

Abstract
The potential contribution of HLA-A alleles to viremic control in chronic HIV type 1 (HIV-1) infection has been relatively understudied compared with HLA-B. In these studies, we show that HLA-A*7401 is associated with favorable viremic control in extended southern African cohorts of >2100 C-clade–infected subjects. We present evidence that HLA-A*7401 operates an effect that is independent of HLA-B*5703, with which it is in linkage disequilibrium in some populations, to mediate lowered viremia. We describe a novel statistical approach to detecting additive effects between class I alleles in control of HIV-1 disease, highlighting improved viremic control in subjects with HLA-A*7401 combined with HLA-B*57. In common with HLA-B alleles that are associated with effective control of viremia, HLA-A*7401 presents highly targeted epitopes in several proteins, including Gag, Pol, Rev, and Nef, of which the Gag epitopes appear immunodominant. We identify eight novel putative HLA-A*7401–restricted epitopes, of which three have been defined to the optimal epitope. In common with HLA-B alleles linked with slow progression, viremic control through an HLA-A*7401–restricted response appears to be associated with the selection of escape mutants within Gag epitopes that reduce viral replicative capacity. These studies highlight the potentially important contribution of an HLA-A allele to immune control of HIV infection, which may have been concealed by a stronger effect mediated by an HLA-B allele with which it is in linkage disequilibrium. In addition, these studies identify a factor contributing to different HIV disease outcomes in individuals expressing HLA-B*5703.
Human leukocyte Ag class I genotype is a major determinant of HIV-1 viremic control and progression to AIDS (1–5). Much of this effect is mediated by the presentation of HIV-specific epitopes by different HLA class I molecules for recognition of virus-infected cells by CD8+ T cells (6–12). HLA-B is the class I locus with the strongest influence on median viral load in chronic infection (13), and HLA-B*5703 is the allele associated with most effective suppression of viremia in C-clade–infected subjects (6, 14–17). However, an effect on viremic control has also previously been identified in association with some HLA-A alleles (18–21), and with the HLA-Cw class I locus (3, 22, 23).
The starting point of these studies was the observation that HLA-A*7401 is associated with the lowest median HIV-1 viral load of all the 23 HLA-A alleles expressed at ≥0.5% phenotypic frequency in a large cohort of HIV-1–infected subjects from Durban, South Africa (3). Furthermore, in this C-clade–infected cohort, we noted that HLA-A*7401 is associated with the selection of escape mutations in Gag, Pol, and Nef (3, 5). These two strands of evidence both suggest that HLA-A*7401–restricted CD8+ T cell responses may contribute to disease control in chronic HIV-1 infection and are in keeping with recently published data documenting an association between HLA-A*7401 and disease control in Tanzania and Kenya (19, 24).
The role of HLA-A*7401 in viremic control in Durban is potentially confounded by its occurrence in linkage disequilibrium with HLA-B*5703 (23, 25). This raises the possibility that the apparent benefits to viremic control seen among subjects with HLA-A*7401 are, in fact, all mediated by a linked HLA-B*5703–restricted response. Alternatively, it is possible that the impact of HLA-A*7401 is independent of HLA-B*5703 but obscured by the stronger effects of the HLA-B allele. Indeed, although HLA-B*5703 is the allele most strongly associated with successful control of viremia in sub-Saharan Africa populations, considerable variation in viral set point is seen among subjects possessing this allele (23). We therefore hypothesized that some of the stratification in disease outcome among subjects with HLA-B*5703 might be accounted for by the presence or absence of HLA-A*7401 in the haplotype.
HLA-A*7401 alleles belong to the HLA-A19 serotype, which comprises HLA-A*29, -A*30, -A*31, -A*32, and -A*74, and has been included within the HLA-A*03 supertype (26, 27). Although HLA-A*7401 is prevalent in sub-Saharan African populations, occurring at a phenotypic frequency of ~10%, [http://www.allelefrequencies.net (28)], the peptide binding motif for HLA-A*7401 has yet to be defined.
The aims of this study were, therefore, as follows: first, to investigate the contribution of HLA-A*7401 to control of HIV-1 infection, both alone and in combination with HLA-B*5703 in southern African populations; and second, to investigate the means by which HLA-A*7401 might contribute to viremic control via the presentation of HLA-A*7401–restricted CD8+ T cell epitopes.
Materials and Methods
Study cohorts
We studied a total of 2126 treatment-naive, adult subjects with chronic HIV-1 C-clade infection from southern Africa. We recruited subjects via five cohorts as follows: 1) Durban, South Africa (n = 1218), as previously described (3, 13, 16, 29); 2) Bloemfontein, South Africa [n = 261; in this cohort, HLA types were ascertained only for subjects with CD4+ T cell counts <100 or >500 cells/mm3, as previously described (30)]; 3) Kimberley, South Africa (n = 31 postnatal mothers); 4) Gaborone, Botswana (n = 514 antenatal women) (10); and 5) southern African subjects attending outpatient HIV clinics in the Thames Valley area of the U.K. (n = 102) originating from Botswana, Malawi, South Africa, and Zimbabwe (n = 1, 11, 17, and 73, respectively). Ethics approval was given by University of KwaZulu-Natal Review Board and the Massachusetts General Hospital Review Board (Durban cohort); the University of the Free State Ethics Committee (Kimberley and Bloemfontein cohorts); the Office of Human Research Administration, Harvard School of Public Health and the Health Research Development Committee, Botswana Ministry of Health (Gaborone cohort); and the Oxford Research Ethics Committee (Durban, Kimberley, and Thames Valley cohorts). Study subjects from all cohorts gave written informed consent for their participation. Viral load in chronic infection was measured using the Roche Amplicor version 1.5 assay; CD4+ T cell counts were measured by flow cytometry (data for each cohort are shown in Table I).
Table I.
Summary of 2126 C-clade HIV-1–infected study subjects from five southern African study cohorts
| Cohort Location | No. of Subjects | Number (%) with HLA-A*7401a | Viral Load (RNA Copies/ml Plasma)
|
CD4 T Cell Count (Cells/mm3)
|
||||
|---|---|---|---|---|---|---|---|---|
| Number | Median | IQR | Number | Median | IQR | |||
| Durban, South Africa | 1218 | 135 (11.1) | 1218 | 38,200 | 7315–154,250 | 1159 | 376 | 239–519 |
| Bloemfontein, South Africab | 261 | 21 (8.0) | 163 | 84,000 | 13,000–250,000 | 261 | 76 | 32–548 |
| Kimberley, South Africa | 31 | 1 (3.2) | 14 | 47,000 | 8800–295,000 | 29 | 326 | 274–477 |
| Gaborone, Botswana | 514 | 46 (8.9) | 471 | 19,100 | 3920–78,200 | 415 | 342 | 220–476 |
| Thames Valley, southern Africa | 102 | 13 (12.7) | 99 | 5090 | 942–21,412 | 98 | 445 | 326–562 |
| All Southern Africa | 2126 | 216 (10.2) | 1965 | 29,500 | 5715–133,500 | 1962 | 360 | 208–520 |
The 4-digit HLA type was determined in 181/216 subjects (84%); in all 181 of these (100%), the 4-digit type was HLA-A*7401. In the remaining 35 cases typed to 2 digits, the 4-digit type was undetermined.
Bloemfontein data are for 261 HLA-typed subjects with CD4+ T cell count < 100 or > 500 cells/mm3. In an extended Bloemfontein cohort (including subjects without HLA types), median CD4+ cell count was 229 cells/mm3, with interquartile range (IQR) of 122–366 cells/mm3 (n = 885), and median viral load was 76,000, with IQR of 20,000–21,0000 cells/mm3 (n = 500).
HLA typing
High-resolution HLA typing was performed from genomic DNA by single-stranded conformation polymorphism PCR. As in previous studies (3, 5, 23), for certain alleles in the Durban cohort, including HLA-A*74, the four-digit type was not resolved in a minority of study subjects. In the Durban cohort, 100/138 (72%) subjects with HLA-A*74 were typed to 4 digits, and 100/100 (100%) of these subjects typed to 4 digits had HLA-A*7401. Of 81 HLA-A*74+ subjects from the Thames Valley, Botswana, Kimberley, and Bloemfontein cohorts typed to 4-digits, again 100% had HLA-A*7401 confirmed by high-resolution typing. To test the generality of these findings, we also examined a random sample of 90 individuals of African descent with HLA-A*74 recently typed by the National Institutes of Health, and found that >97% had HLA-A*7401 (M. Carrington, unpublished observations). In this study, we have therefore classified all HLA-A*74+ subjects together as HLA-A*7401.
