

Abstract
The traditional view of p53 activation includes three steps—p53 stabilization, DNA binding, and transcriptional activation. However, recent studies indicate that each step of p53 activation is more complex than originally anticipated. Moreover, both genetic studies in mice and in vitro studies with purified components suggest that the classical model may not be sufficient to explain all aspects of p53 activation in vivo. To reconcile these differences, we propose that antirepression, the release of p53 from repression by factors such as Mdm2 and MdmX, is a key step in the physiological activation of p53.
Introduction
The year 2009 celebrates the 30th anniversary of the discovery of p53 (Lane and Crawford, 1979; Linzer and Levine, 1979; DeLeo et al., 1979) and the 20th anniversary of its characterization as a tumor suppressor (Baker et al., 1989; Finlay et al., 1989). Mutations that perturb p53 function, often in its DNA-binding domain, or disruptions to p53’s upstream or downstream regulatory network, have been found in more than half of all cancer cases and are present in cancer-prone families with Li-Fraumeni syndrome (Hainaut and Hollstein, 2000; Vogelstein et al., 2000).
p53 has been called a “cellular gatekeeper” (Levine, 1997) or “the guardian of the genome” (Lane, 1992) because of its central role in coordinating the cellular responses to a broad range of cellular stress factors. p53 functions as a node for organizing whether the cell responds to various types and levels of stress with apoptosis, cell cycle arrest, senescence, DNA repair, cell metabolism, or autophagy. p53-controlled transactivation of target genes is an essential feature of each stress response pathway, although some effects of p53 may be independent of transcription (Vogelstein et al., 2000; Vousden and Lane, 2007; Marchenko and Moll, 2007). As a transcription factor that both activates and represses a broad range of target genes, p53 demands an exquisitely complicated network to control and fine-tune responses to the various stress signals encountered by cells (Brooks and Gu, 2003; Laptenko and Prives, 2006). p53 is regulated by an array of posttranslational modifications both during normal homeostasis and in stress-induced responses. More than 36 different amino acids within p53 have been shown to be modified in various biochemical and cell culture studies (Figure 1) (Kruse and Gu, 2008b).
Figure 1. Overview of p53 Posttranslational Modifications.

More than 36 amino acids of p53 are reported to be modified. The major sites of p53 phosphorylation (P), ubiquitination (Ub), and acetylation (Ac) are shown with the corresponding major modifying enzymes and signals. Furthermore, additional phosphorylation and acetylation sites, as well as major sites of methylation (Me), sumoylation (S), neddylation (N8), glycosylation (O-Glc), and ribosylation (ADP), are indicated.
Classical models for the activation of p53 focus on three simple and rate-limiting steps: p53 stabilization induced by ATM/ATR-mediated phosphorylation, sequence-specific DNA binding, and target gene activation by interacting with the general transcriptional machinery (Figure 2). However, as discussed in this review, the diversity and number of modifications, as well as recent studies from knockin mutant mice, suggest a certain degree of redundancy among these modifications. These findings challenge the importance of a number of the traditional regulatory events of p53 activation (Iwakuma and Lozano, 2007; Marine et al., 2006; Wahl, 2006; Brooks and Gu, 2006) and raise questions that cannot be sufficiently explained by the three steps of the classical model. What is the exact mechanism controlling p53 stabilization in vivo? Does p53 require additional activation to induce binding to DNA or is p53 already bound to DNA yet unable to induce transactivation in vivo? Does p53 require activation, or is it intrinsically active in vivo? How are decisions of cell fate controlled in vivo in response to different types and severities of stress? To incorporate recent findings, we revisit the different stages of p53 activation here. We propose that antirepression is a key mechanism for p53 regulation in vivo. Introducing this step for p53 activation into existing models may help to explain the events leading to p53 activation in vivo.
Figure 2. Classical Model of p53 Activation.
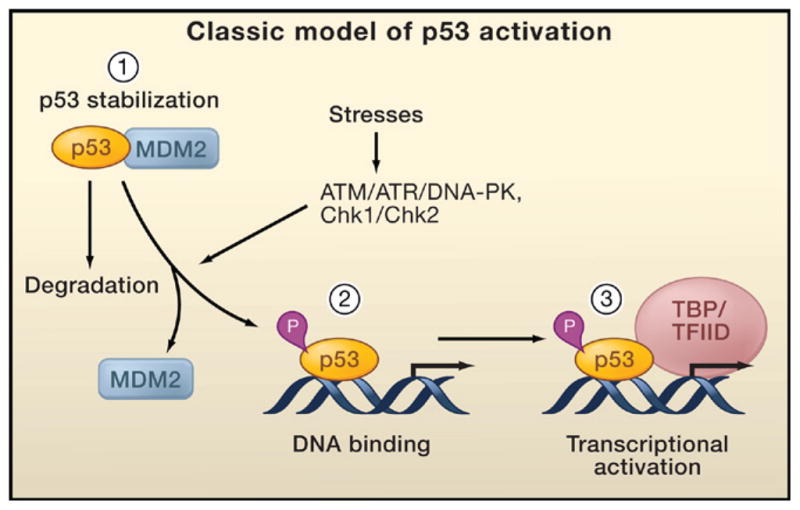
The classical model for p53 activation generally consists of three sequential activating steps: (1) stress-induced stabilization mediated by phosphorylation (P), (2) DNA binding, and (3) recruitment of the general transcriptional machinery. During normal homeostasis, p53 is degraded after Mdm2-mediated ubiquitination (left), while stress signal-induced p53 phosphorylation by ATM, ATR, and other kinases stabilizes p53 and promotes DNA binding. DNA-bound p53 then recruits the transcriptional machinery to activate transcription of p53 target genes.
Mechanisms of p53 Stabilization
The Classical Model
Phosphorylation of p53 is classically regarded as the first crucial step of p53 stabilization. p53 can be modified by phosphorylation by a broad range of kinases, including ATM/ATR/DNA-PK, and Chk1/Chk2 (see Figure 1). Phosphorylation of serine residues within the N-terminal p53 transactivation domain was among the first posttranslational modifications of p53 identified and has been extensively investigated in in vitro biochemical assays, in tissue culture studies, and recently by using site-specific knockin animals. N-terminal phosphorylation at Ser15 (mouse Ser18) and Ser20 (mouse Ser23) have been generally thought to stabilize p53 by inhibiting the interaction between p53 and Mdm2 (Figure 2). Ser15 and Ser20 are phosphorylated after DNA damage and other types of stress by ATM, ATR, DNA-PK, Chk1, and Chk2 (Appella and Anderson, 2001; Shieh et al., 1997; Shieh et al., 2000). Although it remains unresolved which of the different kinases perform the phosphorylation of these N-terminal regulatory sites in response to the varying stress signals, and to which extent N-terminal phosphorylation affects the p53-MDM2 interaction, the general consensus remains that their phosphorylation occurs rapidly in response to various stress stimuli to activate p53.
However, knockin mouse models with individual mutations of these N-terminal sites question the importance of the individual modifications in p53 stabilization in vivo. Studies with a Ser18Ala point mutant knockin mouse (S18A) indicate that at least some of the in vitro effects may not translate directly to a whole organism (Chao et al., 2003; Sluss et al., 2004). Although some mild effects on p53 levels in the knockin cells are observed, stress-induced p53 stabilization is comparable in thymocytes, splenocytes, and fibroblasts derived from wild-type and mutant knockin mice. The consistency in p53 levels in these studies argues against a dominant role for S18 phosphorylation in the regulation of the p53-MDM2 interaction in vivo, as a failure to phosphorylate S18 should promote this interaction and therefore induce p53 ubiquitination and degradation. Similar results are obtained with p53-serine 23 to alanine (S23A) point mutant knockin animals (MacPherson et al., 2004; Wu et al., 2002). Interestingly, more severe defects on both the levels and function of p53 were found in the S18/23A double-mutant mice, lending support to the physiological importance of these two key phosphorylation sites (Chao et al., 2006a). Nevertheless, these defects are limited to certain tissues and mostly for DNA-damage-induced apoptosis. For example, no obvious effects were observed in mouse embryonic fibroblasts or other tissues from the S18/23A double-mutant mice. These results suggest that under specific conditions, p53 phosphorylation is required for p53 stabilization. However, the fact that no differences in stability were observed in other tissues or in embryonic fibroblasts argues that phosphorylation may not be a universal requirement for p53 stabilization. Indeed, results from a wide range of studies have established that p53 can be activated regardless of its phosphorylation status (Ashcroft et al., 1999; 2000; Blattner et al., 1999; Wu et al., 2002). Together, these genetic and biochemical studies emphasize the notion that p53 stabilization in vivo requires a far more sophisticated regulatory network than can be provided by p53 phosphorylation alone.
