

Abstract
TLR9 is a key determinant of the innate immune responses in both infectious and sterile injury. Specific antagonism of TLR9 is of great clinical interest to reduce tissue damage in a wide range of pathologies, and has been approached by modification of nucleic acids, the recognized ligand for TLR9. Such oligonucleotide-derived pharmacotherapeutics have limitations in specificity for nucleic acid receptors, significant potential for immunologic recognition with generation of innate and adaptive immune responses, and limited bioavailability. We have identified enantiomeric analogues of traditional (−)-morphinans as having TLR9 antagonist properties on reporter cell lines. One of these analogues (COV08-0064) is demonstrated to be a novel small molecule antagonist of TLR9 with greater specificity for TLR9 than oligo based antagonists. COV08-0064 has wide bioavailability, including the subcutaneous and per oral route. It specifically inhibits the action of TLR9 antagonists on reporter cells lines and the production of cytokines by TLR9 agonists from primary cells. It also has efficacy in limiting TLR9 mediated sterile inflammation in in vivo models of acute liver injury and acute pancreatitis. The identification of a morphinan based novel small molecule structure with TLR9 antagonism is a significant step in expanding therapeutic strategies in the field of sterile inflammatory injury.
Introduction
TLR9 has been identified as a key determinant of innate immune responses to both infectious and sterile inflammatory injury (1–3). TLR9 is an endosomal receptor for double-stranded DNA in the extracellular compartment and has greatest affinity for unmethylated and CpG rich DNA (4, 5). Unmethylated and CpG DNA is less prevalent in mammalian genomic DNA, yet the ability of mammalian DNA to induce TLR9 mediated inflammation has been demonstrated in multiple pathologies (5).
Receptor ligand interaction has been investigated through homology modeling, mutational analysis and use of chemically modified ligands (6, 7). These investigations have led to antagonism of TLR9 function through competitive inhibition at ligand binding sites employing nucleic acids known as inhibitory oligonucleotides (ODN) or immunoregulatory DNA sequences (IRS) with chemical modification of the oligophosphate backbone to prevent degradation by nucleases (8–11). These nucleic acid based antagonists of TLR9 also function as antagonists of TLR7, an endosomal receptor for single stranded RNA of mammalian and prokaryotic origin (10, 12). Though TLR7 activation parallels TLR9 immune response in many experimental models, the interaction of TLR7 and TLR9 activation and suppression is considerably more complex (13, 14). Inhibition of TLR9 signaling has also been achieved through use of nucleic acid binding polymers and other nucleic acid binding agents, including antimalarials such as chloroquine and imidazoquinolines (15). These agents do not have TLR9 specific effects, either binding and masking other nucleic acid TLR ligands or, in the case of imidazoquinolines, stimulating TLR7 and TLR8 directly. Recently, an oral inhibitor of nucleic acid sensing endosomal TLRs, E6446, has shown efficacy in mitigating inflammatory injury in a murine model of cerebral malaria (16).
Beyond lack of specificity for TLR9, nucleic acid based antagonists have immunoregulatory functionality independent of TLR9 (17). Similarly, DNA vaccines do not require the presence of host TLR9 for generation of potent adaptive immune responses (18). Additionally, TLR9 independent sensors of cytolosic DNA have been identified, specifically absent in melanoma 2 (AIM2) and IFI16 (19–21). The bioavailability of nucleic acid derived TLR9 antagonists is well characterized in vivo in parenteral administration. However, significant bioavailability after oral administration is less clear and appears unlikely to date.
We have identified a new small molecule TLR9 antagonist COV08-0064 with a novel structure based on the (+)-morphinan scaffold. The (+)-morphinan class of compounds refers to the enantiomeric analogues of traditional (−)-morphinans such as (−)-Naltrexone and (−)-Naloxone. Compound COV08-0064 emerged as a potential lead candidate from a broad TLR9 antagonist screen of a library of such functionalized (+)-morphinan derivatives. The pharmacokinetics and TLR specificity of COV08-0064 for a range of TLRs is determined. Additionally, in vivo efficacy for TLR9 antagonist function is established in two models of TLR9 dependent sterile inflammation, acetaminophen hepatotoxicity and caerulein hyperstimulation acute pancreatitis.
Materials and Methods
TLR9 antagonist screen and other TLR reporter screens
TLR9 antagonist screen was performed in a commercially available HEK-Blue TLR9 cell line expressing a TLR9 responsive NF-κB inducible secreted placental alkaline phosphatase (SEAP). Specifically, small molecule candidates were screened for inhibition of SEAP activity induced by the TLR9 ligand ODN2006 as determined by QUANTI-Blue colorimetric assay (Invivogen, San Diego, CA). Candidates with strong TLR9 antagonism in this screen were subsequently screened for TLR agonist and antagonist activity using HEK-Blue TLR reporter cell lines for TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, TLR9, and the NF-kB transgene alone treated with, respectively, heat-killed Listeria monocytogenes (HKLM), poly (I:C), Escherichia coli K12 lipopolysaccharide (LPS), flagellin, CL097, CL075, ODN2006, and TNF-α (Invivogen, San Diego, CA).