Amplification and sequencing of proviral DNA
Gag sequences were generated from genomic DNA extracted from PBMCs, amplified by nested PCR to obtain population sequences, as previously described (16, 31). Sequencing for the Durban and Bloemfontein cohorts was undertaken using the Big Dye Ready Reaction Terminator Mix (V3) (Applied Biosystems, U.K.) and manually aligned using Se_Al software, as previously described (31, 32). Sequencing of subjects from the Thames Valley, Botswana, and Kimberley cohorts was performed by Macrogen (South Korea). Sequences were analyzed using Sequencher v4.8 (Gene Codes Corporation). Online sequences are available at GenBank, FJ198407-FJ199088 (Durban) and FJ497801-FJ497950 (Botswana).
HLA-A*7401 motif prediction and confirmation; HLA–peptide binding studies
Amino acid sequences for HLA-A*7401 and related HLA-A alleles were downloaded from the International Immunogenetics Project (http://www.ebi.ac.uk/imgt/hla/). HLA-A*7401 optimal epitopes were initially predicted using motif inference based on the comparison of HLA-A*7401 with HLA-A alleles that share sequence homology for the B and F pockets of the peptide binding groove (33). These two pockets determine the preferred residues bound at the “anchor” positions of the epitope (corresponding to position two [P2] and the carboxyl-terminal position [PC]) (34). The validity of the predicted HLA-A*7401 peptide binding motif was tested by two methods. Optimal epitopes were first determined using IFN-γ ELISPOT assays to test recognition of peptides that corresponded to the predicted optimal epitope, and of peptides differing by one amino acid in length from each predicted optimal. We used Los Alamos HIV databases (epitopes “A-list”; http://www.hiv.lanl.gov) to search for previously published HLA-A*7401–restricted epitopes.
Binding of these predicted optimal peptides to HLA-A*7401 was tested in HLA–peptide–binding studies, undertaken using a luminescent oxygen channeling immunoassay (LOCI), as previously described (35).
IFN-γ ELISPOT assays
We tested ex vivo PBMCs from 1010 study subjects from the Durban cohort against a panel of 410 overlapping peptides (OLPs) spanning the entire C-clade HIV-1 proteome to screen for IFN-γ ELISOIT responses, as previously described (29). A total of 119 of these subjects had HLA-A*7401 (91 of these 119 [76%] were confirmed to be HLA-A*7401 at 4-digit resolution; the remainder were typed to only 2 digits). Fisher’s exact test was performed to identify responses to OLPs that were significantly associated with expression of HLA-A*7401. Analysis was also repeated in the absence of the 28 subjects for whom HLA-A*74 was typed to only 2-digit resolution to remove any potential bias from subjects not confirmed to have HLA-A*7401 as the high-resolution type. We tested putative optimal epitopes by ELISPOT using ex vivo PBMCs from HLA-A*7401+ subjects recruited through the Thames Valley and Durban cohorts.
Cell staining and flow cytometry
Cell staining was undertaken from cryopreserved PBMCs using anti–CD3-Pacific orange (Invitrogen), anti–CD8-Alexa Fluor 700 (BD Biosciences), and HLA-A*7401-RR9 tetramer conjugated to PE. Dead cells were gated out using “live/dead” viability kit (Invitrogen).
Statistical analysis
We used Fisher’s exact test to screen for LD between HLA class I alleles, using the online tool available at http://www.hiv.lanl.gov/content/immunology/hla/hla_linkage.html, corrected for multiple comparisons using a Bonferroni approach. To identify MHC class I haplotypes, we used PyPop software [Python for Population Genomics, http://www.pypop.org (36)] (Supplemental Table I). Other statistical analysis was undertaken using Prism GraphPad software v. 5.0a.
To assess whether any possible pairs of HLA class I alleles might perform better than a single allele in mediating disease control in chronic infection, we devised a novel statistical test for additive effects using pooled data from a total of 2126 C-clade–infected adult subjects from southern Africa. Each HLA combination was tested to see whether an additive model for two HLA alleles together, irrespective of linkage disequilibrium, performed better in predicting disease control than a model that considered only the most predictive of each single HLA allele. For the purposes of this test, each allele was collapsed to a two-digit resolution only, apart from exceptions in which it is known that the four-digit types cross supertype boundaries or have a strong bearing on outcome (HLA-A*68XX, HLA-B*15XX, and HLA-B*58XX) (13, 27).
For disease control based on the log of the absolute viral load and the log of the CD4+ T cell count, we used the log likelihood of linear-regression–based models to look for additive effects. In particular, we defined the null model for a given HLA pair as the one of the two models, or (defined next), that had the maximum likelihood under a maximum likelihood parameter setting. Intuitively, it is a model where at most only one HLA at a time can have an effect within an individual patient; the HLA chosen is the one with the larger overall effect in such a model. Thus, the null model was either , where X′ = 0 for people with Y = 1, and otherwise X′ = X, or , where Y′ = 0 for people with X = 1, and otherwise Y′ = Y. The alternative model used is given by p1(phen| X, Y) = normal(w0 + wXX + wyY, σ8).
The variable “phen” is, for example, the viral load; X and Y are binary variables representing two HLA alleles; wX and wY are parameters in the model for the effect of each allele; σ8 is the variance parameter of the normal distribution; and w0 is an offset parameter. Parameters were fit independently for each model. The test statistic for an HLA pair was the difference in log likelihood between these null and alternative models after fitting each by maximum likelihood. The p values were obtained by 50,000 permutations of one HLA allele in the test. In addition, any pair of alleles for which one of the alleles did not have a univariate correlation in the direction of control (as defined by the phenotype of interest) was assigned a test statistic of zero. Note that two alleles in perfect linkage disequilibrium (LD) (or anti-LD) could not come up as having an additive effect with this test statistic. For binary outcomes (disease control based on criteria of viral load ≤ 2000 RNA copies/ml plasma and CD4+ T cell count > 250 cells/mm3), we used an analogous test based on logistic regression.
To account for possible differences between cohorts that might cause artifact either by enhancing or obscuring additive effects between HLA alleles (i.e., confounding), we also corrected for geographical origin of the subject by adding a set of binary cohort covariates to each model (one for recruitment in each of Durban, Kimberley, Bloemfontein, or Gaborone; we removed Thames Valley subjects who originate from a wide variety of locations). An LRT test showed these cohort covariates to be highly significant (e.g., p = 2 × 10−59) in predicting the log CD4+ T cell count, so these covariates were used in all models. Note that all HLA pairs coming out as significant in these tests had parameter weights in the direction of control in the alternative model, indicating that better predictive power was obtained because both alleles were contributing to control rather than one negating the effect of the other.
Results
HLA-A*7401 is common in African populations and is found in linkage disequilibrium with HLA-B*5703 in South Africa
HLA-A*74 was found in 216 subjects from a total of 2126 southern African subjects (phenotypic frequency, 10.2%) (Table I). These were confirmed to be HLA-A*7401 in 181/216 cases (84%); the remaining cases were typed only to 2-digit resolution as HLA-A*74. Thus, no four-digit HLA-A*74 allele other than HLA-A*7401 was identified in any of our cohorts.
We confirmed LD between HLA-A*7401 and HLA-B*5703 among South African subjects from Durban, Kimberley, and Bloemfontein (n = 1510, p = 1.1 × 10−9, Fisher’s exact test) (Fig. 1A). In the epidemiologically unlinked study population of adults in Botswana (n = 514), we did not identify statistically significant LD between these two alleles (Fig. 1A, 1B). The apparent lack of LD between HLA-A*7401 and HLA-B*5703 in Botswana does not appear to be attributable to a smaller sample size than our South African cohorts, because in the Botswana cohort, only 1/46 (2.2%) HLA-A*7401+ subjects also carried HLA-B*5703, whereas in the South African cohorts, 25/157 (15.9%) HLA-A*7401+ subjects also carried HLA-B*5703. The HLA-A*74-B*57 haplotype has previously been identified in African Americans [http://www.allelefrequencies.net (28); both alleles resolved to two digits only in this source (3)], but to our knowledge has not been reported in other African populations to date.
FIGURE 1.
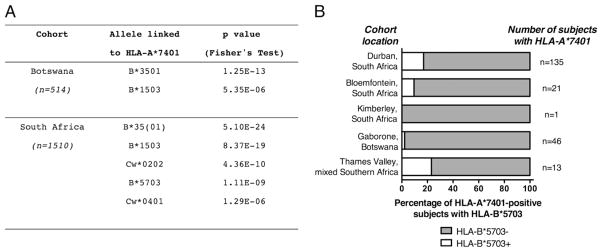
LD between HLA-A*7401 and other class I alleles in southern African populations. A, All HLA-B and HLA-Cw alleles that are significantly linked to HLA-A*7401 in a pooled cohort from South Africa (Durban, Bloemfontein, and Kimberley cohorts, n = 1510) and in Botswana (n = 514) are shown. The p values were calculated by Fisher’s exact test and corrected for multiple comparisons using the Bonferroni approach, using the online tool at: http://www.hiv.lanl.gov/content/immunology/hla/hla_linkage.html. No significant linkage associations were identified for HLA-A*7401 among Zimbabweans recruited via the Thames Valley cohort (n = 73, data not shown). All subjects with HLA-B*35 are pooled together as HLA-B*35(01). B, Percentage of HLA-A*7401+ subjects with HLA-B*5703 in each of five southern African cohorts. Statistically significant LD between these alleles is present only in the Durban cohort.