The Many Layers of Mdm2 Regulation
The tight control of cellular p53 levels is primarily achieved through its ubiquitin-mediated proteasomal degradation (Brooks and Gu, 2006; Michael and Oren, 2003). Three independent studies identified the mouse double minute protein 2 (Mdm2) as the principal endogenous E3-ligase with high specificity for p53 (Haupt et al., 1997; Honda et al., 1997; Kubbutat et al., 1997). Interestingly, although the level of p53 is elevated in the absence of Mdm2, p53 is still degraded in the cells of Mdm2 null mice (Ringshausen et al., 2006), suggesting the existence of alternative, Mdm2-independent pathways for p53 degradation in vivo. Indeed, the recently discovered E3-ligases COP1 (Dornan et al., 2004), Pirh2 (Leng et al., 2003), Arf-BP1 (Chen et al., 2005), and others have clearly been shown to contribute to the efficient control of p53 levels in tissue culture and in in vitro biochemical experiments. Nevertheless, the precise roles of Mdm2-independent degradation in stress-induced p53 stabilization remain to be elucidated.
There are multiple layers of regulation that connect Mdm2 function with p53 stability during stress responses. A prominent physiological regulator of Mdm2 is the tumor suppressor ARF (Lowe and Sherr 2003). ARF interferes with the Mdm2-p53 interaction, thereby acting to stabilize and activate p53. The low steady-state levels of ARF in normal cells are dramatically induced upon oncogenic stress, which suppresses abnormal cell proliferation by triggering p53-dependent growth arrest or apoptosis (Sherr, 2006). Interestingly, the mechanisms by which ARF activates p53 function are complex. On one hand, ARF is predominately a nucleolar protein. Elegant work has shown that ARF can stabilize nucleoplasmic p53 by binding Mdm2 and sequestering it in the nucleolus (Weber et al., 1999). Yet, equally convincing studies have also shown that nucleoplasmic forms of ARF activate p53 by directly inhibiting the ubiquitin ligase activity of Mdm2 (Llanos et al., 2001; Honda and Yasuda., 1999). Moreover, in addition to suppressing Mdm2-mediated effects on p53, ARF may modulate the activity of other E3 ligases such as ARF-BP1, and our recent studies demonstrate that the ARF/ARF-BP1 interaction is involved in both p53-dependent and p53-independent functions of ARF (Chen et al., 2005). Thus, although the precise molecular mechanisms by which ARF mediates these activities need further elucidation, it is clear that ARF has an essential role in p53 regulation.
The other prominent regulator of Mdm2 activity is MdmX (also known as Mdm4). Like Mdm2, MdmX is a critical negative regulator of p53 (Marine and Jochemsen, 2005). The direct roles played by MdmX on p53 activity are discussed in more detail below; here, we focus on the ability of MdmX to regulate the function of Mdm2. Mdm2 and MdmX interact with each other via their C-terminal RING domains. Interestingly, MdmX stabilizes both Mdm2 and p53, yet MdmX also promotes the E3-ligase activity of Mdm2 (Linares et al., 2003; Poyurovsky et al., 2007; Uldrijan et al., 2007). The ability of MdmX to promote Mdm2 function, as well as its own direct impact on p53, suggests that MdmX, like Mdm2, may be a potential therapeutic target for regulating p53 activity (Shangary and Wang, 2009). In addition, the transcription factor YY1 and the ribosomal proteins L5, L11, and L23 interact with Mdm2 and regulate its function. YY1 promotes the ubiquitination of p53 in vivo and in vitro by strengthening the interaction between p53 and Mdm2 (Sui et al., 2004). Conversely, binding of the ribosomal proteins L5, L11, and L23 to Mdm2 inhibits Mdm2 function and plays a crucial role in p53 activation upon ribosomal stress (Dai et al., 2004; Lohrum et al., 2003; Zhang et al., 2003).
The complexity of the regulation of p53 stability through Mdm2 becomes even more apparent when considering the activity of the deubiquitinating enzyme HAUSP. Mdm2 is an unstable protein, primarily due to autoubiquitination or its ubiquitination by undefined E3 ligases (Fang et al., 2000; Stommel and Wahl, 2004; Itahana et al., 2007). HAUSP not only deubiquitinates p53 (Li et al., 2002a), but also Mdm2 (Cummins et al., 2004; Li et al., 2004). The protein DAXX is an important component of this multiprotein complex that regulates HAUSP-mediated deubiquitination of Mdm2 (Tang et al., 2006a), and recent work now shows that the assembly of this complex is in part regulated by the protein RASSF1A (Song et al., 2008). RASSF1A disrupts the interaction between Mdm2, DAXX, and HAUSP, thereby promoting Mdm2 ubiquitination, and consequently resulting in p53 stabilization. As this example demonstrates, an ever growing number of proteins continue to be defined that affect the levels or activities of Mdm2 and thereby influence p53 levels.
Mdm2 is also subject to regulation by phosphorylation and acetylation. Various phosphorylation sites have been described, and depending on the site and the modifying kinase, these modifications can inhibit or activate the activity of Mdm2 (reviewed in Meulmeester et al., 2005). ATM and c-Abl inhibit Mdm2 activity via the phosphorylation of Ser395 (S395) and Tyr394, respectively, whereas phosphorylation of Ser166 and Ser186 promote its E3-ligase activity (Meek, 2004; Mayo and Donner, 2002). Conversely, dephosphorylation of Mdm2 by WIP1 stabilizes Mdm2 and facilitates p53 ubiquitination and degradation (Lu et al., 2007). Mdm2 activity is also inhibited by CBP/p300-mediated acetylation, resulting in increased p53 levels (Wang et al., 2004). This provides an interesting level of coordinated p53 regulation, as acetylation of the p53 C terminus also blocks at least some of the major p53 ubiquitination sites used by Mdm2 for p53 degradation. The six C-terminal lysines of p53 are acetylated by CBP/p300 (Gu and Roeder, 1997). Ubiquitination and acetylation are mutually exclusive modifications, and competition between these modifications is thought to affect p53 stability. p53 acetylation levels are markedly enhanced in response to stress, promoting p53 stabilization and activation (Brooks and Gu, 2003; Luo et al., 2001; Sakaguchi et al., 1998). Purified acetylated p53 cannot be ubiquitinated by Mdm2 in vitro, and ubiquitination is significantly reduced upon induction of acetylation (Ito et al., 2001; Li et al., 2002b). More recent work has shown that the acetylation of eight different lysine residues on p53 prevents the interaction between p53 and Mdm2 (Tang et al., 2008). Therefore, both phosphorylation and acetylation of p53 can inhibit the Mdm--p53 interaction, suggesting a critical role of posttranslational modifications in affecting the p53/Mdm2 feedback loop.
Due to the importance of the Mdm2-p53 interaction, inhibition of this event with small molecules is regarded as having therapeutic potential. A number of different strategies have been employed to screen for and develop small molecules that bind specifically to the N-terminal region of Mdm2 that interacts with p53 (Shangary and Wang, 2009). The most promising candidates described so far have been Nutlin-3A (Vassilev et al., 2004) and MI-219 (Shangary et al., 2008), and analogs of these lead compounds are being developed or have already entered preclinical development or early clinical trials. As expected, inhibition of the Mdm2-p53 interactions by small molecules results in increased p53 levels. The significance of controlling the ubiquitin ligase activity of Mdm2 is further bolstered by work studying the effects of a site-specific (C462A) knockin mutation of Mdm2 (Itahana et al., 2007). Notably, HLI98, a promising inhibitor of Mdm2 E3 ligase activity, also activates p53 (Yang et al., 2005). Nevertheless, the outcome of raising p53 levels and subsequent p53-mediated apoptosis is very dependent on the cell or tissue type. It will be of great interest to determine whether these compounds when used in combination therapies might selectively target cancer cells in the clinic.
