number of signaling pathways as Nf2 downstream effectors, whether these effector pathways mediate Nf2 function in vivo has not been well established. In this study, we reveal that a primary function of Nf2 during brain development is to limit the expansion of the neural progenitor population. We show that, upon Nf2 loss, NPCs at the cortical hem, hippocampus and neocortex retain their early progenitor properties longer and overexpand, resulting in severe malformation of the hippocampus in the adult brain. We further provide multiple lines of evidence indicating that a major molecular function of Nf2 in brain NPCs is to inhibit Yap/Taz. First, brain tissues lacking Nf2 have increased Yap/Taz protein levels, enhanced nuclear localization of these proteins, and upregulation of their target genes. Second, transgenic mice with elevated YAP in their NPCs closely phenocopy Nf2 mutants. Last and foremost, although loss of Yap alone does not impair hippocampal development, it strongly suppresses the hippocampal defects of Nf2 mutants. Our study not only provides novel insights into Nf2 function during mammalian brain development, but might also shed light on the pathogenesis of ependymomas. Previous studies suggest that Nf2 loss promotes tumorigenesis by destabilizing cell-cell junctions (Lallemand et al., 2003). Our study and those of others (Mizuno et al., 2012; Striedinger et al., 2008) raise the possibility that upregulation of YAP/TAZ activity might be a significant contributor to tumor development.
Nf2 is required for cell-cell junction formation in several cell types (Gladden et al., 2010; Lallemand et al., 2003). In fact, a previous study showed that Nf2 is required for the formation of NPC apical junctions during mouse brain development (McLaughlin et al., 2007). Our Nf2 mutants (Nf2F/F;Emx1-Cre and Nf2F/F;Nes-Cre), however, do not have detectable NPC junctional defects, even though we used the same Nf2 floxed allele as the previous study. The difference could be due to the timing of Nf2 deletion: the Cre line used by McLaughlin et al. is active as early as E8.5, whereas the lines in our study are turned on after E9.5. McLaughlin et al. proposed that Nf2 is required specifically for the assembly, but not the maintenance, of the junctional complex (McLaughlin et al., 2007); thus, Nf2 loss after the junctions have been established would have no effect. In agreement with this proposal, a study using epidermal keratinocytes showed that Nf2 is required for junctional maturation by directly interacting with the junctional component α-catenin and the polarity protein Par3 and thereby promoting the polarization of junctional components, which is probably why Nf2 is no longer required after the junctions have been established (Gladden et al., 2010). Although this might be a parsimonious explanation, it should be noted that it is unclear when NPC apical junctions are fully established during neural tube development, how apical junctions are distributed when NPCs divide, and whether new junctions form in the expanding neuroepithelium after neural tube closure. Answers to these questions will help us to better understand the role of Nf2 in the formation of NPC junctions.
The absence of NPC junctional defects allowed us to specifically examine the role of Nf2 in progenitor self-renewal and differentiation. We showed that Nf2 promotes the developmental progression and limits the expansion of NPCs. It might seem counterintuitive that hippocampus size and neocortex thickness were reduced in adult Nf2 mutants even though NPC numbers increased during embryonic development. It is possible that certain homeostasis mechanisms eliminated the cells that failed to undergo differentiation at the correct time. We indeed observed increased cell death in Nf2 mutant brains at embryonic and early postnatal stages. In addition, the morphogenetic defects associated with the hippocampal development of Nf2 mutants are likely to play a major role in hippocampus size reduction. First, dentate gyrus growth is largely achieved by local proliferation of progenitor cells after they reach the dentate primordium (Altman and Bayer, 1990). Failure of Nf2 mutants to establish the proper dentate primordium, owing to the reduction in hippocampal fissure size, disorganization of the dentate glial scaffold and consequently impaired migration of dentate precursors, is likely to have prevented further growth of the dentate gyrus. Moreover, the migration of dentate precursors and simultaneous tissue folding probably provide more space for the expansion of Ammon’s horn. Impairment of these processes in Nf2 mutants thus limited the elongation of Ammon’s horn, which was much shorter in mutants than in control mice. It is important to note that, although the reduction in CR cell number could explain why the hippocampal fissure was smaller in Nf2 mutants, it is unlikely to have caused the radial glial fibers to become overabundant and disorganized because loss of CR cells actually results in fewer radial glial fibers (Meyer et al., 2004).