To confirm the haplotypes in which HLA-A*7401 is transmitted, we analyzed HLA data for Durban, Bloemfontein, and Gaborone using PyPop software [http://www.pypop.org (36)] (Supplemental Table I). In keeping with our analysis of LD (Fig. 1), this demonstrates that HLA-A*7401 is transmitted on a haplotype with HLA-B*1503, HLA-B*3501, and HLA-B*5703 in South Africa. The association of HLA-A*7401 with HLA-B*1503 and HLA-B*3501 is also seen in Botswana, but in this population, there is no HLA-A*7401-B*5703 haplotype. In addition, these data show a weaker haplotype of HLA-A*7401-B*4201 in all three cohorts, but the LD between HLA-A*7401 and this allele was not statistically significant.
HLA-A*7401 is statistically associated with lowered viral load in chronic C-clade HIV-1 infection
To assess the relation between HLA-A*7401 and viral load in chronic infection, we compared subjects with and without HLA-A*7401 in four different cohorts: Durban, Gaborone, Bloemfontein, and the Thames Valley cohorts (excluding Kimberley because of small numbers). In each case, HLA-A*7401 was associated with a reduction in viral load in chronic infection [p = 0.0007 (Fig. 2A), p = 0.02, p = 0.2, and p = 0.05, respectively, Mann–Whitney U test]. The trend toward lowered viremia in association with HLA-A*7401 in Bloemfontein may not have reached statistical significance at least in part because of the small number of HLA-A*7401 subjects in Bloemfontein (n = 8). In all southern African subjects, HLA-A*7401 was strongly associated with lowered viremia (median viral set point, 14600 versus 32450 RNA copies/ml in HLA-A*7401+ versus HLA-A*7401−; n = 1965 subjects with viral load data available; p < 0.0001, Mann–Whitney U test) (Fig. 2B).
FIGURE 2.

Viral load (VL) and CD4+ T cell count in the presence and absence of HLA-A*7401 in adults with chronic HIV-1 C-clade infection in southern African cohorts. A, VL in Durban, South Africa. B, VL in pooled southern Africa cohort (Durban, South Africa; Gaborone, Botswana; Thames Valley subjects from southern Africa [Botswana, Malawi, South Africa, and Zimbabwe]). C, CD4+ T cell count in Gaborone, Botswana. D, CD4+ T cell count in pooled southern Africa cohort (as above). Median VL (RNA copies/ml plasma) or CD4+ T cell count (cells/mm3) for each group is stated. Boxes show median, 25th and 75th percentile; whiskers 10th–90th percentile. The p values by Mann–Whitney U test.
In subjects from Gaborone (where HLA-A*7401 is not in linkage disequilibrium with HLA-B*5703), CD4+ T cell counts were statistically higher in subjects with HLA-A*7401 (p = 0.0005, Mann–Whitney U test) (Fig. 2C). This illustrates that HLA-A*7401 may mediate a protective effect that is independent of linkage disequilibrium with HLA-B*5703. Associations between absolute CD4+ T cell count and expression of HLA-A*7401 did not reach statistical significance in the Durban, Bloemfontein, or Thames Valley cohorts (data not shown). However, in pooled data from all subjects, there was a significant association between the presence of HLA-A*7401 and higher CD4+ T cell count (median CD4+ cell count, 409 versus 350 in HLA-A*7401+ versus HLA-A*7401−; n = 1962 subjects with CD4+ count available; p = 0.001, Mann–Whitney U test) (Fig. 2D).
HLA-A*7401 and HLA-B*5703 have an additive effect in lowering viral load in chronic C-clade HIV-1 infection
The relation between the presence of HLA-A*7401 and improved disease control of HIV-1 may be confounded by haplotype, whereby the observed effect of HLA-A*7401 on viral load arises only as a consequence of linkage with favorable alleles, in particular, with HLA-B*5703 in South African subjects (Fig. 1A). To mitigate this haplotype effect, we next investigated the impact of HLA-A*7401 on viral load after removing subjects with HLA-B*5703 from the analysis. Among subjects with HLA-A*7401, there remained an association with lowered viremia in the absence of HLA-B*5703 (p = 0.002 in the pooled cohort, p = 0.09 in the Durban cohort, Mann–Whitney U test) (Fig. 3A, 3B). Strikingly, however, the combination of HLA-A*7401 and HLA-B*5703 resulted in significantly lower viral loads than in the presence of either allele alone, suggesting an additive effect of these alleles in mediating control of disease (Fig. 3A, 3B), as seen previously (23). Additional evidence for the independent role of HLA-A*7401 in lowering viremia, irrespective of the presence of HLA-B*5703, comes from the Botswana cohort in which there is no LD between these alleles (Fig. 1), but HLA-A*7401 is still associated with favorable outcomes (see earlier and Fig. 2C).
FIGURE 3.

Effect of combinations of HLA-A*7401 and HLA-B*5703 on viral load. A and B, Viral load (RNA copies/ml plasma) of C-clade–infected adult study subjects from all study subjects (A) and Durban (B), in the presence and absence of HLA-B*5703 and HLA-A*7401. Boxes show median, 25th and 75th percentile; whiskers 10th–90th percentile. The p values were by Mann–Whitney U test. C and D, Subjects with HLA-B*5703 ranked by viral load and divided by quartiles into four interquartile (IQ) pools for all study subjects (n = 97) (C) and Durban (n = 55) (D). The proportion of each of the four subgroups with HLA-A*7401 is shown, demonstrating that HLA-A*7401 is enriched in the IQ pool with the lowest viral loads. The p values were by Fisher’s exact test.
As HLA-A*7401 is also transmitted in strong LD with HLA-B*1503 and HLA-B*3501 in both Botswana and South Africa (Fig. 1A), we also sought to exclude either of these alleles as the driving force behind the apparent benefit of HLA-A*7401. There was no relation between HLA-B*1503 or HLA-B*3501 and lowered viremia in this cohort (data not shown); indeed, in Durban, HLA-B*1503 was actually statistically associated with higher viral loads (p = 0.03, Mann–Whitney U test; data not shown). The overall relation between HLA-A*7401 and lowered viral load is, therefore, not likely to be attributable to linkage with either of these alleles.
To further investigate the possible additive effect of HLA-A*7401 on disease control in subjects with HLA-B*5703, we ranked HLA-B*5703+ subjects from the whole pooled southern African cohort (n = 97) and from Durban (n = 55) according to viral load. We found significant enrichment of HLA-A*7401 in subjects with viral loads less than the median in each case (p = 0.015 in the whole cohort, p = 0.003 in Durban, Fisher’s exact test). Division of these HLA-B*5703+ subjects into four pools by viral load quartiles confirmed enrichment of HLA-A*7401 in subjects with the lowest viral loads (Fig. 3C, 3D). This phenomenon is not associated with the presence of other favorable HLA-B alleles, such as HLA-B*5702, -B*5801, or -B*8101, in subjects with the lowest viral loads (data not shown), suggesting that HLA-A*7401 may indeed be responsible for mediating the effect.
To investigate whether the beneficial effect of HLA-A*7401 occurs significantly in combination with HLA alleles other than HLA-B*5703, we undertook a computational analysis of all 2126 southern African subjects. Using this newly developed test, we sought evidence for additive effects of any combination of two HLA alleles (collapsed to two-digit resolution), irrespective of LD in improving disease control compared with the effect of any one allele alone. After correction for cohort location, we identified statistically significant additive effects between HLA-A*74 and two favorable HLA-B alleles, HLA-B*57 and HLA-B*81, in mediating control of viral load (Table II). No significant effect of HLA-A*74 was determined in conjunction with any other allele in maintenance of CD4+ T cell count. As discussed earlier, HLA-A*7401 is the only four-digit HLA-A*74 allele detected in these populations, so these results are applicable specifically to the effect of HLA-A*7401.
Table II.
Additive effects between HLA-A*74 and other HLA class I alleles in mediating good HIV-1 disease control
| Criterion for Disease Control | HLA-1 | HLA-2 | p Value | q Value |
|---|---|---|---|---|
| Absolute viral load | A*74 | B*57 | 2.00E-05 | 0.023 |
| A*74 | B*81 | 2.00E-05 | 0.023 | |
| Viral load < 2000 copies/ml | A*74 | B*57 | 2.00E-05 | 0.023 |
| A*74 | B*81 | 2.00E-05 | 0.023 | |
| Absolute CD4+ T cell count | A*74 | NS | ||
| CD4 T cell count > 250 cells/mm3 | A*74 | NS |
Analysis of HLA type, viral load (n = 1972), and CD4 count (n = 1871) in adults with chronic HIV-1 infection from pooled subjects from southern Africa (South Africa, Botswana, Zimbabwe, and Malawi). All associations between HLA-A*74 and any other allele (q < 0.2) are shown.
