

Abstract
Aberrant regulation of glycogen synthase kinase-3 (GSK-3) is implicated in Alzheimer’s disease (AD), but the mechanisms involved remain elusive. Our recent study shows that GSK-3 impairs lysosomal acidification and that inhibition of GSK-3 re-acidified lysosomes in brains of AD mice. This effect was accompanied by reductions in β-amyloid pathology and amelioration of cognitive deficits. Presenilin-1 (PS1) is an essential factor in lysosomal acidification. To determine whether the inhibition of GSK-3 restores lysosomal malfunction caused by dysfunctional PS1, we treated MEF cells deficient in presenilin proteins (MEF-PS1/2−/−) with a selective substrate competitive GSK-3 inhibitor, L803-mts. L803-mts enhanced the acidic lysosomal pool in MEF-PS1/2−/− cells and increased levels of activated cathepsin D in the lysosomes. We conclude that GSK-3 and PS1 operate via similar mechanisms to disrupt lysosomal acidification. Importantly, these data indicate that GSK-3 inhibitors have potential in treatment of conditions associated with defective PS1.
Keywords: Alzheimer’s disease, Aβ pathology, GSK-3, lysosome, presenilin-1
Lysosomes are the primary degradative components responsible for clearing intracellular waste products and damaged proteins, their proper activity is vital for the well-being of the cell.1 The enzymatic degradation that occurs in lysosomes is highly dependent on the lysosomal acidic pH, which is maintained by the vacuolar ATPase (v-ATPase) proton pumps. Impaired lysosomal activity was initially observed in heredity lysosome storage diseases, and recent studies have demonstrated a tight link between lysosomes and neurodegenerative diseases. Of particular interest is the role of lysosomes in Alzheimer’s disease (AD): A decline in lysosomal activity is observed in aging brains, and defects in lysosomal acidification are associated with typical AD pathology of fibrillogenic β amyloid (Aβ) deposits.2-5
Accumulation of Aβ plaques is a key hallmark in AD pathogenesis. The 40- or 42-residue Aβ peptides that make up the plaques are generated by sequential proteolysis of the amyloid precursor protein (APP) by β-secretase, βeta-site APP Cleaving Enzyme 1 (BACE1), and presenilin-dependent γ-secretase.6,7 Reducing the accumulation of Aβ deposition is thus believed to be a useful therapeutic strategy. Disruption in lysosomal acidification resulted in enhanced Aβ pathology and reduced cognitive ability in AD mouse models,2-5 giving rise to the hypothesis that restoring lysosomal acidity reverse AD symptoms.
Glycogen synthase kinase-3 (GSK-3) is an evolutionary conserved serine/threonine kinase expressed as two isozymes, GSK-3α and GSK-3β. GSK-3 is emerging an important drug target in AD therapy. Excessive phosphorylation of GSK-3 targets such as the microtubule-associated protein tau, collapsin response mediator proteins (CRMPs) and β-catenin is implicated in mechanisms contributing to AD pathogenesis.8-10 Indeed, treatment with GSK-3 inhibitors reverses AD symptoms in various animal models.11 An initial study connected GSK-3α isozyme with Aβ production via enhanced γ-secretase-mediated APP proteolysis.12 To gain further insights into the role of GSK-3 in Aβ pathology, we used the “5XFAD” mouse model. These mice co-express a total of five familial AD mutations in APP and presenilin-1 (PS1) and develop massive cerebral Aβ loads.13 We treated these mice nasally with L803-mts, a selective, substrate-competitive GSK-3 inhibitor developed in our laboratory. We found that treatment with L803-mts reduces Aβ pathology and ameliorates cognitive deficits.14 We also showed that L803-mts restores the lysosomal acidification that was severely impaired in the brains of the 5XFAD mice.14 This effect was independent of autophagy indicating that lysosomes play a major role in the catabolic disposal of Aβ loads under these conditions.
Recent studies implicated PS1 in controlling lysosomal acidification.15 We asked whether inhibition of GSK-3 can “repair” lysosomal malfunction caused by dysfunctional PS1. We treated MEF cells deficient in presenilin proteins (MEF-PS1/2−/−)16 with L803-mts. After the treatment cells were stained with LysoTracker Red, a dye that accumulates in acidified organelles, and imaged by confocal microscopy. L803-mts increased the number of acidified lysosomes and intensity of staining as compared with control untreated cells (Fig. 1). We next examined the levels of Cathepsin D (CatD), a principle lysosomal protease that is activated in the acidified lysosomal environment. Immunofluorescence analysis with anti-CatD antibody showed a low level, diffuse signal in the untreated cells. In contrast, L803-mts increased CatD signal (Fig. 1). To examine whether CatD was more active in L803-mts treated cells, cells were stained with pepstatin A BODIPY, which binds specifically to the active form of CatD. The BODIPY signal was enhanced by L803-mts, confirming that L803-mts restored lysosomal acidification in these cells. We conclude that GSK-3 and PS1 likely operate via similar mechanisms that impair lysosomal acidification, perhaps through disrupted glycosylation of v-ATPase V0a1 subunit; this glycosylation is critically important for v-ATPase assembly in the lysosome membrane.15 Another important conclusion from this study is that GSK-3 inhibition should provide benefit in treating conditions associated with defective PS1.