We provided molecular and genetic evidence that Yap, probably with Taz playing a partially redundant role, is the main downstream effector of Nf2 in brain NPCs. However, we do not fully understand how Nf2 inhibits Yap/Taz. These proteins are the key effectors of the Hippo pathway, which is a conserved pathway regulating organ growth and tumorigenesis (Halder and Johnson, 2011; Yu and Guan, 2013). The Hippo pathway inhibits Yap/Taz by phosphorylating them on multiple serines, resulting in their sequestration in the cytoplasm and degradation. Interestingly, although Nf2 loss in NPCs led to increased Yap/Taz protein levels and nuclear localization in vivo and increased protein stability in vitro, Yap phosphorylation levels did not seem to be affected. It is possible that the change was too small to be reliably detected by our methods or that phosphorylation sites other than that recognized by the only available antibody were affected. Alternatively, Nf2 might regulate Yap/Taz through a phosphorylation-independent mechanism. Recent work has shown that angiomotin proteins can inhibit Yap/Taz through a direct angiomotin-Yap/Taz interaction that is independent of Yap/Taz phosphorylation status (Chan et al., 2011; Zhao et al., 2011), and that the tension of the actomyosin cytoskeleton can regulate Yap nuclear translocation without affecting its phosphorylation (Dupont et al., 2011). Nf2 has been shown to associate with the actin network and to affect its organization (reviewed by Curto and McClatchey, 2008); thus, Nf2 might regulate Yap/Taz nuclear localization via the actin cytoskeleton. A previous study found that Nf2 loss in glial cells results in increased proliferation (Houshmandi et al., 2009), a finding consistent with ours. Interestingly, upregulation of Src activity was found to be responsible, whereas we did not detect any change in the amount of total or active Src (phospho-Y416-Src) in E13.5 Nf2 cKO brain lysates (data not shown). These results suggest that Nf2 could function through distinct pathways in different cell types, even when the end effect is the same.
Although Yap/Taz can promote the expansion of several types of stem cells (reviewed by Barry and Camargo, 2013), the transcriptional program mediating this effect is unclear. In our profiling experiments aimed at detecting early changes associated with Nf2 loss or YAP overexpression, we did not find significant changes in the mRNA levels of cell cycle regulators or factors well known to be involved in NPC self-renewal and differentiation. However, we did observe upregulation of several genes involved in cell-cell junctions (Cldn12, Pkp2) and extracellular matrix (ECM) interactions (Thbs1), and of genes encoding ECM components (Sparc) and transmembrane proteins that may be involved in ECM interactions (Steap1, Podxl) (see supplementary material Table S3). Because cell adhesions and ECM interactions play essential roles in stem cell self-renewal (Fietz et al., 2012; Marthiens et al., 2010), it is plausible that a direct role of Yap/Taz in NPC maintenance is to regulate genes involved in cell-cell and cell-matrix interactions. Understanding not only how Yap/Taz are regulated but also the transcriptional targets and cellular processes that they regulate will provide significant insight into the mechanisms that control organ development, tissue homeostasis and tumorigenesis.
Supplementary Material
Acknowledgments
We thank Fernando Camargo and Thijn Brummelkamp for providing the TetO-YAP1 line; Steven Kernie for the Nes-rtTA line; INSERM (France) for the Nf2F line; Andrew McMahon, Elizabeth Grove, Juan Pedro Martinez-Barbera and Gabriele Kardon for in situ probes; Randy Johnson (MD Anderson Cancer Center, TX) for discussions; Sharon Frase, Vickie Frohlich and the staff at the St Jude Imaging Center for assistance; Jim Morgan, David Solecki and Young-Goo Han for helpful discussions and comments on the manuscript; and Cherise Guess for editorial assistance.