NS, not significant.
Prediction of the HLA-A*7401 binding motif
Having established the association between expression of HLA-A*7401 and improved disease control in HIV-1, we next investigated the likely mechanism for this effect. To seek evidence of CD8+ T cell epitopes restricted by this allele, we predicted the peptide binding motif for HLA-A*7401 from the residues that form the B and F pockets into which peptide residues at epitope positions two and PC would bind. This approach has previously been validated for other HLA alleles prevalent in sub-Saharan African populations, such as HLA-B*8101 and HLA-Cw*1801, for which the peptide binding motif has not been formally defined by sequencing of eluted peptides (33).
The amino acid sequence determining the B pocket of the peptide binding groove of HLA-A*7401 allele is identical to that of HLA-A*3201 and closely related to that of HLA-A*0101 (37) (Table III, top half). HLA-A*0101 preferentially binds epitopes bearing a small aliphatic residue, such as Thr/Ser/Ile/Met, at position 2 (33, 37, 38), but differs from the HLA-A*7401 allele by expressing a Met residue at position 67 of the molecule. In HLA-A*7401, the smaller m.w. Val residue at position 67 may provide a more capacious B pocket able to accommodate somewhat larger aliphatic residues. At the F pocket (where the C-terminal position [PC] of the epitope is accommodated), HLA-A*7401 is identical to HLA-A*3301, -A*6601, and -A*6801 (Table III, bottom half), predicting preferential binding of the basic residues Arg or Lys (27, 33, 39).
Table III.
Sequences of HLA-A*7401 and related HLA-A alleles at the B and F pockets of the peptide binding groove and predicted residues bound at these positions
| Residues Determining B Pocket
|
(Predicted) Residue at P2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 9 | 24 | 25 | 34 | 45 | 63 | 66 | 67 | 70 | 99 | ||
| HLA-A consensus | Y | F | A | V | V | M | E | N | V | H | Y | |
| HLA-A*0101 | — | — | — | — | — | — | — | — | M | — | — | T/S/I/M |
| HLA-A*3201 | — | — | — | — | — | — | — | — | — | — | — | (I/M) |
| HLA-A*7401 | — | — | — | — | — | — | — | — | — | — | — | (L/I/M/Q) |
| Residues Determining F Pocket
|
(Predicted) Residue at PC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 77 | 80 | 81 | 84 | 95 | 116 | 123 | 143 | 146 | 147 | ||
| HLA-A consensus | D | T | L | Y | I | D | Y | T | K | W | |
| HLA-A*3303 | — | — | — | — | — | — | — | — | — | — | R |
| HLA-A*6601 | — | — | — | — | — | — | — | — | — | — | R/K |
| HLA-A*6801 | — | — | — | — | — | — | — | — | — | — | R/K |
| HLA-A*7401 | — | — | — | — | — | — | — | — | — | — | (R/K) |
Identification of putative HLA-A*7401-restricted epitopes in HIV-1
Having predicted a peptide binding motif for HLA-A*7401, we identified regions of the HIV-1 proteome-containing potential HLA-A*7401–restricted epitopes using two approaches. First, we screened IFN-γ ELISPOT responses from 1010 Durban subjects. We identified two statistically significant IFN-γ responses made by 119 subjects with HLA-A*7401: these were to OLP-19 in p24 Gag (p = 1.7 × 10−4) and OLP-100 in Rev (p = 1.8 × 10−5; Fisher’s exact test) (Fig. 4, Table IV). Even after exclusion of the minority of subjects with HLA-A*74 typed to only two digits (n = 28), both of these associations remained statistically significant (p = 2.5 × 10−4 and p = 1.8 × 10−5, respectively). To define the optimal epitope within each of these OLPs, we identified a putative HLA-A*7401–restricted epitope based on the earlier predicted binding motif for HLA-A*7401 (L/I/M/Q at P2 and R/K at PC) (Table III). Within OLP-19 (IVQNLQGQMVHQAISPR), the peptides GR11 (GQMVHQAISPR, p24-Gag 8–18) and QR10 (QMVHQAISPR, p24-Gag 9–18) fitted this motif. Within the Rev 17mer OLP-100 (RWRARQRQIHSISERIL), two putative overlapping HLA-A*7401–binding peptides were identified: Rev-RR9 (RQIHSISER, Rev 50–58) and Rev-RR11 (RQRQIHSISER, Rev 48–58).
FIGURE 4.

Proportion of subjects with and without HLA-A*7401 making IFN-γ ELISPOT responses to HIV-1 C-clade OLPs. IFN-γ ELI-SPOT responses to OLP-19 and OLP-100 are significantly enriched among subjects with HLA-A*7401 (Durban cohort; n = 1010; p values by Fisher’s exact test). Subjects with HLA-B*1510 are removed from the analysis of responses to OLP-100 to avoid potential confounding because of the presence of an overlapping HLA-B*1510 epitope IL-9 (IHSISERIL, Rev 52–60) (29, 56), accounting for higher than expected rate of responses in the HLA-A*7401− population (method as previously described [13]). n = number making response/total number of subjects.
Table IV.
Putative HLA-A*7401–restricted epitopes predicted using motif inference (32) combined with analysis of IFN-γ ELISPOT assays
| Protein | OLP Number | C-clade OLP Sequence | p Valuea | Predicted HLA-A*7401 Epitope
|
||
|---|---|---|---|---|---|---|
| P2↓ | C Terminus ↓ | Epitope Designation | ||||
| p24-Gag | 19 | I V Q N L Q G Q M V H Q A I S P R | 1.7 × 10−4 | Q M V H Q A I S P | R | (Gag-QR10) |
| G Q M V H Q A I S P | R | (Gag-GR11) | ||||
| Rev | 100b | R W R A R Q R Q I H S I S E R I L | 1.8 × 10−5 | R Q I H S I S E | R | (Rev-RR9) |
| R Q R Q I H S I S E | R | (Rev-RR11) | ||||
Two C-clade 17-mer OLPs significantly associated with IFN-γ ELISPOT responses in subjects expressing HLA-A*7401 are shown (Durban and Thames Valley cohorts, as shown in Fig. 4; p values by Fisher’s exact test). Arrows indicate the anchor positions at which the optimal binds the class I allele (position 2 and C terminus); these residues are shown in bold and underlined.
The p values both remain statistically significant if analysis repeated after removal of minority of subjects with HLA-A*74 typed to only two-digit resolution: OLP-19, p = 2.5 × 10−4; OLP-100, p = 1.8 × 10−5.
Responses to OLP-100 are corrected by removal of subjects with HLA-B*1510 from analysis, because of presence of HLA-B*1510 epitope IL9 (IHSISERIL, Rev 52–60; ).
The second approach we used to identify HIV-specific HLA-A*7401–restricted epitopes involved reference to previously determined HLA-A*7401–associated HIV amino acid polymorphisms within Gag, Pol, and Nef population sequences from the Durban cohort (3). Six sites of HIV polymorphism associated with HLA-A*7401 were identified (3, 40) (Table V). Based on the predicted HLA-A*7401 binding motif, we used these sites of point mutation to identify further putative HLA-A*7401–restricted epitopes, p17-KR9 (p17 12–20, KLDKWEKIR), Nef-SR9 (SQARRHLAR, Nef 188–196), Protease-NR10 (NLAFPQGEAR, Pol 6–14), RT-QR9 (QIYPGIKVR, RT 269–277) and the overlapping RT-SR10 (SQIYPGIKVR, RT 268–277), and RT-SK9 (SLTETTNQK, RT 467–475) (Table V). None of the corresponding OLPs (OLPs 2, 91, 145, 202, 227/228) had significant IFN-γ ELISPOT responses associated with the expression of HLA-A*7401 (data not shown), possibly because of loss of the CD8+ T cell response after early selection of escape. Of the ELISPOT responses significantly associated with HLA-A*7401, the Gag-GR11 response appears immunodominant, occurring in >10% of subjects with HLA-A*7401 (Fig. 4).
Table V.
