

Abstract
Autophagy plays a critical role in cellular quality control and is involved in removing damaged or excess organelles. Dysfunctional mitochondria are quickly cleared from the cell by autophagosomes before they can cause damage to the cell. Parkin is an E3 ubiquitin ligase that selectively ubiquitinates proteins on dysfunctional mitochondria, thereby marking those mitochondria for degradation by autophagosomes. In our recent study, we investigated the functional role of Parkin in the myocardium and discovered that Parkin is dispensable in the adult heart under normal conditions. Instead, our findings suggest that Parkin plays an important role in clearing damaged mitochondria in myocytes during stress. Here, we report that Parkin deficiency results in the accumulation of abnormal mitochondria in myocytes with age.
Keywords: mitochondria, autophagy, parkin, heart failure, aging
Autophagy plays an important role in cellular quality control as it degrades protein aggregates and dysfunctional organelles that can be harmful to the cell.1 In the heart, mitochondria are responsible for providing myocytes with energy in the form of ATP to sustain contraction. However, in response to stress, mitochondria can change to organelles that promote cell death, as they become producers of reactive oxygen species and release pro-apoptotic proteins such as cytochrome c. To prevent unnecessary death, a dysfunctional mitochondrion can be selectively removed from the cell through mitochondrial autophagy, or mitophagy.2 The specific targeting of mitochondria for autophagic degradation has recently been shown to involve a family of proteins found to be mutated in familial Parkinson disease (PD).3,4 One such protein, Parkin, is an E3 ubiquitin ligase that rapidly translocates to dysfunctional mitochondria where it promotes ubiquitination of mitochondrial proteins, which serves as a signal for mitophagy.5,6
We recently demonstrated that Parkin plays an important role in adapting to stress in the myocardium. We discovered that 12-week old Parkin-deficient mice (Parkin−/−) have normal cardiac function under baseline conditions.7 At this age, Parkin−/− mice also have normal cardiac mitochondrial function, although mitochondria appear smaller and clustered. However, in response to myocardial infarction (MI), these mice suffer exaggerated cardiac damage and increased mortality compared with wildtype (WT) mice. The increased severity of tissue damage in Parkin−/− mice is associated with rapid accumulation of swollen, dysfunctional mitochondria in the infarct border zone that is consistent with a defect in mitochondrial clearance. Taken together, these results suggest that a Parkin-independent, alternative pathway of mitophagy is present and may account for the normal appearance and function of mitochondria in 12-week-old Parkin−/− mice at baseline.7 This pathway may become overwhelmed following MI, allowing dysfunctional mitochondria to accumulate.
Continuous turnover of mitochondria via autophagy is important in maintaining a healthy population of mitochondria in cells. Impaired autophagy results in accumulation of dysfunctional mitochondria and is linked to aging and development of heart failure.8-10 In our study, we found that mitochondria in hearts of 3-month-old Parkin−/− mice were disorganized and often found in large clusters with many small, round mitochondria.7 Interestingly, the change in morphology did not affect mitochondrial function. Here, we used electron microscopy to examine mitochondria in hearts of 6-month-old WT and Parkin−/− mice. Similarly to the 3-month-old mice, mitochondria from aged Parkin−/− mice were disarrayed, smaller and clustered, but still maintained cristae density and appeared generally normal (Fig. 1). However, at this age, we also observed the appearance of abnormal mitochondria containing electron-dense macromolecules in the matrix in Parkin−/− hearts. These findings suggest that alternative pathways of mitochondrial clearance can compensate for loss of Parkin under normal conditions. Our findings also suggest that clearance occurs at a slower rate and therefore abnormal mitochondria start to accumulate with age. The reduced rate of clearance might also account for the accumulation of dysfunctional mitochondria in the heart after MI.7 It is not surprising that there are redundant pathways involved in mitochondrial clearance; this ensures that there is no disruption in the removal of dysfunctional and potentially harmful mitochondria in cells.