Footnotes
Funding
This work was supported by American Lebanese Syrian Associated Charities, Basil O’Connor Starter Scholar Research Award [5-FY10-488] from the March of Dimes Foundation; by the Whitehall Foundation [2012-05-106 to X.C.]; and by the National Institutes of Health [P30CA021765 to G.N.; HL-077439, HL-111665, HL093039 and U01-HL-100401 to E.N.O.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
X.C. conceived the project. X.C. and A.L. designed the experiments. X.C., A.L., Y.H. and J.P. performed the experiments and analyzed the results. G.N. analyzed the microarray data. E.N.O. and M.G. generated the YapF and Nf2F mouse lines, respectively. X.C. and A.L. wrote the manuscript.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.096537/-/DC1
References
- Akhmametyeva E. M., Mihaylova M. M., Luo H., Kharzai S., Welling D. B., Chang L. S. (2006). Regulation of the neurofibromatosis 2 gene promoter expression during embryonic development. Dev. Dyn. 235, 2771–2785 [DOI] [PubMed] [Google Scholar]
- Altman J., Bayer S. A. (1990). Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J. Comp. Neurol. 301, 365–381 [DOI] [PubMed] [Google Scholar]
- Barry E. R., Camargo F. D. (2013). The Hippo superhighway: signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr. Opin. Cell Biol. 25, 247–253 [DOI] [PubMed] [Google Scholar]
- Baser M. E., R Evans D. G., Gutmann D. H. (2003). Neurofibromatosis 2. Curr. Opin. Neurol. 16, 27–33 [DOI] [PubMed] [Google Scholar]
- Benhamouche S., Curto M., Saotome I., Gladden A. B., Liu C. H., Giovannini M., McClatchey A. I. (2010). Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 24, 1718–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 [Google Scholar]
- Breuhahn K., Schirmacher P. (2010). A cellular view of Nf2 in liver homeostasis and tumorigenesis. Dev. Cell 19, 363–364 [DOI] [PubMed] [Google Scholar]
- Camargo F. D., Gokhale S., Johnnidis J. B., Fu D., Bell G. W., Jaenisch R., Brummelkamp T. R. (2007). YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054–2060 [DOI] [PubMed] [Google Scholar]
- Cao X., Pfaff S. L., Gage F. H. (2008). YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 22, 3320–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caronia G., Wilcoxon J., Feldman P., Grove E. A. (2010). Bone morphogenetic protein signaling in the developing telencephalon controls formation of the hippocampal dentate gyrus and modifies fear-related behavior. J. Neurosci. 30, 6291–6301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. W., Lim C. J., Chong Y. F., Pobbati A. V., Huang C., Hong W. (2011). Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J. Biol. Chem. 286, 7018–7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curto M., McClatchey A. I. (2008). Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. Br. J. Cancer 98, 256–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Río J. A., Martínez A., Fonseca M., Auladell C., Soriano E. (1995). Glutamate-like immunoreactivity and fate of Cajal-Retzius cells in the murine cortex as identified with calretinin antibody. Cereb. Cortex 5, 13–21 [DOI] [PubMed] [Google Scholar]
- Diep D. B., Hoen N., Backman M., Machon O., Krauss S. (2004). Characterisation of the Wnt antagonists and their response to conditionally activated Wnt signalling in the developing mouse forebrain. Brain Res. Dev. Brain Res. 153, 261–270 [DOI] [PubMed] [Google Scholar]
- Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 [DOI] [PubMed] [Google Scholar]
- Fietz S. A., Lachmann R., Brandl H., Kircher M., Samusik N., Schröder R., Lakshmanaperumal N., Henry I., Vogt J., Riehn A., et al. (2012). Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc. Natl. Acad. Sci. USA 109, 11836–11841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galceran J., Miyashita-Lin E. M., Devaney E., Rubenstein J. L., Grosschedl R. (2000). Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 127, 469–482 [DOI] [PubMed] [Google Scholar]
- Giovannini M., Robanus-Maandag E., van der Valk M., Niwa-Kawakita M., Abramowski V., Goutebroze L., Woodruff J. M., Berns A., Thomas G. (2000). Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 14, 1617–1630 [PMC free article] [PubMed] [Google Scholar]
- Gladden A. B., Hebert A. M., Schneeberger E. E., McClatchey A. I. (2010). The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev. Cell 19, 727–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski J. A., Talley T., Qiu M., Puelles L., Rubenstein J. L., Jones K. R. (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 22, 6309–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz M., Huttner W. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788 [DOI] [PubMed] [Google Scholar]
- Grove E. A., Tole S. (1999). Patterning events and specification signals in the developing hippocampus. Cereb. Cortex 9, 551–561 [DOI] [PubMed] [Google Scholar]
- Grove E. A., Tole S., Limon J., Yip L., Ragsdale C. W. (1998). The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 125, 2315–2325 [DOI] [PubMed] [Google Scholar]
- Halder G., Johnson R. L. (2011). Hippo signaling: growth control and beyond. Development 138, 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfuss E., Galli R., Heins N., Götz M. (2001). Characterization of CNS precursor subtypes and radial glia. Dev. Biol. 229, 15–30 [DOI] [PubMed] [Google Scholar]
- Houshmandi S. S., Emnett R. J., Giovannini M., Gutmann D. H. (2009). The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2- and Src-dependent manner. Mol. Cell. Biol. 29, 1472–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N., Thatte J., Braciale T., Ley K., O’Connell M., Lee J. K. (2003). Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics 19, 1945–1951 [DOI] [PubMed] [Google Scholar]