Putative HLA-A*7401–restricted epitopes predicted using motif inference (32) combined with analysis of HLA-A*7401 associations with sequence polymorphisms
| Protein | HXB2 Position of Polymorphism | Durban Consensus Sequencea
|
Reversion | p Value | q Value | Predicted HLA-A*7401 Epitope
|
||
|---|---|---|---|---|---|---|---|---|
| A*7401 Polymorphism ▼ | P2 | C Terminus | Epitope Designation | |||||
| p17 Gag | 12 | G A R A S I L R G E K L D K W E K I R L R | R | 2.71E-14 | 0.00 | S I L R G E K L D | K | (Gag-SK10) |
| p17 Gag | 20 | G E K L D K W E K I R L R P G G K K H Y M | R | 6.47E-10 | 0.00 | K L D K W E K I | R | (Gag-KR9) |
| Protease | 9 | F F R E N L A F P Q G E A R E F P S E | 4.83E-05 | 0.02 | N L A F P Q G E A | R | (Prot-NR10) | |
| RT | 277 | A S Q I Y P G I K V R Q L C K L L R G A K | 3.13E-04 | 0.13 | Q I Y P G I K V S Q I Y P G I K V |
R R |
(RT-QR9) (RT-SR10) |
|
| RT | 476 | I V S L T E T T N Q K T E L Q A I Q L A L | 6.03E-04 | 0.15 | S L T E T T N Q | K | (RT-SK9) | |
| Nef | 192 | K W K F D S S L A R R H L A R E L H P E Y | R | 4.95E-05 | 0.00 | S L A R R H L A | R | (Nef-SR9) |
Six sites of HLA-A*7401–associated polymorphism identified from lineage-corrected analysis of sequences from a total of 710 Durban subjects (q < 0.2), as previously published (3). The position of the polymorphism is marked ▼, with consensus sequence 10 aa upstream and downstream of the site of polymorphism. Arrows indicate anchor positions (shown in bold and underlined) as in Table IV. R indicates polymorphisms that are predicted to revert to wild-type after transmission to an HLA-mismatched recipient [methods as previously described (17, 57)].
Sequences based on Durban consensus sequence from Gag (n = 446) and Nef (n = 436) subjects.
Confirmation of optimal HLA-A*7401 epitopes in HIV-1 Gag, RT, and Rev
To confirm the optimal epitope, we used IFN-γ ELISPOT assays to quantify CD8+ T cell responses to the optimal peptide and variant truncations of the peptide in individual subjects selected on the basis that they made a response to the relevant OLP. Using this method, we acquired data to support the optimal epitope in three of our predicted HLA-A*7401–restricted epitopes: p24-Gag-GR11 (p24 Gag 8–18, GQMVHQAISPR), RT-QR9 (RT 269–277, QIYPGIKVR), and Rev-RR9 (Rev 50–58, RQIHSISER) (Fig. 5A–C, respectively). In the case of Rev-RR9, we also synthesized the peptide–MHC class I tetramer to confirm the HLA restriction of the response (Fig. 5D).
FIGURE 5.

In vitro data to support three new HLA-A*7401–restricted epitopes: Gag-GR11, RT-QR9, and Rev-RR9. A, Magnitude of IFN-γ ELISPOT responses to Gag-GR11 (GQMVHQAISPR), the alternative epitope QR10 (QMVHQAISPR), and other peptide truncations in a chronically infected adult subject from South Africa (subject ID SK362: HLA-A*0301/A*7401/B*1503/B*1510/Cw*0210/Cw*0401). B, Magnitude of IFN-γ ELISPOT responses to RT-QR9 (QIYPGIKVR) and peptide truncations in a chronically infected adult subject from Zimbabwe (subject ID H005: HLA-A*7401/A*3004/B*3501/B*5802/Cw*0401/Cw*0602). C, Magnitude of IFN-γ ELISPOT responses to Rev-RR9 (RQIHSISER), the alternative epitope RR11 (RQRQIHSISER), and the OLP containing these epitopes (OLP-100) in a chronically infected adult subject from Uganda (subject ID R070: HLA-A*0201/A*7401/B*4016/B*4901/Cw*0701/Cw*0802). D, FACS plot gated on live CD3+ population of PBMCs from subject R070, stained with MHC class I tetramer HLA-A*7401-Rev-RR9 conjugated to PE fluorochrome, showing a tetramer-positive population of CD3+ CD8+ T lymphocytes.
We also used LOCI (35) to confirm binding of HLA-A*7401 to optimal peptides Gag-GR11, RT-QR9, and Rev-RR9 (Fig. 6). These binding studies also add to the evidence for two other putative HLA-A*7401–restricted epitopes, p17-KR9 and Nef-SR9. There was no substantial difference in the strength of binding of overlapping variants of our three confirmed epitopes (p24-Gag epitopes p24-GR11/QR10, RT-SR10/QR9, and Rev-RR11/RR9) (Fig. 6), suggesting that the optimal peptide for presentation is not necessarily determined by differences in HLA class I binding.
FIGURE 6.

Binding of HLA-A*7401 to putative optimal epitopes, including overlapping variants of epitopes in p17, RT, and Rev and sequence variants of the epitope in Nef. Binding affinity KD (nM) of HLA-A*7401 to optimal peptides assessed by LOCI (35). Strength of binding is classified on a log scale in accordance with previous methods (35).
Together, these ELISPOT, tetramer, and binding data strongly support three new HLA-A*7401–restricted epitopes (Gag-GR11, RT-QR9, and Rev-RR9) and point to two further likely epitopes (Gag-KR9 and Nef-SR9). These data also substantiate our proposed motif for HLA-A*7401 as L/I/M/Q at P2. At the PC of the epitope, R appears to be the preference (our two putative epitopes bearing K at this position, p17-SK10 and RT-SK9, were both low binders to HLA-A*7401). We also tested binding of the only HLA-A*7401 HIV-1 epitope that has previously been reported by Los Alamos HIV databases (http://www.hiv.lanl.gov), Prot-IV9 (Pol 58–67, ITLWQRPLV) (41); this does not match our motif prediction at the PC, and was also a low binder.
In the presence of HLA-A*7401, the HLA-B*5703 Gag footprint is diminished and HLA-B*5703 -restricted IFN-γ ELISPOT responses are preserved
To investigate the way in which HLA-A*7401 may ameliorate disease progression in subjects with HLA-B*5703, we investigated the impact of HLA-A*7401 on the selection of polymorphisms and on IFN-γ ELISPOT responses associated with HLA-B*5703. HLA-B*5703 is strongly associated with the selection of five mutations in p24 Gag, at positions A146, I147, A163, S165, and T242, within and flanking the epitopes ISW9 (p24 Gag 147–155, ISPRTLNAW), KF11 (p24 Gag 162–172, KAFSPEVIPMF), and TW10 (p24 Gag 240–249, TSTLQEQIAW) (3, 6, 16, 17).
In 64 subjects with HLA-B*5703, there was a trend toward preservation of the wild-type amino acid in the presence of HLA-A*7401 at 4 of the 5 sites of polymorphism (I147, A163, S165, and T242), although this only reached statistical significance at S165 (p = 0.004, Fisher’s exact test; Fig. 7A). The exception was A146, flanking the ISW9 epitope. However, selection at this position may occur as a consequence of many different alleles because of the extensive overlap of CD8+ T cell epitopes at this position (including HLA-B*3910, HLA-B*1510, HLA-B*5801, and B*4801) (10). Subjects with HLA-A*7401 were also less likely than HLA-A*7401− subjects to have mutations at all five sites (p = 0.03, Fisher’s exact test) (Fig. 7A).
FIGURE 7.

Relation between HLA-A*7401 status and frequency of HLA-B*5703–restricted CD8+ T cell mutations/IFN-γ ELISPOT responses. A, Frequency of HLA-B*5703–selected Gag polymorphisms [defined by previous studies (3, 6, 16, 17)], in 64 subjects with HLA-B*5703 according to the presence/absence of HLA-A*7401. p24-Gag sequences from Bloemfontein (n = 8), Durban (n = 38), Gaborone (n = 11), and Thames Valley cohorts (n = 7), with sequence data available for TW10 (n = 63), KF11 (n = 64), ISW9 (n = 63), and all three epitopes (n = 62). B, Frequency of IFN-γ ELISPOT response to three HLA-B*5703 epitopes in p24-Gag in 46 subjects with HLA-B*5703 according to the presence or absence of HLA-A*7401. The p values were by Fisher’s exact test. Data from Durban (n = 31) and Thames Valley cohorts (n = 15).
To determine whether the presence of HLA-A*7401 has an impact on HLA-B*5703 responses to Gag epitopes, we studied ELISPOT data from 46 HLA-B*5703+ southern African subjects from the Durban and Thames Valley cohorts. Although responses to individual OLPs containing HLA-B*5703 epitopes were not statistically associated with the presence or absence of HLA-A*7401, we found overall preservation of an IFN-γ response to these OLPs in the presence of HLA-A*7401 (p = 0.006 for response to any OLP containing an HLA-B*5703 epitope, Fisher’s exact test) (Fig. 7B). The trend toward maintenance of these IFN-γ ELISPOT responses is in keeping with the sequence data showing preservation of wild-type epitopes in the presence of HLA-A*7401.