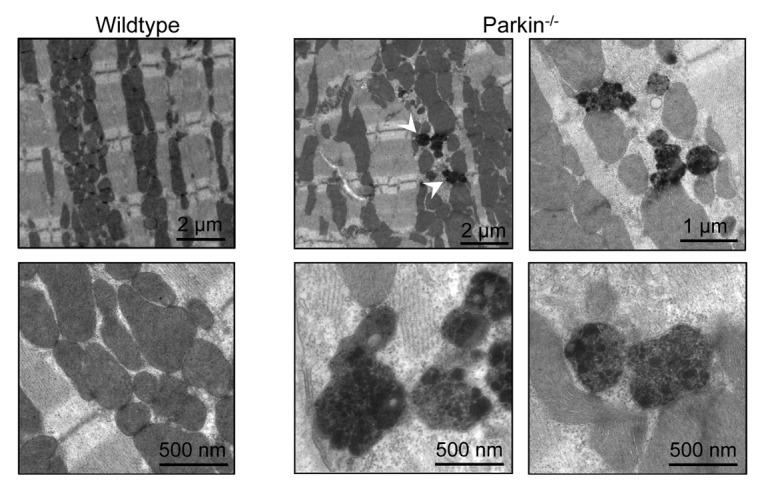
Figure 1. Transmission electron micrographs of heart sections from 6-month-old mice. Images are representative of > 15 photos taken from 2 animals per group. Arrowheads signify abnormally dense mitochondria.
The propensity for Parkin−/− mice to suffer from more severe infarcts during MI and the accumulation of damaged mitochondria over time may have serious implications for human patients with mutations in the PARK2 gene encoding Parkin. Aside from the obvious risk of developing early onset PD, these patients may be at a higher risk for fatal myocardial infarction, a risk which most likely increases with age as abnormal mitochondria accumulate in the myocytes. An early epidemiological study found a correlation between PD and increased risk of ischemic heart disease (IHD).11 PD patients have a greater than 2 fold risk of death from IHD and have significantly higher mortality than control patients. Additionally, mutations in the PARK2 gene may be present in as high as 18% of PD cases with onset before age 40,12 and may account for 50% of early-onset autosomal recessive PD cases.13 Moreover, although more than 100 disease-causing mutations in the PARK2 gene have been identified to date,14 the structure and function of cardiac mitochondria in PD patients have not been evaluated. Dysfunctional mitochondria have been identified in the brains15 and skeletal muscle of PD patients.16 Of greater relevance, impaired mitochondrial function has been found in leukocytes of patients with mutations in PARK2.17 Clearly, a more in-depth investigation is necessary to ascertain whether mutations in the PARK2 gene are associated with elevated mortality due to heart-related issues. This information could be critical in improving longevity and quality of life for Parkinson disease patients.
Material and Methods
Transmission electron microscopy
TEM was performed as previously described.7 Briefly, hearts were harvested from six-month-old mice, fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer, post-fixed in 1% osmium tetroxide, and then treated with 0.5% tannic acid, 1% sodium sulfate, cleared in 2-hydroxypropyl methacrylate and embedded in LX112 (Ladd Research). Sections were mounted on copper slot grids coated with parlodion and stained with uranyl acetate and lead citrate for examination on a Philips CM100 electron microscope (FEI).
Acknowledgments
This work was supported by NIH grants R01HL087023 and R01HL101217.
Disclosure of Potential Conflicts of Interests
No potential conflicts of interests were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cib/article/24511
References
- 1.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–21. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 4.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 5.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–26. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 9.Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 2009;1:425–37. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res. 2012;110:1125–38. doi: 10.1161/CIRCRESAHA.111.246108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-Shlomo Y, Marmot MG. Survival and cause of death in a cohort of patients with parkinsonism: possible clues to aetiology? J Neurol Neurosurg Psychiatry. 1995;58:293–9. doi: 10.1136/jnnp.58.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poorkaj P, Nutt JG, James D, Gancher S, Bird TD, Steinbart E, et al. parkin mutation analysis in clinic patients with early-onset Parkinson [corrected] disease. Am J Med Genet A. 2004;129A:44–50. doi: 10.1002/ajmg.a.30157. [DOI] [PubMed] [Google Scholar]
- 13.Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, et al. French Parkinson’s Disease Genetics Study Group. European Consortium on Genetic Susceptibility in Parkinson’s Disease Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–7. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 14.Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31:3038–62. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/S0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 16.Bindoff LA, Birch-Machin MA, Cartlidge NE, Parker WD, Jr., Turnbull DM. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J Neurol Sci. 1991;104:203–8. doi: 10.1016/0022-510X(91)90311-T. [DOI] [PubMed] [Google Scholar]
- 17.Müftüoglu M, Elibol B, Dalmizrak O, Ercan A, Kulaksiz G, Ogüs H, et al. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord. 2004;19:544–8. doi: 10.1002/mds.10695. [DOI] [PubMed] [Google Scholar]