The six C-terminal lysines of p53 are the predominant sites for Mdm2-mediated ubiquitination (Lohrum et al., 2001) (Figure 1). Although the in vitro data certainly demonstrate the importance of the six C-terminal lysines of p53 for Mdm2-mediated ubiquitination, knockin studies in which the equivalent lysines are mutated in analogous fashion to arginines (p53-6KR knockin mice) do not dramatically alter p53 protein levels (Feng et al., 2005; Krummel et al., 2005). Analysis of p53 protein stability in embryonic stem cells, embryonic fibroblasts, and thymocytes derived from p53-6KR mutant knockin animals reveals normal p53 stabilization both before and after DNA damage (Feng et al., 2005). Furthermore, the half-life of an analogous p53-7KR mutant (a mouse p53 construct with the seven C-terminal lysines mutated) showed no difference when compared to wild-type p53 in embryonic fibroblasts and thymocytes derived from p53-7KR knockin mice (Krummel et al., 2005). Together, these two studies support the theory that the C-terminal lysines are not essential for efficient p53 degradation in vivo and that additional E3-ligases, as well as ubiquitination of additional p53 lysines, are required for effective p53 regulation. Recent in vitro data suggest that lysine residues located in the DNA-binding domain and in the N-terminal region may be ubiquitinated (Chan et al., 2006). Thus, the sites of Mdm2-mediated ubiquitination are not as specific as originally anticipated. It is very likely that the C terminus of human p53 is preferentially targeted by Mdm2 for ubiquitination, but Mdm2 is still able to degrade p53 in vivo in the absence of these sites.
The levels of nuclear p53 are also modulated by degradation-independent ubiquitination. Mdm2-mediated p53 monoubiquitination, occurring when Mdm2 activity levels are low, promotes p53 nuclear export and accumulation of cytoplasmic p53 (Lohrum et al., 2001; Li et al., 2003; Marchenko et al., 2007). Although originally thought of as a passive way to block the nuclear function of p53, accumulating evidence suggests that cytoplasmic localization of p53 plays an active role in p53-mediated functions such as apoptosis and autophagy. Once in the cytoplasm, p53 can localize to the mitochondria, where it induces apoptosis (Marchenko and Moll, 2007). p53 interacts with antiapoptotic members of the Bcl family such as Bcl-XL and Bcl-2 at the outer mitochondrial membrane to promote the oligomerization of the proapoptotic factors Bak and Bax. These in turn drive the formation of pores in the mitochondrial membrane, resulting in the release of cytochrome c and other apoptotic activators from the mitochondria (Chipuk et al., 2004; Mihara et al., 2003; Tomita et al., 2006). Alternatively, p53 can directly interact with Bak, releasing Bak from its inhibitory interaction with Mcl1 (Leu et al., 2004). Further evidence shows that changes in subcellular localization of p53 can have profound effects on the cell. Recently, cytoplasmic p53 has been shown to regulate autophagy (Tasdemir et al., 2008). An additional factor that regulates p53 localization is MSL2, another E3-ligase for p53, which can promote its cytoplasmic localization independent of Mdm2 (Kruse and Gu, 2008a). Significantly, this RING-E3 does not affect p53 levels or Mdm2 mediated-p53 monoubiquitination, and targets different lysines (K351, K357) for ubiquitination than Mdm2 (Figure 1). It will be interesting to determine whether MSL2 regulates apoptosis or other roles ascribed to cytoplasmic p53.
Together, the studies using knockin mice with mutations in p53’s phosphorylation sites and C-terminal lysine residues indicate that p53 stabilization in vivo involves many different mechanisms. None of the sites tested is able to recapitulate in vivo the dramatic effects on p53 stabilization observed in in vitro biochemical and tissue culture studies. Overall, the model for p53 stabilization in vivo has shifted from a primary emphasis on p53 phosphorylation to a greater appreciation for the multiple layers of Mdm2 regulation. However, it remains incompletely understood how p53 is stabilized in vivo in different tissues and in response to different stress signals.
Sequence-Specific DNA Binding
An early breakthrough for the p53 field was the discovery that p53 is a sequence-specific DNA-binding protein (Bargonetti et al., 1991; Kern et al., 1991). The DNA consensus sequence of p53 response elements was subsequently identified by in vitro assays (El-Deiry et al., 1992) and has been recently refined by chromatin immunoprecipitation (ChIP) sequencing in human cells (Wei et al., 2006). The consensus sequence contains two inverted pentameric sequences with the pattern 5′-RRRC(A/T)|(A/T)GYYY-3′. The importance of sequence-specific DNA binding is underscored by the fact that the vast majority of tumor-associated p53 mutations occur within its DNA-binding domain (Hainaut and Hollstein, 2000), which consists of a beta sandwich with a series of loops and short helices (Cho et al., 1994). Transactivation requires p53 to interact with promoter regions of p53 target genes. In the classical model for p53 activation, the second canonical step is the binding of stress-activated and posttranslationally modified p53 to specific response elements in the promoters of target genes (Figure 2). As discussed below, it is unclear whether DNA binding by p53 is dependent on stress signals or other modes of regulation. Early studies focused on the ability of p53 to bind DNA in a sequence-specific manner. DNA binding was thought to occur via the conserved central core domain, whereas the C-terminal region was thought to act as a negative modulator that had to be modified to allow sequence-specific DNA binding. Phosphorylation, ubiquitination, methylation, sumoylation, neddylation, and acetylation all target and modify the C-terminal region of p53, thereby affecting the ability of p53 to bind DNA (Appella and Anderson, 2001; Brooks and Gu, 2003). In addition to affecting DNA binding via the core domain, these modifications may affect C terminus-mediated DNA binding, direct other regulatory events such as cofactor recruitment, and provide sequence specificity.
The ability of stress-induced, allosteric activation of unmodified p53 to bind sequence specific DNA (Hupp et al., 1992) has recently been questioned by a variety of studies. Experiments using larger DNA molecules or chromatin as targets demonstrate that activation of p53 is not required for p53 to bind to DNA (Cain et al., 2000; Kim et al., 1999). Furthermore, analysis of wild-type and mutant p53 variants using nuclear magnetic resonance show that presumed latent and active p53 have an extremely similar molecular structures and that the C-terminal region does not interact with the central DNA-binding region (Ayed et al., 2001). It is currently believed that both the core DNA-binding domain and the C-terminal domain of p53 possess DNA-binding activities, with the former primarily providing sequence specificity whereas the latter recognizes structural features of target DNA.
In light of its ability to bind DNA both based on sequence specificity and on DNA structure and topology, the question of whether stress is required to induce DNA binding of p53 becomes even more intriguing. A number of recent studies now provide additional evidence that a significant portion of p53 is bound to DNA in unstressed cells. An unbiased global ChIP assay demonstrates that the C-terminal domain facilitates p53 binding to genomic DNA, and only 5% of p53-bound genomic sequence isolated by ChIP contains the p53 consensus sites (Liu et al., 2004). Furthermore, a microarray analysis of p53 binding to genomic DNA also shows that only 2% of p53 binding events are associated with p53 consensus sites (Cawley et al., 2004). In addition to this nonspecific binding to DNA, ChIP assays show that p53 is present at promoters of its transcriptional targets during normal homeostasis (Kaeser and Iggo, 2002; Szak et al., 2001). Quantitative ChIP assays demonstrate that p53 is present at the promoters of the p21 and Mdm2 genes from wild-type, unstressed p53 cells. These findings provide further support for a model in which p53 is capable of binding DNA in unstressed cells but remains inactive probably because of repression by Mdm2 and MdmX.
Overall, there is no doubt that the binding of p53 to DNA is essential for p53 function, yet it may not be a rate-limiting step in the p53 response as originally thought (Hupp et al., 1992). DNA binding received tremendous attention early on, in part due to the poor DNA binding activity of p53 in vitro and the known tumor-derived mutants that affect DNA binding. However, recent work has shown that in many cases p53 is already bound to the p53 target promoters without activating transcription, although it remains possible that acetylation is important for enhancement of DNA binding on some specific p53 proapoptotic targets (Sykes et al., 2006).