Discussion
These studies add to recently published literature (19, 23) substantiating the beneficial influence of HLA-A*7401 on disease control in adult subjects with HIV-1 infection, both alone and in tandem with HLA-B*5703. HLA-A*7401 is common in black African populations (occurring at a 10.2% phenotypic frequency in our extended cohort of >2100 southern African subjects), but has been relatively understudied compared with many HLA-B alleles.
The transmission of HLA-A*7401 in LD with HLA-B*5703 in the Durban cohort highlights the difficulties of appreciating the influence of a single allele. Although our calculations to verify LD do not, of course, demonstrate, in an individual subject, on which haplotype the HLA-A*7401 and HLA-B*5703 alleles are located, they show that statistically these alleles are likely to arise on the same haplotype when coexpressed in a particular individual, and this is verified by haplotype analysis using PyPop (36) (Supplemental Table I). Given the well-established benefits of HLA-B*5703, it would be easy to dismiss the apparent association between HLA-A*7401 and low viremia as simply caused by linkage with HLA-B*5703. However, in addition to our data demonstrating a significant HLA-A*7401–restricted response through CD8+ T cell epitopes, we also present two further strands of evidence for an independent role for HLA-A*7401. First, the favorable effect of this allele on viremic control observed in a Botswana cohort (in which these two alleles are not transmitted in LD) suggests a true independent benefit of HLA-A*7401. Second, we have identified a consistent additive effect of HLA-A*74 and HLA-B*57 through a novel computational approach to the analysis of all possible pairs of two alleles at the A, B, and C loci in >2100 subjects.
Our results demonstrate that HLA-A*7401 is associated with lower viral loads and higher CD4+ T cell counts in several southern African cohorts. Although these associations do not reach statistical significance in every cohort, the trends are broadly consistent in different settings. There are several possible explanations for the lack of strong correlation with CD4+ T cell counts in cohorts other than Gaborone. First, viral load and CD4+ T cell count are correlated, but not always strongly (typical correlation coefficients reported are of r2 = 0.28) (42), so an association between HLA-A*7401 and lowered viremia does not necessarily translate into a statistically significant association with higher CD4+ T cell count, especially where cohort size is small. Indeed, this same observation has been made in a previous study of HLA associations with viral set point or absolute CD4+ T cell count (23), in which some alleles were significantly associated with low viral set point, for example, but not with high absolute CD4+ T cell count. An alternative possible explanation of this observation might be that certain HLA alleles have a stronger impact on CD4 count than on viral load, or vice versa, for reasons unknown.
Using motif prediction undertaken according to previously validated methodology (33), in combination with ELISPOT and sequence data, we identified eight potential sites of HLA-A*7401–restricted CD8+ T cell epitopes in HIV-1 Gag, Pol, Rev, and Nef (Tables IV, V). Putative epitopes at five of these sites are intermediate or good binders to HLA-A*7401. In particular, the presence of epitopes in Gag may contribute to viremic control in two specific ways. First, the presence of Gag epitopes suggests the potential for an HLA-A*7401–restricted response to emerge early in the course of acute infection because of the abundance of Gag protein at the initiation of infection (43). Second, of the six sequence polymorphisms we identified in association with HLA-A*7401, three have previously been predicted to revert to wild-type after transmission to an HLA-mismatched host (Table V), suggesting a fitness cost imposed by the mutation (3). In these instances, we predict that selection of the mutation itself may contribute to suppression of viremia in subjects with HLA-A*7401 (3) through an effect on viral infectivity and/or replicative capacity. This effect has previously been shown in mutations in HLA-B*57 epitopes that contribute to a reduced fitness virus (6, 17) and are therefore associated with clinical control of disease (44–46). In particular, the presence of two reverting sites in Gag highlights specifically that HLA-A*7401 may contribute to viremic control through the selection of costly mutations in the highly structurally constrained Gag protein (17, 47–49).
To date, only one putative HIV-1 epitope restricted by HLA-A*7401 is listed by Los Alamos HIV databases “A-list” (http://www.hiv.lanl.gov), Prot-IV9 (Pol 58–67 ITLWQRPLV) (41). However, this epitope sequence was determined through prediction, and the HLA restriction has not been demonstrated. The binding studies shown in this article suggest this peptide is a very low binder of HLA-A*7401 (Fig. 6), and together with two of the putative epitopes presented in this article (Prot-NR10 and RT-SK9), it is therefore less likely to be a commonly targeted HLA-A*7401–restricted epitope. A further two putative overlapping HLA-A*7401–restricted epitopes have previously been proposed on the basis of an HLA-A*7401–associated polymorphism at position Gag-441 (24). One of these did fit the HLA-A*7401 binding motif proposed in this article, with Lys at the PC; however, the other lacked a positively charged residue at PC. No HLA-A*7401 binding data were provided for these proposed epitopes, and these peptide sequences did not meet the more rigorous criteria for epitopes set out in the Los Alamos Immunology Database “A-list.” These criteria include titration curves showing recognition of the optimal epitope at serial peptide dilutions compared with four additional peptides, each differing from the optimal by one amino acid, either longer or shorter by one amino acid at the N or C termini, respectively (see Fig. 5A, 5B), or unequivocal staining with a peptide–MHC tetramer (see Fig. 5D). Peptides have been described that do not precisely fit the established peptide binding motif for the respective HLA molecule (50), but these are the exception rather than the rule. In contrast, more typically, a peptide that does not fit the peptide binding motif (such as Prot-IV9; Fig. 6) proves not to bind once the peptide–MHC binding assays are undertaken.
Notably, the approach we adopted using HLA-associated polymorphisms to identify location of an epitope successfully enabled us to identify optimal HLA-A*7401–restricted epitopes, but this approach is challenging because, by definition, few HLA-A*7401+ individuals make responses to epitopes where escape mutations are so readily selected. Despite screening cryopreserved PBMCs from 15 HLA-A*7401+ subjects from the Thames Valley cohort against OLPs and optimal peptides for putative epitopes restricted by this allele, we identified only two responses: one each in subjects H005 and R070 (Fig. 5). On a follow-up visit (after an interval of 23 mo), H005 had lost the IFN-γ response to RT-QR9, and we were therefore unable to proceed with further restriction of this epitope. These findings suggest that HLA-A*7401 CD8+ T cell responses may be short-lived (arising early in the course of infection and being lost as a consequence of immune escape), or may be subdominant compared with greater magnitude responses restricted by HLA-B alleles, and therefore difficult to detect using this IFN-γ ELISPOT screening method. However, despite the challenges of confirming HLA-A*7401–restricted responses, in this article, we present in vitro data (binding studies and tetramer data) that support three new epitopes: Gag-GR11 (GQMVHQAISPR), RT-QR9 (QIYPGIKVR), and Rev-RR9 (RQIHSISER).
In these in vitro experiments, HLA-A*7401 constructs were used, confirming the binding of this allele (resolved to high resolution) to predicted peptides. Although ELISPOT responses to a particular peptide cannot necessarily be attributed to one specific allele (in this case, HLA-A*7401), the combined approach of analysis of ELISPOT and sequence data, motif inference, in vitro binding studies, and tetramer binding is a robust method of highlighting putative novel epitopes.
The mechanism by which the additive effect of HLA-A*7401 and HLA-B*5703 arises is uncertain. The preservation of wild-type HLA-B*5703 epitopes and the related maintenance of an HLA-B*5703–restricted CD8+ T cell response in the presence of HLA-A*7401 may be the cause or effect of lowered viremia. Several potential mechanisms may explain the favorable effect of this haplotype. First, there may simply be an additive effect of multiple CD8+ T cell responses consequent on the dual presentation of epitopes restricted by both alleles on the surface of infected cells, particularly given the Gag-directed responses restricted by both alleles. The preservation of wild-type sequence in HLA-B*5703–restricted Gag epitopes in the presence of HLA-A*7401 suggests the possibility that subdominant responses from the HLA-A allele delay HLA-B–selected escape mutations, allowing preservation of responses to epitopes restricted by HLA-B*5703. Further protection might be afforded by HLA-A*7401–peptide complexes as killer Ig-like receptor ligands (51), activating a favorable NK response. Finally, we cannot exclude the presence of another confounding variable, either within or outside the MHC locus, that is transmitted in linkage with HLA-A*7401, which is responsible for enhanced viremic control.
It is well-known that HLA-B*5703 and other HLA-B*57 alleles are strongly associated with control of HIV. The mechanism for this control may hinge on the presentation of multiple Gag epit-opes (3, 8, 52), the selection of escape mutations that impose a cost to viral fitness (3, 15, 17, 48), and the interaction with KIR3DS1 to activate a favorable NK cell response (53, 54). However, it remains unclear why some people with HLA-B*57 have better immune control than others. One possibility is that effects resulting from LD between HLA-B*57 and other alleles may contribute to the B*57 effect. The data presented in this article demonstrate that, for HLA-A*7401, this is indeed the case. As cohort sizes increase, it will become possible to dissect out the haplotypic effects arising from the presence of several distinct HLA and non-HLA alleles on the same chromosome.