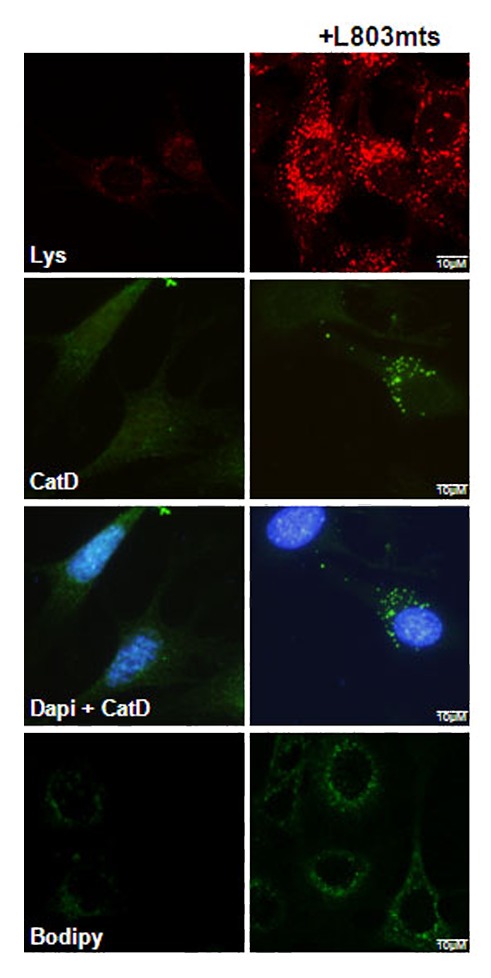
Figure 1. Inhibition of GSK-3 restores impaired lysosomal acidification caused by disrupted PS proteins. MEF-PS1/2−/− cells were treated with L803-mts (40 μM, 6 h) and screened by the following lysosomal markers: live-cell imaging of cells stained with Lysotracker-Red (Lys, top panel); fixed cells immunostained with CatD antibody (middle panels); and live-cell imaging of cells stained with pepstatin A BODIPY (bottom panel).
Additional work that was published in parallel to our publication demonstrated the role of GSK-3 in regulating Aβ pathology, but suggested different mechanisms that involved either reduction in β-site APP cleaving enzyme-1, BACE1, expression17 or enhancement in APP processing via lysosome biogenesis.18 Altogether, GSK-3 is clearly a prominent factor that contributes to accumulation of Aβ loads in the AD brain. The mechanisms involved are likely dependent on the cellular context including the levels and/or activities of additional factors that contribute to Aβ pathology such as APP, PS1 and lysosomes.
Glossary
Abbreviations:
- GSK-3
glycogen synthase kinase-3
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Aβ
beta amyloid
- PS1
presenilin-1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cib/article/25179
References
- 1.Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal. 2012;17:766–74. doi: 10.1089/ars.2011.4405. [DOI] [PubMed] [Google Scholar]
- 2.Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. 2011;7:788–9. doi: 10.4161/auto.7.7.15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler D, Hwang J, Estick C, Nishiyama A, Kumar SS, Baveghems C, et al. Protective effects of positive lysosomal modulation in Alzheimer’s disease transgenic mouse models. PLoS ONE. 2011;6:e20501. doi: 10.1371/journal.pone.0020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller-Steiner S, et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron. 2008;60:247–57. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuervo AM, Dice JF. When lysosomes get old. Exp Gerontol. 2000;35:119–31. doi: 10.1016/S0531-5565(00)00075-9. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 7.De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev. 2010;90:465–94. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- 8.Hernández F, Avila J. The role of glycogen synthase kinase 3 in the early stages of Alzheimers’ disease. FEBS Lett. 2008;582:3848–54. doi: 10.1016/j.febslet.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, et al. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008;172:786–98. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–95. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–9. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 13.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–40. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of GSK-3 Ameliorates beta-Amyloid(A-beta) Pathology and Restores Lysosomal Acidification and mTOR Activity in the Alzheimer's Disease Mouse Model. In vivo and In vitro Studies. J Biol Chem. 2013;288:1295–306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herreman A, Van Gassen G, Bentahir M, Nyabi O, Craessaerts K, Mueller U, et al. gamma-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J Cell Sci. 2003;116:1127–36. doi: 10.1242/jcs.00292. [DOI] [PubMed] [Google Scholar]
- 17.Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123:224–35. doi: 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-β precursor protein. Mol Cell Biol. 2012;32:4410–8. doi: 10.1128/MCB.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]