Insights into Promoter-Specific Activation
Early studies suggest that p53 promotes transcription activation by simply interacting with general transcription factors such as TFIID/TAFs or components of SRB/mediator complexes (Thut et al., 1995; Gu et al., 1999). This simple model for transcriptional activation belies the complexity of achieving promoter specificity. In broad terms, promoter specific transactivation could be achieved by the interaction of p53 with specific repressors and activators. The interaction of repressors and activations might be regulated by the posttranslational modification of p53, or interacting proteins might promote changes in p53 posttranslational modification to achieve promoter-specific transactivation. Although there are many proteins reported to bind and regulate p53, very few have been shown to be physically recruited to the promoters of p53 targets, and genetic evidence identifying them as bona fide regulators of p53 is typically lacking. To provide a structure and an overview for this very complex topic, we discuss the roles of p53 repressors and activators and the impact of various p53 modifications in detail below.
p53 Repression by MdmX and Mdm2
Mdm2 and MdmX are structurally related proteins, and physiological levels of both proteins are required in a nonredundant manner to balance p53 activity during embryonic development (Marine et al., 2006). The functions of p53 are downregulated by the Mdm2 oncoprotein, at least in part by ubiquitin-mediated proteolysis (Brooks and Gu, 2006). The central role of Mdm2 in this process is best illustrated by studies carried out in mice, where inactivation of p53 was shown to completely rescue the embryonic lethality caused by loss of Mdm2 function (Jones et al., 1995; Montes de Oca Luna et al., 1995). Notably, MdmX knockout mice die despite having functional Mdm2, and this lethality is also rescued by inactivation of p53 (Parant et al., 2001; Migliorini et al., 2002). Thus, MdmX is as critical as Mdm2 in repressing p53 function. In addition to promoting p53 degradation, accumulating evidence indicates that Mdm2 and MdmX form a protein complex with p53 on the promoters of specific p53 target genes (Minsky and Oren, 2004; Ohkubo et al., 2006; Tang et al., 2008).
Initial studies indicate that MdmX represses p53-mediated transcriptional activation (Marine and Jochemsen, 2005). Despite its sequence homology with Mdm2 and the presence of a RING domain, MdmX does not have intrinsic E3-ligase activity for p53 but instead inhibits p53-induced transcription. The interplay between p53, Mdm2, and MdmX is an extremely interesting topic, and new studies with mutant mice highlight their intricate relationship (Iwakuma and Lozano, 2007; Marine et al., 2006; Wahl, 2006), arguing that the major role of MdmX lies in its ability to repress but not degrade p53. Mdm2 and MdmX can both be recruited to p53 promoter regions and act via a multitude of mechanisms to repress transcription of p53 target genes (Tang et al., 2008). The various results from animal studies, together with in vitro and tissue culture data, have led to models that describe the distinct yet combinatorial roles played by these two negative regulators of p53 (Marine et al., 2006; Wahl, 2006). Mdm2 and MdmX may determine whether a cell responds to p53 activation with growth arrest or apoptosis, but the molecular mechanism of these differential effects remains unknown (Barboza et al., 2008). It is possible that Mdm2 and MdmX are recruited by p53 to different target promoters to execute distinct functions.
p53 Acetylation and Promoter-Specific Activation
Histone acetyltransferases (HATs) provide an important layer of p53 regulation, particularly in transcription (Brooks and Gu, 2003). The covalent linkage of an acetyl group to a lysine residue was first discovered on histones, and histone acetylation is now known to have a major impact on transcriptional regulation (Jenuwein and Allis, 2001). However, histones are not the only proteins that can be acetylated; p53 was the first nonhistone substrate shown to be functionally regulated by acetylation and deacetylation (Gu and Roeder, 1997; Luo et al., 2000). p53 is acetylated by the histone acetyltransferase CBP/p300, and CBP/p300 mutations are found in several types of human tumors (Goodman and Smolik, 2000; Iyer et al., 2004). p53 acetylation is critically important both for the efficient recruitment of cofactors and for the activation of p53 target genes in vivo. Once localized at promoter regions, CBP/p300 can enhance transcription by acetylating histones in the vicinity of target genes, thereby establishing a more accessible chromatin conformation, and by bridging transcription factors to the pol II holoenzyme (Goodman and Smolik, 2000). In addition, the acetyltransferases Tip60 and hMof are cofactors that can be recruited by p53 and are able to acetylate p53 as well as histones (Tang et al., 2006b; Sykes et al., 2006). The acetyl-transferase activity of Tip60 is implicated in both DNA repair and apoptosis, but, most significantly, evidence suggests that Tip60 is a p53 activator by a large-scale RNA interference screen (Berns et al., 2004).
p53 acetylation is elevated in response to stress, and levels of acetylation correlate well with p53 activation and stabilization (Luo et al., 2000; 2001; Vaziri et al., 2001; Ito et al., 2001; Knights et al., 2006). The transcription phenotypes described for the C-terminal acetylation-deficient p53-6KR knockin mice show that p53-dependent gene expression in embryonic stem cells and thymocytes is impaired in a promoter-specific manner after DNA damage. This finding supports the theory that C-terminal acetylation helps to activate p53 dependent trans-activation after DNA damage (Feng et al., 2005). However, the transcription profiles in cells from the 6KR knockin mice show some cell type specificity, with the most dramatic differences observed in embryonic stem cells, a more selective defect in thymocytes, and no significant differences in embryonic fibro-blasts. Similarly, embryonic fibroblasts generated from the p53-7KR knockin mice did not show substantial differences in p53-mediated cell cycle control or apoptosis (Krummel et al., 2005). Together, these two knockin studies suggest that C-terminal acetylation shows some cell type-specific regulatory effects, but does not have a major general impact on apoptosis or cell cycle control. In addition to CBP/p300, p300/CBP associated factor (PCAF) is another transcriptional coactivator of p53, which acetylates p53 at lysine K320 (K317 in mice) (Sakaguchi et al., 1998; Liu et al., 1999). Analysis of K317 acetylation in a mouse model shows that a K317R mutant p53 protein is stabilized and accumulates at a rate comparable to wild-type p53 in embryonic fibroblasts and thymocytes (Chao et al., 2006b). Although the expression profile of p53 target genes is largely normal in K317R mouse embryonic fibroblasts, the authors surprisingly find an increase in the expression of proapoptotic genes in thymocytes. There is also an increase in the apoptotic response upon irradiation in thymocytes and epithelial cells from the small intestine, indicating that the effects of K320 acetylation by PCAF are far more complicated than originally anticipated.
Although the C-terminal lysine mutant knockin animal models raise some concerns over the importance of C-terminal acetylation, recent studies show that p53 acetylation is not limited to this domain (Sykes et al., 2006; Tang et al., 2006b). Both studies show that lysine K120 is acetylated, providing an example of transcriptional regulation mediated by acetylation that requires the DNA-binding core domain. In contrast to other sites of p53 modification, K120 is conserved in all species that contain functional p53 genes, implying that this modification may have an evolutionarily conserved role. K120 is acetylated by hMOF and TIP60, acetyltransferases of the MYST family that share no homology with the CBP/p300 or PCAF acetyltransferases. K120 acetylation is essential for p53-mediated activation of the proapoptotic target genes puma and bax, whereas expression of p21 and Mdm2 are not affected. Furthermore, K120 modification affects neither p53 stability nor p53-DNA binding. K120 acetylation occurs after DNA damage, as well as after activation of the p19ARF oncogenic stress pathway (Mellert et al., 2007; Tang et al., 2006b). K164 is another newly identified site of acetylation by CBP/p300. Like K120, K164 is located within the DNA-binding domain (Tang et al., 2008), and both residues are recurrent sites for p53 mutation in human cancer (Hainaut and Hollstein, 2000). Simultaneous loss of acetylation at K120, K164, and the six C-terminal lysines completely abolishes the ability of p53 to activate p21 and suppress cell cycle progression (Tang et al., 2008). Interestingly, separate acetylation defects at K120, K164, or the 6KR sites can be compensated by acetylation of the other sites, enabling p53-mediated transcription of p21. Importantly, an acetylation-defective p53-8KR mutant retains the ability to function as a DNA-binding transcription factor and induces the p53-Mdm2 feedback loop, establishing promoter specificity for this regulatory event. The complete inability of the p53-8KR mutants to induce cell cycle and apoptotic regulators demonstrates that acetylation is absolutely required for key aspects of p53-mediated tumor suppression. Both K120 and K164 residues, as well as the majority of the p53 core domain, are highly conserved, and it will be of great interest to see what effect a p53 K120R or K164R mutation would have in a knockin animal model.