In addition to HLA-A*7401, there may be other examples of HLA-A alleles that are associated with good viremic control but in which the benefit is obscured by the influence of HLA-B alleles with which they are linked. One such example is HLA-A*6601. This occurs in LD with HLA-B*5802 in Durban (p value for linkage = 3.5 × 10−7, Fisher’s exact test), an allele associated with poor viremic control and rapid progression to AIDS (13, 55). When ranked according to median viremia in chronic infection, HLA-A*6601 appears to be among the worst performing of HLA-A alleles; however, when subjects with HLA-B*5802 are removed from the analysis, a significant association between HLA-A*6601 and good control of viremia emerges (23).
In summary, this work represents investigation of HLA-A*7401 and the HLA-A*7401-B*5703 haplotype. Although this allele has recently been recognized to be associated with favorable viremic control (19, 23, 24), the mechanism for this phenomenon has not previously been studied. Despite the strong favorable effect mediated by HLA-B*5703 on control of viremia (3, 6, 13, 16, 23), we show in this article that the HLA-A*7401-B*5703 haplotype is more favorable than HLA-B*5703 alone. Our data do not demonstrate a single explanation for this effect, but we present compelling evidence for an independent CD8+ T cell response restricted by HLA-A*7401. These findings highlight the potential for HLA class I alleles to work in tandem to influence HIV-1 disease control, and demonstrate the importance of studying HLA class I alleles that arise in different populations; HLA-A*7401 is not found in white subjects, but is common, and appears to exert considerable influence, in black African cohorts. Furthermore, our data highlight the importance of a broad CD8+ T cell response, especially targeting Gag peptides, and suggest the utility of combining multiple CD8+ T cell responses in a potential vaccine.
Supplementary Material
Acknowledgments
This work was supported by the United Kingdom Medical Research Council (to P.C.M.), the Oxford Radcliffe Hospitals Medical Research Fund (to P.C.M), National Institutes of Health Grant 2RO1AI46995, National Institute of Allergy and Infectious Diseases Contract HHSN272200900045C, the South African AIDS Vaccine Initiative, the Wellcome Trust (to A.L. and P.J.R.G.), the National Cancer Institute, National Institutes of Health Contract HHSN261200800001E, and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. P.J.R.G. is an Elizabeth Glaser Pediatric AIDS Foundation Scientist.
Abbreviations used in this article
- HIV-1
HIV type 1
- LD
linkage disequilibrium
- LOCI
luminescent oxygen channeling immunoassay
- OLP
overlapping peptide
- P2
position two of epitope
- PC
C-terminal position of epitope
Footnotes
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Brumme ZL, Tao I, Szeto S, Brumme CJ, Carlson JM, Chan D, Kadie C, Frahm N, Brander C, Walker B, et al. Human leukocyte antigen-specific polymorphisms in HIV-1 Gag and their association with viral load in chronic untreated infection. AIDS. 2008;22:1277–1286. doi: 10.1097/QAD.0b013e3283021a8c. [DOI] [PubMed] [Google Scholar]
- 2.Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthews PC, Prendergast A, Leslie A, Crawford H, Payne R, Rousseau C, Rolland M, Honeyborne I, Carlson J, Kadie C, et al. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. J Virol. 2008;82:8548–8559. doi: 10.1128/JVI.00580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Brien SJ, Gao X, Carrington M. HLA and AIDS: a cautionary tale. Trends Mol Med. 2001;7:379–381. doi: 10.1016/s1471-4914(01)02131-1. [DOI] [PubMed] [Google Scholar]
- 5.Rousseau CM, Daniels MG, Carlson JM, Kadie C, Crawford H, Prendergast A, Matthews P, Payne R, Rolland M, Raugi DN, et al. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype c proteome: immune escape and viral load. J Virol. 2008;82:6434–6446. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford H, Prado JG, Leslie A, Hué S, Honeyborne I, Reddy S, van der Stok M, Mncube Z, Brander C, Rousseau C, et al. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J Virol. 2007;81:8346–8351. doi: 10.1128/JVI.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geldmacher C, Currier JR, Herrmann E, Haule A, Kuta E, McCutchan F, Njovu L, Geis S, Hoffmann O, Maboko L, et al. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-sero-positive patients. J Virol. 2007;81:2440–2448. doi: 10.1128/JVI.01847-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goulder PJ, Bunce M, Krausa P, McIntyre K, Crowley S, Morgan B, Edwards A, Giangrande P, Phillips RE, McMichael AJ. Novel, cross-restricted, conserved, and immunodominant cytotoxic T lymphocyte epitopes in slow progressors in HIV type 1 infection. AIDS Res Hum Retroviruses. 1996;12:1691–1698. doi: 10.1089/aid.1996.12.1691. [DOI] [PubMed] [Google Scholar]
- 9.Honeyborne I, Prendergast A, Pereyra F, Leslie A, Crawford H, Payne R, Reddy S, Bishop K, Moodley E, Nair K, et al. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J Virol. 2007;81:3667–3672. doi: 10.1128/JVI.02689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature. 2009;458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leslie A, Price DA, Mkhize P, Bishop K, Rathod A, Day C, Crawford H, Honeyborne I, Asher TE, Luzzi G, et al. Differential selection pressure exerted on HIV by CTL targeting identical epitopes but restricted by distinct HLA alleles from the same HLA supertype. J Immunol. 2006;177:4699–4708. doi: 10.4049/jimmunol.177.7.4699. [Published erratum appears in 2006 J Immunol. 177: 8878.] [DOI] [PubMed] [Google Scholar]
- 12.Payne RP, Kløverpris H, Sacha JB, Brumme Z, Brumme C, Buus S, Sims S, Hickling S, Riddell L, Chen F, et al. Efficacious early antiviral activity of HIV Gag- and Pol-specific HLA-B 2705-restricted CD8+ T cells. J Virol. 2010;84:10543–10557. doi: 10.1128/JVI.00793-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 14.Altfeld M, Addo MM, Rosenberg ES, Hecht FM, Lee PK, Vogel M, Yu XG, Draenert R, Johnston MN, Strick D, et al. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS. 2003;17:2581–2591. doi: 10.1097/00002030-200312050-00005. [DOI] [PubMed] [Google Scholar]
- 15.Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, Tang J, Farmer P, Ndung’u T, Lakhi S, et al. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med. 2009;206:909–921. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 17.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol. 2006;80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawashima Y, Satoh M, Oka S, Shirasaka T, Takiguchi M. Different immunodominance of HIV-1-specific CTL epitopes among three subtypes of HLA-A*26 associated with slow progression to AIDS. Biochem Biophys Res Commun. 2008;366:612–616. doi: 10.1016/j.bbrc.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 19.Koehler RN, Walsh AM, Saathoff E, Tovanabutra S, Arroyo MA, Currier JR, Maboko L, Hoelscher M, Robb ML, Michael NL, et al. Class I HLA-A*7401 is associated with protection from HIV-1 acquisition and disease progression in Mbeya, Tanzania. [Published erratum appears in 2011 J. Infect. Dis. 203: 749.] J Infect Dis. 2010;202:1562–1566. doi: 10.1086/656913. [DOI] [PubMed] [Google Scholar]
- 20.Schaubert KL, Price DA, Frahm N, Li J, Ng HL, Joseph A, Paul E, Majumder B, Ayyavoo V, Gostick E, et al. Availability of a diversely avid CD8+ T cell repertoire specific for the subdominant HLA-A2-restricted HIV-1 Gag p2419-27 epitope. J Immunol. 2007;178:7756–7766. doi: 10.4049/jimmunol.178.12.7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang J, Shao W, Yoo YJ, Brill I, Mulenga J, Allen S, Hunter E, Kaslow RA. Human leukocyte antigen class I genotypes in relation to heterosexual HIV type 1 transmission within discordant couples. J Immunol. 2008;181:2626–2635. doi: 10.4049/jimmunol.181.4.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leslie A, Matthews PC, Listgarten J, Carlson JM, Kadie C, Ndung’u T, Brander C, Coovadia H, Walker BD, Heckerman D, Goulder PJ. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J Virol. 2010;84:9879–9888. doi: 10.1128/JVI.00320-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semeniuk CA, McKinnon L, Peters HO, Gubbins M, Mao X, Ball TB, Luo M, Plummer FA. Multiple T-cell epitopes overlap positively-selected residues in the p1 spacer protein of HIV-1 gag. AIDS. 2009;23:771–777. doi: 10.1097/QAD.0b013e32832995e0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catano G, Kulkarni H, He W, Marconi VC, Agan BK, Landrum M, Anderson S, Delmar J, Telles V, Song L, et al. HIV-1 disease-influencing effects associated with ZNRD1, HCP5 and HLA-C alleles are attributable mainly to either HLA-A10 or HLA-B*57 alleles. PLoS ONE. 2008;3:e3636. doi: 10.1371/journal.pone.0003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naugler C. Origins and relatedness of human leukocyte antigen class I allele supertypes. Hum Immunol. 2010;71:837–842. doi: 10.1016/j.humimm.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1–15. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Middleton D, Menchaca L, Rood H, Komerofsky R. New allele frequency database. Tissue Antigens. 2003;64:403–407. doi: 10.1034/j.1399-0039.2003.00062.x. [DOI] [PubMed] [Google Scholar]