- Kriegstein A., Alvarez-Buylla A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A., Zimmer A., Schnütgen F., Brüning G., Spener F., Müller T. (1994). The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development 120, 2637–2649 [DOI] [PubMed] [Google Scholar]
- Lallemand D., Curto M., Saotome I., Giovannini M., McClatchey A. I. (2003). NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 17, 1090–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A., Oliver G. (2007). Prox1 expression patterns in the developing and adult murine brain. Dev. Dyn. 236, 518–524 [DOI] [PubMed] [Google Scholar]
- Li G., Kataoka H., Coughlin S. R., Pleasure S. J. (2009). Identification of a transient subpial neurogenic zone in the developing dentate gyrus and its regulation by Cxcl12 and reelin signaling. Development 136, 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Cooper J., Karajannis M. A., Giancotti F. G. (2012). Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 13, 204–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangale V. S., Hirokawa K. E., Satyaki P. R., Gokulchandran N., Chikbire S., Subramanian L., Shetty A. S., Martynoga B., Paul J., Mai M. V., et al. (2008). Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science 319, 304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthiens V., Kazanis I., Moss L., Long K., Ffrench-Constant C. (2010). Adhesion molecules in the stem cell niche - more than just staying in shape? J. Cell Sci. 123, 1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey A. I., Saotome I., Ramesh V., Gusella J. F., Jacks T. (1997). The Nf2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes Dev. 11, 1253–1265 [DOI] [PubMed] [Google Scholar]
- McLaughlin M. E., Kruger G. M., Slocum K. L., Crowley D., Michaud N. A., Huang J., Magendantz M., Jacks T. (2007). The Nf2 tumor suppressor regulates cell-cell adhesion during tissue fusion. Proc. Natl. Acad. Sci. USA 104, 3261–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G., Cabrera Socorro A., Perez Garcia C. G., Martinez Millan L., Walker N., Caput D. (2004). Developmental roles of p73 in Cajal-Retzius cells and cortical patterning. J. Neurosci. 24, 9878–9887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misson J. P., Edwards M. A., Yamamoto M., Caviness V. S., Jr (1988). Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res. Dev. Brain Res. 44, 95–108 [DOI] [PubMed] [Google Scholar]
- Mizuno T., Murakami H., Fujii M., Ishiguro F., Tanaka I., Kondo Y., Akatsuka S., Toyokuni S., Yokoi K., Osada H., et al. (2012). YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 31, 5117–5122 [DOI] [PubMed] [Google Scholar]
- Muzio L., Mallamaci A. (2005). Foxg1 confines Cajal-Retzius neuronogenesis and hippocampal morphogenesis to the dorsomedial pallium. J. Neurosci. 25, 4435–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira E., Yuasa S. (2005). Neuronal generation, migration, and differentiation in the mouse hippocampal primoridium as revealed by enhanced green fluorescent protein gene transfer by means of in utero electroporation. J. Comp. Neurol. 483, 329–340 [DOI] [PubMed] [Google Scholar]
- Ogawa M., Miyata T., Nakajima K., Yagyu K., Seike M., Ikenaka K., Yamamoto H., Mikoshiba K. (1995). The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 14, 899–912 [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N., Gerfin-Moser A. (1993). A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100, 431–440 [DOI] [PubMed] [Google Scholar]
- Striedinger K., VandenBerg S. R., Baia G. S., McDermott M. W., Gutmann D. H., Lal A. (2008). The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia 10, 1204–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian L., Remedios R., Shetty A., Tole S. (2009). Signals from the edges: the cortical hem and antihem in telencephalic development. Semin. Cell Dev. Biol. 20, 712–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. D., Poppleton H., Fuller C., Su X., Liu Y., Jensen P., Magdaleno S., Dalton J., Calabrese C., Board J., et al. (2005). Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8, 323–335 [DOI] [PubMed] [Google Scholar]
- Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P. C., Bock R., Klein R., Schütz G. (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23, 99–103 [DOI] [PubMed] [Google Scholar]
- Xie Z., Ma X., Ji W., Zhou G., Lu Y., Xiang Z., Wang Y. X., Zhang L., Hu Y., Ding Y. Q., et al. (2010). Zbtb20 is essential for the specification of CA1 field identity in the developing hippocampus. Proc. Natl. Acad. Sci. USA 107, 6510–6515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M., Kim Y., Sutherland L. B., Qi X., McAnally J., Schwartz R. J., Richardson J. A., Bassel-Duby R., Olson E. N. (2011). Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 4, ra70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F. X., Guan K. L. (2013). The Hippo pathway: regulators and regulations. Genes Dev. 27, 355–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T. S., Dandekar M., Monteggia L. M., Parada L. F., Kernie S. G. (2005). Temporally regulated expression of Cre recombinase in neural stem cells. Genesis 41, 147–153 [DOI] [PubMed] [Google Scholar]
- Zhang N., Bai H., David K. K., Dong J., Zheng Y., Cai J., Giovannini M., Liu P., Anders R. A., Pan D. (2010). The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev. Cell 19, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Pasolli H. A., Fuchs E. (2011). Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 108, 2270–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J. D., Wang C. Y., Chinnaiyan A. M., et al. (2008). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Li L., Lu Q., Wang L. H., Liu C. Y., Lei Q., Guan K. L. (2011). Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 25, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.