The functional consequences of p53 acetylation suggest that the timing of acetylation of the different p53 regions may be important for accurate p53 regulation and cell fate determination. It is very likely that distinct mechanisms activate different types of p53 targets during the course of the stress response. For each p53-dependent stress response, at least three different classes of target genes can be turned on by p53 to execute p53-mediated function at different levels (Figure 3). The first class of p53 target genes protects cells from excessive p53 activation; these include Mdm2, Pirh2, and others. Given that activated p53 is potentially dangerous to cell viability, the feedback mediated by Mdm2 is probably an early step in the p53-mediated stress response. p53 modification (at least, in the case of acetylation) and coactivator recruitment (e.g., Tip60) are not required for this step (Tang et al., 2006b, 2008). The second class of p53 target genes induces growth arrest, and these genes are particularly important for cells to initiate productive DNA repair. The most important target of this class is p21. p53-dependent recruitment of coactivators such as CBP/p300 and Tip60/hMOF is absolutely required for p21 activation (Sykes et al., 2006; Tang et al., 2006b). Nevertheless, p21 induction is probably redundantly regulated via the modification of p53 at multiple sites. Although the complete loss of p21 induction in the absence of p53 acetylation in the 8KR mutant cells demonstrates that this event is indispensable for p53-mediated growth arrest, a partial modification of p53 is sufficient to induce p21 activation (Tang et al., 2006b, 2008). The third class of p53 target genes induces apoptosis, including PUMA, BAX, NOXA, and others. Given that this step initiates an irreversible apoptotic response, the requirement for full activation of proapoptotic targets is stringent. For example, all the key coactivators have to be recruited to the target promoters; specific modification (e.g., K120 acetylation) is required, and p53 is likely modified at many different sites (Figure 3). In addition, the corepressors such as Mdm2 and MdmX must be completely removed from these target promoters.
Figure 3. p53 Acetylation and Target Gene Regulation.
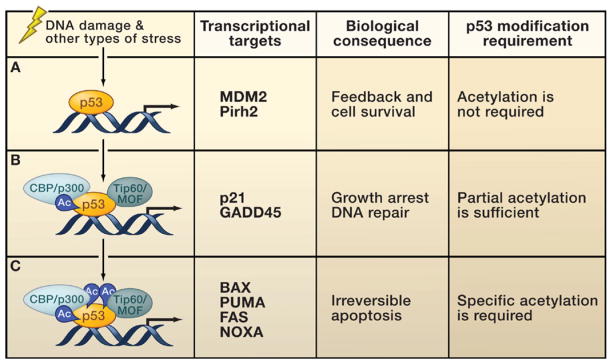
Upon stress-induced p53 activation, different sets of p53 target genes have different requirements for p53 posttranslational modifications.
(A) A number of promoters can be activated by un-acetylated p53. This class of p53 target genes protects cells from excessive p53 activation. These target genes include Mdm2, Pirh2, and others.
(B) The activation of genes involved in DNA repair and cell cycle control requires recruitment of specific histone acetyltransferases (HATs) and partial acetylation (Ac), acting at least in part by antirepression.
(C) Full acetylation of p53 is required for the activation of proapoptotic genes. Activation of these targets induces a program to ensure efficient apoptosis.
Conversely, p53 can be deacetylated and acetylation-dependent upregulation of p53 transactivation can be reversed by distinct histone deacetylase (HDAC) complexes containing HDAC1 (Luo et al., 2000) or Sir2α/Sirt1 (Luo et al., 2001; Vaziri et al., 2001). Deacetylation represses p53-dependent transcriptional activation, apoptosis, and growth arrest. p53 deacetylation by the HDAC1 complex strongly represses p53 transcriptional activity and modulates growth arrest and apoptosis (Luo et al., 2000). Sirt1-mediated p53 deacetylation represses p53-dependent apoptosis in response to DNA damage and oxidative stress, whereas expression of a Sirt1 dominant-negative point mutant increases the sensitivity of cells to stress (Luo et al., 2001). This notion is further supported by the observation that p53 is hyperacetylated in Sirt1-deficient mice and cells from these mice display increased radiation-induced apoptosis (Cheng et al., 2003). Sirt1-mediated deacetylation of p53 is suppressed by a putative tumor suppressor, DBC-1 (deleted in breast cancer gene 1), in human cells (Zhao et al., 2008; Kim et al., 2008) and is upregulated by oncogenic viral protein HPV E7 (Allison et al., 2009). A small molecule, tenovin-6, has been recently identified as a potent SIRT1 inhibitor that effectively induces p53 activation and cell growth repression in cancer cells (Lain et al., 2008). Furthermore, Sirt1 colocalizes with p53 in promyelocytic leukemia (PML) nuclear bodies and undergoes a conformational change when bound to acetylated p53 to antagonize the increase in p53 acetylation induced by PML (Pearson et al., 2000). p53 deacetylation may provide a quick-acting mechanism to stop p53 function when transcriptional activation of target genes is no longer required. Targeted deacetylation occurs very quickly amidst a global equilibrium of genomic acetylation and deacetylation. Restoring a steady-state level of p53 targets after completion of DNA repair is crucial to allow cells to return to normal homeostasis. Deacetylation could serve as an important step to allow Mdm2-mediated p53 degradation, given that acetylation of p53 inhibits p53 ubiquitination by Mdm2 in vivo and in vitro (Li et al., 2002b).
p53 Transcriptional Activation: Methylation, Sumoylation, and Neddylation
It is not only acetylation and ubiquitination that compete for p53’s C-terminal lysines. Methylation, sumoylation, and neddylation all occur at specific sites, and the exact interplay of these modifications may contribute to p53 promoter specificity. The many possible variations of these modifications of p53 may act like a barcode, resulting in specific p53 responses.
To this point, three different methyltransferases have been shown to be able to methylate C-terminal lysine residues of p53. Set7/9-mediated monomethylation of p53 at lysine K372 promotes p53 activity, in particular the ability of p53 to transactivate p21 (Chuikov et al., 2004). Monomethylation of p53 at K370 and K382 by Smyd2 and Set8/PR-Set7, respectively, represses p53 activity (Huang et al., 2006; Shi et al., 2007). Regulated dimethylation of K370 and K382 provides a binding site for the DNA repair factor 53BP1, and DNA damage increases p53 dimethylation and therefore promotes the interaction of p53 and 53BP1. Interestingly, the demethylase LSD1 specifically demethylates K370, thereby removing at least one anchoring point for 53BP1 (Huang et al., 2007). Further support for the notion that methylation contributes to the regulation of the p53 response comes from evidence that both p53 (Jansson et al., 2008) and the surrounding histones (An et al., 2004) are arginine methylated. An et al. demonstrate that arginine methylation by PRMT1 and acetylation by p300 and CARM1 entails both direct modification of p53 as well as the controlled modification of the surrounding histones to regulate the expression of p53 responsive genes. Jansson et al. demonstrate that arginine methylation of p53 by PRMT5 regulates target gene specificity of p53 by altering promoter binding.
Modification of p53 by the ubiquitin-like modifications SUMO and Nedd8 further add to the competition for the C-terminal lysines. The exact functional consequences of p53 sumoylation at K386 are still somewhat unclear. Although some studies report that sumoylation promotes p53 transcriptional activity (Melchior and Hengst, 2002), others demonstrate that sumoylation promotes the localization of p53 to the cytoplasm (Carter et al., 2007). The role of C-terminal neddylation is interesting but not well defined. Mdm2-mediated neddylation of K370, K372, and K373 (Xirodimas et al., 2004) and FBXO11-mediated neddylation of K320 and K321 (Abida et al., 2007) appear to inhibit p53-mediated transcriptional activation. It remains to be determined under what circumstances methylation, sumoylation, and neddylation are required for p53 activation in vivo.