- 29.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 30.Huang KH, Goedhals D, Fryer H, van Vuuren C, Katzourakis A, De Oliveira T, Brown H, Cassol S, Seebregts C, McLean A, et al. Bloemfontein-Oxford Collaborative Group. Prevalence of HIV type-1 drug-associated mutations in pre-therapy patients in the Free State, South Africa. Antivir Ther (Lond) 2009;14:975–984. doi: 10.3851/IMP1416. [DOI] [PubMed] [Google Scholar]
- 31.Leslie A, Kavanagh D, Honeyborne I, Pfafferott K, Edwards C, Pillay T, Hilton L, Thobakgale C, Ramduth D, Draenert R, et al. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J Exp Med. 2005;201:891–902. doi: 10.1084/jem.20041455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frater AJ, Brown H, Oxenius A, Günthard HF, Hirschel B, Robinson N, Leslie AJ, Payne R, Crawford H, Prendergast A, et al. Effective T-cell responses select human immunodeficiency virus mutants and slow disease progression. J Virol. 2007;81:6742–6751. doi: 10.1128/JVI.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Honeyborne I, Rathod A, Buchli R, Ramduth D, Moodley E, Rathnavalu P, Chetty S, Day C, Brander C, Hildebrand W, et al. Motif inference reveals optimal CTL epitopes presented by HLA class I alleles highly prevalent in southern Africa. J Immunol. 2006;176:4699–4705. doi: 10.4049/jimmunol.176.8.4699. [DOI] [PubMed] [Google Scholar]
- 34.Parham P. Immunology. Deconstructing the MHC. Nature. 1992;360:300–301. doi: 10.1038/360300a0. [DOI] [PubMed] [Google Scholar]
- 35.Harndahl M, Justesen S, Lamberth K, Røder G, Nielsen M, Buus S. Peptide binding to HLA class I molecules: homogenous, high-throughput screening, and affinity assays. J Biomol Screen. 2009;14:173–180. doi: 10.1177/1087057108329453. [DOI] [PubMed] [Google Scholar]
- 36.Lancaster AK, Single RM, Solberg OD, Nelson MP, Thomson G. PyPop update—a software pipeline for large-scale multilocus population genomics. Tissue Antigens. 2007;69(Suppl 1):192–197. doi: 10.1111/j.1399-0039.2006.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsh SG, Parham EP, Barber LD. The HLA Facts Book. Academic Press; London: 2000. [Google Scholar]
- 38.Sette A, Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 1999;50:201–212. doi: 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- 39.Oxenius A, Jakobsen BK, Easterbrook PJ, Boulter JM, Tun T, Waters A, Agudelo J, Barnardo M, Phillips RE, Price DA. Complete mapping of a novel HLA A*6801-restricted HIV-1 Tat epitope directly with a rapid modified enzyme-linked immunospot assay. AIDS. 2002;16:1285–1287. doi: 10.1097/00002030-200206140-00012. [DOI] [PubMed] [Google Scholar]
- 40.Bhattacharya T, Daniels M, Heckerman D, Foley B, Frahm N, Kadie C, Carlson J, Yusim K, McMahon B, Gaschen B, et al. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science. 2007;315:1583–1586. doi: 10.1126/science.1131528. [DOI] [PubMed] [Google Scholar]
- 41.Kaul R, Dong T, Plummer FA, Kimani J, Rostron T, Kiama P, Njagi E, Irungu E, Farah B, Oyugi J, et al. CD8(+) lymphocytes respond to different HIV epitopes in seronegative and infected subjects. J Clin Invest. 2001;107:1303–1310. doi: 10.1172/JCI12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cozzi Lepri A, Katzenstein TL, Ullum H, Phillips AN, Skinhøj P, Gerstoft J, Pedersen BK. The relative prognostic value of plasma HIV RNA levels and CD4 lymphocyte counts in advanced HIV infection. AIDS. 1998;12:1639–1643. doi: 10.1097/00002030-199813000-00011. [DOI] [PubMed] [Google Scholar]
- 43.Sacha JB, Chung C, Rakasz EG, Spencer SP, Jonas AK, Bean AT, Lee W, Burwitz BJ, Stephany JJ, Loffredo JT, et al. Gag-specific CD8+ T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. J Immunol. 2007;178:2746–2754. doi: 10.4049/jimmunol.178.5.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chopera DR, Woodman Z, Mlisana K, Mlotshwa M, Martin DP, Seoighe C, Treurnicht F, de Rosa DA, Hide W, Karim SA, et al. CAPRISA 002 Study Team. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 2008;4:e1000033. doi: 10.1371/journal.ppat.1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, Derdeyn CA, Tang J, Kaslow RA, Bansal A, et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med. 2008;205:1009–1017. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thobakgale CF, Prendergast A, Crawford H, Mkhwanazi N, Ramduth D, Reddy S, Molina C, Mncube Z, Leslie A, Prado J, et al. Impact of HLA in mother and child on disease progression of pediatric human immunodeficiency virus type 1 infection. J Virol. 2009;83:10234–10244. doi: 10.1128/JVI.00921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borghans JA, ølgaard AM, de Boer RJ, Keşmir C. HLA alleles associated with slow progression to AIDS truly prefer to present HIV-1 p24. PLoS ONE. 2007;2:e920. doi: 10.1371/journal.pone.0000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boutwell CL, Rowley CF, Essex M. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J Virol. 2009;83:2460–2468. doi: 10.1128/JVI.01970-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneidewind A, Brockman MA, Sidney J, Wang YE, Chen H, Suscovich TJ, Li B, Adam RI, Allgaier RL, Mothé BR, et al. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J Virol. 2008;82:5594–5605. doi: 10.1128/JVI.02356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goulder PJ, Reid SW, Price DA, O’Callaghan CA, McMichael AJ, Phillips RE, Jones EY. Combined structural and immunological refinement of HIV-1 HLA-B8-restricted cytotoxic T lymphocyte epitopes. Eur J Immunol. 1997;27:1515–1521. doi: 10.1002/eji.1830270630. [DOI] [PubMed] [Google Scholar]
- 51.Thananchai H, Gillespie G, Martin MP, Bashirova A, Yawata N, Yawata M, Easterbrook P, McVicar DW, Maenaka K, Parham P, et al. Cutting Edge: Allele-specific and peptide-dependent interactions between KIR3DL1 and HLA-A and HLA-B. J Immunol. 2007;178:33–37. doi: 10.4049/jimmunol.178.1.33. [DOI] [PubMed] [Google Scholar]
- 52.Streeck H, Lichterfeld M, Alter G, Meier A, Teigen N, Yassine-Diab B, Sidhu HK, Little S, Kelleher A, Routy JP, et al. Recognition of a defined region within p24 gag by CD8+ T cells during primary human immunodeficiency virus type 1 infection in individuals expressing protective HLA class I alleles. J Virol. 2007;81:7725–7731. doi: 10.1128/JVI.00708-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, Miller JS, Carrington M, Altfeld M. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol. 2009;83:6798–6805. doi: 10.1128/JVI.00256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Trowsdale J, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 55.Ngumbela KC, Day CL, Mncube Z, Nair K, Ramduth D, Thobakgale C, Moodley E, Reddy S, de Pierres C, Mkhwanazi N, et al. Targeting of a CD8 T cell env epitope presented by HLA-B*5802 is associated with markers of HIV disease progression and lack of selection pressure. AIDS Res Hum Retroviruses. 2008;24:72–82. doi: 10.1089/aid.2007.0124. [DOI] [PubMed] [Google Scholar]
- 56.Gray CM, Mlotshwa M, Riou C, Mathebula T, de Assis Rosa D, Mashishi T, Seoighe C, Ngandu N, van Loggerenberg F, Morris L, et al. CAPRISA 002 Acute Infection Study Team. Human immunodeficiency virus-specific gamma interferon enzyme-linked immunospot assay responses targeting specific regions of the proteome during primary subtype C infection are poor predictors of the course of viremia and set point. J Virol. 2009;83:470–478. doi: 10.1128/JVI.01678-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carlson JM, Brumme ZL, Rousseau CM, Brumme CJ, Matthews P, Kadie C, Mullins JI, Walker BD, Harrigan PR, Goulder PJ, Heckerman D. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLOS Comput Biol. 2008;4:e1000225. doi: 10.1371/journal.pcbi.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.