Regulation of p53 by the ASPP Family, 53BP1, and Other Cofactors
There are a number of proteins that affect p53 activity through their selective interaction with the tumor suppressor. The ASPP (ankyrin-repeat-, SH3-domain- and proline-rich-region-containing proteins) family specifically affects the p53-mediated apoptotic response (Trigiante and Lu, 2006). The ASPP family consists of two proapoptotic mediators, ASPP1 and ASPP2, and the antiapoptotic mediator iASSP. ASPP1 and ASPP2 enhance the proapoptotic function of p53 by selectively promoting the binding of p53 to proapoptotic gene targets such as BAX, PUMA, and PIG3 (Samuels-Lev et al., 2001). Conversely, iASSP prevents the transcriptional activity of p53 bound to proapoptotic promoters (Bergamaschi et al., 2003). However, the exact mechanism underlying the promoter specificity of ASPP1, ASPP2, and iASSP is still unknown. Interestingly, the expression levels of ASPP1/ASPP2 and iASSP correlate with cellular sensitivity or resistance to apoptosis. Due to their specific regulation of the p53 response, targeting the ASPP family may have therapeutic potential.
53BP1 binds p53 and mediates promoter specificity by a yet unknown mechanism (Iwabuchi et al., 1994). It was originally identified together with 53BP2, which was later shown to be a truncated N-terminal splice form of ASPP2 and has recently been shown to be a coactivator of p53 mediated proliferation arrest (Brummelkamp et al., 2006). In addition to its role in the regulation of p53, it also has an important function in DNA repair. The fact that 53BP1 is involved in both DNA damage repair and p53-induced cell cycle arrest suggests that 53BP1 could contribute to the coordination of these responses. With an ever growing number of p53-interacting partners being identified, such as HZF, APAK, and hCAS/CSE1L (Das et al., 2007; Tanaka et al., 2007; Tian et al., 2009), it will be increasingly important to characterize cofactors that affect promoter specificity of p53. Are there specific cofactors and combinations of cofactors that regulate specific subsets of target genes? Identifying the specific targets affected by an interaction between p53 and its cofactors remains critical for advancing our understanding of the p53 response in vivo.
Lessons from the Classical Models for p53 Activation
In broad terms, the traditional model for stress-induced p53 activation consists of a p53 stabilization step that is followed by an intricate regulatory network to induce DNA binding and transcriptional activation of p53 target genes (Figure 2). The stress signal leads to p53 stabilization, primarily via the release of p53 from its interaction with Mdm2. Phosphorylation of p53 inhibits its interaction with Mdm2 and thereby prevents Mdm2-mediated ubiquitination and the resulting proteasomal degradation of p53. Once stabilized, p53 has been thought to be activated by further posttranslational modifications, leading to increased binding of p53 to DNA at p53-specific promoters. After the binding to specific high- or low-affinity promoter sites, p53 would then act as a transcription factor to regulate the expression of its target genes, at least in part via the recruitment of coactivators and corepressors.
This classic model of stabilization and activation continues to provide an explanation for many modes of p53 regulation, especially via posttranslational modifications as observed in in vitro biochemical and in tissue culture systems. However, recent results from genetic studies introducing point mutations in p53 (Toledo and Wahl, 2006) and observations demonstrating the importance of specific modifications (Tang et al., 2008) reveal aspects of p53 regulation that may not be sufficiently explained by the classic model. A refined and extended model is needed to adequately explain our current understanding of p53 activation in vivo.
In vitro and cell culture studies establish that phosphorylation is a key event in both p53 stabilization and regulation of DNA binding (Appella and Anderson, 2001; Hupp et al., 1992). However, as discussed above, point mutant knockin approaches have shown that the dramatic effects observed in vitro are only partially recapitulated in cells and tissues from knockin animals. These observations point toward a regulatory system for p53 in vivo in which individual phosphorylation events may be somewhat redundant, yet by no means insignificant. It is very likely that the loss of phosphorylation at a particular site can be compensated by phosphorylation at different sites, or by other types of modifications such as acetylation. Various types of stress and different tissues may require different modes for p53 regulation. Regulatory redundancy among posttranslational modifications may ensure that p53 is regulated appropriately in response to different stress stimuli and intensities, as well as with the required tissue specificity.
A number of key observations suggest that p53 may be active in the absence of further activation by stress stimuli. As discussed above, both Mdm2 and MdmX knockout mice are embryonic lethal, and the rescue of this phenotype by loss of p53 indicates that p53 is responsible for the early embryonic lethality (Iwakuma and Lozano, 2007; Marine et al., 2006; Wahl, 2006). The severity of the phenotypes of the Mdm2 and MdmX knockout mice suggests that p53 has significant activity. Given the early embryonic lethality, it may be impossible to address whether additional modifications may be required for the activation of p53. Instead of further p53 activation via modifications, the loss of the repressors Mdm2 and MdmX may be sufficient to elevate p53 function, which results in embryonic lethality. This model is supported by data demonstrating that loss of the p53 interaction with either Mdm2 or MdmX, is essential for the activation of cell cycle-specific target genes (Tang et al., 2008). Interestingly, p53 is spontaneously active in tissues lacking mdm2, which leads to p53-dependent pathologies in radiosensitive tissues (Ringshausen et al., 2006). This study also demonstrates that this activity of p53 occurs in the absence of further posttranslational modifications. The notion that p53 does not absolutely require activating modifications is supported by results obtained from purified in vitro systems (An et al., 2004; Espinosa and Emerson, 2001; Gu et al., 1999). These studies raise a fundamental question: what is the main purpose of p53 modification during the stress response? Why is p53 modification required for some biological settings but not others?
p53 Regulation by Antirepression
In light of the studies discussed above, it appears that p53 activation in vivo requires not only stabilization and activation by posttranslational modifications, but also release from a repressed state. Including this step of antirepression in a refined model of p53 activation may provide a more accurate way to explain p53 activation in vivo (Figure 4). Both in vitro and genetic data indicate that p53 is intrinsically active, yet is repressed by its key negative regulators, Mdm2 and MdmX. By starting with the view that p53 is intrinsically active, we may be able to explain differences observed between in vitro and in vivo experiments by the absence or aberrant activity of specific cofactors or modifications. Interestingly, knockin animals that carry the p53QS mutation are embryonic lethal even though p53QS retains the ability to bind DNA (Johnson et al., 2005). However, p53QS loses its ability to interact with both Mdm2 and MdmX, which may contribute to the embryonic lethality similar to that observed in Mdm2 null mice. p53QS has deficiencies in transactivation, yet retains the ability to activate some targets. This may be a consequence of very high levels of p53QS because of loss of the interaction with Mdm2. These findings further support the concept that p53 is constitutively active but continuously repressed. In order to observe the role anti-repression plays in p53 regulation, both positive and negative regulators, as well as p53, need to be present at near physiological levels.
Figure 4. Refined Model for p53 Activation.

Promoter-specific p53 activation in vivo consists of three key steps: (1) p53 stabilization, (2) antirepression, and (3) promoter-specific activation.
(1) Stress-induced p53 stabilization occurs through many different mechanisms, many of which act by affecting the ability of Mdm2 to ubiquitinate p53.
(2) Antirepression describes the release of p53 from the repression mediated by Mdm2 and MdmX. This step requires the acetylation of p53 at key lysine residues and facilitates the activation of specific subsets of p53 targets. Phosphorylation of p53 or treatment with the small molecule Nutlin-3 may have similar effects on antirepression.
(3) For full activation of specific promoters, p53 recruits and interacts with numerous cofactors. These act by modifying p53, the surrounding histones, or other transcription factors. Regulating the activation of specific groups of p53 targets for apoptosis, senescence, cell cycle control, DNA repair, autophagy, metabolism, or aging may require exact combinations of cofactors and posttranslational modifications.
Abbreviations: Ac, acetylation; P, phosphorylation; Me, methylation; N8, neddylation; S, sumoylation.
Overexpression of positive or negative regulators of p53 could mask the effect of antirepression in vivo. This may explain why in vitro experiments using unacetylated p53 show unexpected transcriptional activity in purified systems (An et al., 2004; Espinosa and Emerson, 2001), as negative regulators Mdm2 and MdmX were not present. This could also explain why overexpression of the various kinases that inhibit the p53-Mdm2 interaction lead to a masking of this effect. It will be very interesting to determine to what extent p53 phosphorylation and p53 acetylation contribute to the stabilization and activation of p53 by synergistically acting to inhibit the binding of p53 to its negative regulators. Another intriguing question that arises from the interaction of p53 with Mdm2 and MdmX on gene promoters is whether p53 can be ubiquitinated by Mdm2 while bound to DNA and what role phosphorylation may play in regulating the p53 interaction with Mdm2 and MdmX when p53 is bound to DNA. Although the N-terminus of p53 was originally thought to be the predominant site of the interaction with Mdm2, both the DNA-binding domain (Wallace et al., 2006; Kulikov et al., 2006; Tang et al., 2008; Yu et al., 2006) and the C-terminal domain (Tang et al., 2008) interact with Mdm2. The presence of multiple Mdm2 binding sites in p53 may suggest that there is an interplay between posttranslational modifications that activate p53 by stabilization and that release it from repression.
It is conceivable that antirepression may have varying degrees of significance in vivo, depending on the relative responsiveness of different p53 promoters. Whereas antirepression may be sufficient for the activation of p53 targets that are highly responsive, such as those that block cell cycle progression, antirepression alone may be insufficient to induce apoptosis, which would require additional p53 posttranslational modifications. In this proposed mechanism, a stress signal would initially result in reversible cell cycle arrest, and the more dramatic and irreversible apoptotic response would require additional stimulation. Such regulatory specificity may explain results showing that the small molecule Nutlin-3 acts primarily to activate cell cycle control genes, whereas the transactivation of proapoptotic p53 targets is comparatively weak (Tovar et al., 2006; Vassilev et al., 2004). Inhibition of the p53-Mdm2 interaction leading to p53 stabilization and removal of Mdm2-mediated repression at the promoter may be sufficient to activate cell cycle arrest upon Nutlin-3 treatment, although certain cofactors may be still needed (Brummelkamp et al., 2006). In contrast, the induction of apoptotic p53 targets may require additional signals and posttranslational modifications of p53. This type of differential regulation of specific targets and the concomitant importance of different cofactors reacting to varying stress signals may explain why phenotypes from knockin mice with specific point mutations are modest compared to in vitro results. In a given knockin animal, cells from different tissues may respond to specific stress signals via a multitude of pathways to activate p53. Although particular aspects of the response may be inhibited by the loss of a specific modification site, overall stress responses may remain intact if p53 can still be released from repression and other modifications augment p53 activation to trigger appropriate responses.
Concluding Remarks
Here, based on the latest advances, we have proposed that p53 activation in vivo consists of three major steps: (1) p53 stabilization, (2) antirepression, and (3) promoter-specific activation (Figure 4). In this refinement of the current model, p53 stabilization involves multiple layers of Mdm2 regulation; anti-repression, which is mediated by p53 acetylation and probably phosphorylation; and promoter-specific activation of p53 target genes involving additional posttranslational modifications and cofactor recruitment.
Although the previous 30 years of p53 research have led to an incredibly detailed understanding of the biology of p53, a number of key questions in the study of p53 activation continually reappear in a new form. Are there unified mechanisms that control p53 stabilization in vivo? Do different tissues and stress factors use different pathways for the stabilization of p53? How does the cell coordinate the status of DNA repair and the levels of p53 activation? What is the in vivo significance of ATM- and ATR-mediated phosphorylation in p53 activation? Are the activities of HATs such as CBP/p300 and Tip60/MOF regulated upon stress? How are the many different types of lysine modification of the C-terminal domain coordinated? How many targets of p53 are repressed by the presence of Mdm2 and MdmX? How does Mdm2 or MdmX repress transcriptional activation on DNA? Do promoters for specific p53 responses have similar modes of regulation? For example, are all proapoptotic target genes under the control of one or more specific regulatory mechanisms that are different from mechanisms regulating senescence, autophagy, or metabolic targets? Is there any specific target gene of p53 directly involved in the control of aging or antiaging processes? As in the past, answers to such questions will mean that models for the activation of p53 will need to continually evolve.
Acknowledgments
The work in W.G.’s laboratory is supported by the NIH/NCI, the Leukemia and Lymphoma Society, and the Ellison Medical Foundation.
References
- Abida WM, Nikolaev A, Zhao W, Zhang W, Gu W. FBXO11 promotes the Neddylation of p53 and inhibits its transcriptional activity. J Biol Chem. 2007;282:1797–1804. doi: 10.1074/jbc.M609001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison SJ, Jiang M, Milner J. Oncogenic viral protein HPV E7 upregulates the SIRT1 longivity protein in human cervical cancer cells. Aging. 2009;1:316–327. doi: 10.18632/aging.100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Appella E, Anderson CW. Posttranslational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- Ashcroft M, Kubbutat MH, Vousden KH. Regulation of p53 function and stability by phosphorylation. Mol Cell Biol. 1999;19:1751–1758. doi: 10.1128/mcb.19.3.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol. 2000;20:3224–3233. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayed A, Mulder FA, Yi GS, Lu Y, Kay LE, Arrowsmith CH. Latent and active p53 are identical in conformation. Nat Struct Biol. 2001;8:756–760. doi: 10.1038/nsb0901-756. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- Barboza JA, Iwakuma T, Terzian T, El-Naggar AK, Lozano G. Mdm2 and Mdm4 loss regulates distinct p53 activities. Mol Cancer Res. 2008;6:947–954. doi: 10.1158/1541-7786.MCR-07-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargonetti J, Friedman PN, Kern SE, Vogelstein B, Prives C. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell. 1991;65:1083–1091. doi: 10.1016/0092-8674(91)90560-l. [DOI] [PubMed] [Google Scholar]
- Bergamaschi D, Samuels Y, O’Neil NJ, Trigiante G, Crook T, Hsieh JK, O’Connor DJ, Zhong S, Campargue I, Tomlinson ML, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33:162–167. doi: 10.1038/ng1070. [DOI] [PubMed] [Google Scholar]
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P. DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene. 1999;18:1723–1732. doi: 10.1038/sj.onc.1202480. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Fabius AW, Mullenders J, Madiredjo M, Velds A, Kerkhoven RM, Bernards R, Beijersbergen RL. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol. 2006;2:202–206. doi: 10.1038/nchembio774. [DOI] [PubMed] [Google Scholar]
- Cain C, Miller S, Ahn J, Prives C. The N terminus of p53 regulates its dissociation from DNA. J Biol Chem. 2000;275:39944–39953. doi: 10.1074/jbc.M002509200. [DOI] [PubMed] [Google Scholar]
- Carter S, Bischof O, Dejean A, Vousden KH. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat Cell Biol. 2007;9:428–435. doi: 10.1038/ncb1562. [DOI] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- Chan WM, Mak MC, Fung TK, Lau A, Siu WY, Poon RY. Ubiquitination of p53 at multiple sites in the DNA-binding domain. Mol Cancer Res. 2006;4:15–25. doi: 10.1158/1541-7786.MCR-05-0097. [DOI] [PubMed] [Google Scholar]
- Chao C, Hergenhahn M, Kaeser MD, Wu Z, Saito S, Iggo R, Hollstein M, Appella E, Xu Y. Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J Biol Chem. 2003;278:41028–41033. doi: 10.1074/jbc.M306938200. [DOI] [PubMed] [Google Scholar]
- Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006a;25:2615–2622. doi: 10.1038/sj.emboj.7601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, Wang JY, Anderson CW, Appella E, Xu Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol. 2006b;26:6859–6869. doi: 10.1128/MCB.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428 doi: 10.1038/nature02501. 1 p following 486. [DOI] [PubMed] [Google Scholar]
- Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–7668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Raj L, Zhao B, Bernstein A, Aaronson SA, Lee SW. Hzf, a key modulator of p53 mediated transcription, functions as a critical determinant of cell survival and death upon genotoxic stress. Cell. 2007;130:624–637. doi: 10.1016/j.cell.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci USA. 1979;76:2420–2424. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O’Rourke K, Koeppen H, Dixit VM. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–5395. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Gu W, Malik S, Ito M, Yuan CX, Fondell JD, Zhang X, Martinez E, Qin J, Roeder RG. A novel human SRB/MED-containing cofactor complex, SMCC, involved in transcription regulation. Mol Cell. 1999;3:97–108. doi: 10.1016/s1097-2765(00)80178-1. [DOI] [PubMed] [Google Scholar]
- Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Honda R, Yasuda H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999;18:22–27. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71:875–886. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12:355–366. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20:1331–1340. doi: 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci USA. 1994;91:6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakuma T, Lozano G. Crippling p53 activities via knock-in mutations in mouse models. Oncogene. 2007;26:2177–2184. doi: 10.1038/sj.onc.1210278. [DOI] [PubMed] [Google Scholar]
- Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431–1439. doi: 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, Attardi LD. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet. 2005;37:145–152. doi: 10.1038/ng1498. [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Kaeser MD, Iggo RD. Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proc Natl Acad Sci USA. 2002;99:95–100. doi: 10.1073/pnas.012283399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- Kim E, Rohaly G, Heinrichs S, Gimnopoulos D, Meissner H, Deppert W. Influence of promoter DNA topology on sequence-specific DNA binding and transactivation by tumor suppressor p53. Oncogene. 1999;18:7310–7318. doi: 10.1038/sj.onc.1203139. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartz-beck A, Quong AA, Zhang X, Beerman T, Pestell RG, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci USA. 2005;102:10188–10193. doi: 10.1073/pnas.0503068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. MSL2 promotes MDM2 independent cytoplasmic localization of p53. J Biol Chem. 2008a;284:3250–3263. doi: 10.1074/jbc.M805658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. SnapShot: p53 posttranslational modifications. Cell. 2008b;133:930. doi: 10.1016/j.cell.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kulikov R, Winter M, Blattner C. Binding of p53 to the central domain of Mdm2 is regulated by phosphorylation. J Biol Chem. 2006;281:28575–28583. doi: 10.1074/jbc.M513311200. [DOI] [PubMed] [Google Scholar]
- Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 2006;13:951–961. doi: 10.1038/sj.cdd.4401916. [DOI] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002a;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem. 2002b;277:50607–50611. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13:879–886. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA. 2003;100:12009–12014. doi: 10.1073/pnas.2030930100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lagowski JP, Vanderbeek GE, Kulesz-Martin MF. Facilitated search for specific genomic targets by p53 C-terminal basic DNA binding domain. Cancer Biol Ther. 2004;3:1102–1108. doi: 10.4161/cbt.3.11.1189. [DOI] [PubMed] [Google Scholar]
- Llanos S, Clark PA, Rowe J, Peters G. Stabilization of p53 by p14(ARF) without relocation of MDM2 to the nucleolus. Nat Cell Biol. 2001;3:445–452. doi: 10.1038/35074506. [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH. C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol. 2001;21:8521–8532. doi: 10.1128/MCB.21.24.8521-8532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 auto-regulatory loop. Cancer Cell. 2007;12:342–354. doi: 10.1016/j.ccr.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- MacPherson D, Kim J, Kim T, Rhee BK, Van Oostrom CT, DiTullio RA, Venere M, Halazonetis TD, Bronson R, De Vries A, et al. Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. EMBO J. 2004;23:3689–3699. doi: 10.1038/sj.emboj.7600363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko ND, Moll UM. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle. 2007;6:1718–1723. doi: 10.4161/cc.6.14.4503. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, Jochemsen AG. Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun. 2005;331:750–760. doi: 10.1016/j.bbrc.2005.03.151. [DOI] [PubMed] [Google Scholar]
- Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927–934. doi: 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27:462–467. doi: 10.1016/s0968-0004(02)02166-7. [DOI] [PubMed] [Google Scholar]
- Meek DW. The p53 response to DNA damage. DNA Repair (Amst) 2004;3:1049–1056. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Melchior F, Hengst L. SUMO-1 and p53. Cell Cycle. 2002;1:245–249. [PubMed] [Google Scholar]
- Mellert H, Sykes SM, Murphy ME, McMahon SB. The ARF/oncogene pathway activates p53 acetylation within the DNA binding domain. Cell Cycle. 2007;6:1304–1306. doi: 10.4161/cc.6.11.4343. [DOI] [PubMed] [Google Scholar]
- Meulmeester E, Pereg Y, Shiloh Y, Jochemsen AG. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle. 2005;4:1166–1170. doi: 10.4161/cc.4.9.1981. [DOI] [PubMed] [Google Scholar]
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–5538. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell. 2004;16:631–639. doi: 10.1016/j.molcel.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- Ohkubo S, Tanaka T, Taya Y, Kitazato K, Prives C. Excess HDM2 impacts cell cycle and apoptosis and has a selective effect on p53-dependent transcription. J Biol Chem. 2006;281:16943–16950. doi: 10.1074/jbc.M601388200. [DOI] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- Poyurovsky MV, Priest C, Kentsis A, Borden KL, Pan ZQ, Pavletich N, Prives C. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007;26:90–101. doi: 10.1038/sj.emboj.7601465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 2006;10:501–514. doi: 10.1016/j.ccr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8:781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangary S, Wang S. Small-molecule inhibitors of the MDM--p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell. 2007;27:636–646. doi: 10.1016/j.molcel.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- Sluss HK, Armata H, Gallant J, Jones SN. Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol Cell Biol. 2004;24:976–984. doi: 10.1128/MCB.24.3.976-984.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Song SJ, Kim SY, Oh HJ, Lim DS. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 2008;27:1863–1874. doi: 10.1038/emboj.2008.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004;23:1547–1556. doi: 10.1038/sj.emboj.7600145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui G, Affar ElB, Shi Y, Brignone C, Wall NR, Yin P, Donohoe M, Luke MP, Calvo D, Grossman SR. Yin Yang 1 is a negative regulator of p53. Cell. 2004;117:859–872. doi: 10.1016/j.cell.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szak ST, Mays D, Pietenpol JA. Kinetics of p53 binding to promoter sites in vivo. Mol Cell Biol. 2001;21:3375–3386. doi: 10.1128/MCB.21.10.3375-3386.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Qu LK, Zhang J, Wang W, Michaelson JS, Degenhardt YY, El-Deiry WS, Yang X. Critical role for Daxx in regulating Mdm2. Nat Cell Biol. 2006a;8:855–862. doi: 10.1038/ncb1442. [DOI] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006b;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–650. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thut CJ, Chen JL, Klemm R, Tjian R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science. 1995;267:100–104. doi: 10.1126/science.7809597. [DOI] [PubMed] [Google Scholar]
- Tian C, Xing G, Xie P, Lu K, Nie J, Wang J, Li L, Gao M, Zhang L, He F. KRAB-type zinc-finger protein Apak specifically regulates p53-dependent apoptosis. Nat Cell Biol. 2009 doi: 10.1038/ncb1864. in press. Published online April 19, 2009. [DOI] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, Klein C, Pan H, Kessler H, Pancoska P, Moll UM. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 2006;281:8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigiante G, Lu X. ASPP [corrected] and cancer. Nat Rev Cancer. 2006;6:217–226. doi: 10.1038/nrc1818. [DOI] [PubMed] [Google Scholar]
- Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007;26:102–112. doi: 10.1038/sj.emboj.7601469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Eaton NgE, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Wahl GM. Mouse bites dogma: how mouse models are changing our views of how P53 is regulated in vivo. Cell Death Differ. 2006;13:973–983. doi: 10.1038/sj.cdd.4401911. [DOI] [PubMed] [Google Scholar]
- Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell. 2006;23:251–263. doi: 10.1016/j.molcel.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Wang X, Taplick J, Geva N, Oren M. Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett. 2004;56:195–201. doi: 10.1016/S0014-5793(04)00168-1. [DOI] [PubMed] [Google Scholar]
- Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Wu Z, Earle J, Saito S, Anderson CW, Appella E, Xu Y. Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol. 2002;22:2441–2449. doi: 10.1128/MCB.22.8.2441-2449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, Safiran YJ, Oberoi P, Kenten JH, Phillips AC, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7:547–559. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- Yu GW, Rudiger S, Veprintsev D, Freund S, Fernandez-Fernandez MR, Fersht AR. The central region of HDM2 provides a second binding site for p53. Proc Natl Acad Sci USA. 2006;103:1227–1232. doi: 10.1073/pnas.0510343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]