Pharmacokinetics of COV08-0064 in Rats and Mice
Conscious Sprague-Dawley rats were administered COV08-0064 by intravenous tail vein injection, subcutaneous injection in the dorsum of the neck, intraperitoneal injection, or oral gavage in non-fasted animals as single doses. IV, SC, and IP injections were at 10 mg per kilogram body weight and per oral administration was at 100 mg per kilogram body weight. Serum concentrations of COV08-0064 was assessed by serial sampling from the jugular vein, tail vein, and at time of euthanasia by cardiac puncture at pretreatment and at 5, 15, 30, 60, 90, 120, 180, 240, and 360 minutes post-administration.
Isolation and Treatment of Peritoneal Elicited Cells (PECs)
Thioglycollate elicited peritoneal macrophages were elicited by intraperitonal injection of C57BL/6 male mice with sterile 4% thioglycollate broth (Sigma, St. Louis, MO) and harvest of peritoneal elicited cells after 72 hours. Peritoneal elicited cells (PECs) were plated on 24 well plates at 106 cells per well in 24 well dishes, non-adherent cells removed at 1 hour, and the cells recovered for 2 additional hours prior to treatment with ODN1826, ODN1585, and Imiquimod (R837) (Invivogen, San Diego, CA) with or without COV08-0064 for 3 hours. All compounds were reconstituted in sterile phosphate buffered saline.
Quantitative Polymerase Chain Reaction (Q-PCR) Assessment of Target Genes
RNA was extracted from PECs using an RNeasy Plus Mini Kit from Qiagen (Valenica, CA). Reverse transcription was performed with Transcriptor reverse transcriptase from Roche Applied Science (Indianopolis, In). Quantitative real-time PCR was performed for mRNA expression for pro-IL-1β, Nlrp3, and Gapdh using commercial primer probe sets from Applied Biosystems (Foster City, CA) and the Applied Biosystems 7500 real-time PCR System. Expression of Gapdh was used to standardize the samples. Results were expressed as a ratio to Gapdh standardized expression in untreated PECs for pro-IL-1β, Nlrp3, and respectively.
Caspase-1 activity assays
Pancreatic and liver tissues were flash frozen in liquid nitrogen and then homogenized with a rotor stator homogenizer in cell lysis buffer (Biovision, Milpitas, CA). To 50 µL of sample, 50 µL of 2× reaction buffer (Biovision, Milpitas, CA) was then added. Caspase1 substrate AcYVAD-MCA (Peptides International, Osaka, Japan) was resuspended in DMSO at 20mM and then added to a final concentration of 100µM to each sample in opaque 96 well dishes preheated to 37 °C. The samples were then incubated for 5 minutes in a Synergy II fluorescence plate reader with excitation and emission wavelengths of 380 nm and 460 nm (Biotek, Winooski, VT). Mean velocity of emission fluorescence was calculated from 10 readings over 5 minutes per sample. For liver tissue sample, this was normalized against total protein content in the sample as assessed using a Coomasie Protein Assay Kit (Pierce Thermo Scientific, Rockland, IL). For pancreatic tissue samples, this was normalized to total amylase content as determined using a Phadebas Kit (Amersham Pharmacia, Rochester, NY).
IL-6 and IL-12p40 ELISA assays
Supernatant was collected from peritoneal elicited cells co-treated with ODN1826 and COV08-0064 for 8 hours. Pancreatic and liver tissues were flash frozen in liquid nitrogen and then homogenized with a rotor stator homogenizer in cell lysis buffer (Biovision, Milpitas, CA). IL-6 and IL-12p40 was performed using commercially available assays (eBioscience, San Diego, CA). For pancreatic tissue, values were normalized to total protein content. For liver tissue, values were normalized to total protein content.
Animals
C57BL/6 male mice five to eight weeks of age were purchased from the National Cancer Institute. All experiments and animal handling were performed under approved protocols at the Yale University Institutional Animal Care and Use Committees. Sprague-Dawley rat experiments were performed on-site by Covidien, Mansfield, MA.
Acetaminophen (APAP) induced hepatotoxicity, COV08-0064 treatment, and COV08-0055 treatment
APAP (Sigma-Aldrich, St. Louis, MO) solution was prepared as 20 mg per ml in sterile Dulbecco’s phosphate buffered saline (DPBS). COV08-0055 and COV08-0064 were prepared as 10 mg per ml in DPBS and filter sterilized. Mice were fasted for 15 hours and then treated with COV08-0055 or COV08-0064 at 60 mg per kilogram body weight or DPBS vehicle by intraperitoneal injection. One hour later, mice were administered APAP at 500 mg per kilogram body weight by intraperitoneal injection. Animals were euthanized by isoflurane at 12 hours for collection of serum and liver tissue.
Caerulein hyperstimulation pancreatitis and COV08-0064 treatment
C57BL/6 male mice were administered caerulein sulfate (Sigma-Aldrich, St. Louis, MO) at 50 µg per kilogram body weight by intraperitoneal injection in sterile normal saline for 10 sequential hourly injections. COV08-0064 was prepared as 10 mg per ml in DPBS and filter sterilized. Animals were administered COV08-0064 at 30 mg per kilogram body weight or DPBS vehicle by subcutaneous injection concurrent with the first, fourth, and seventh doses of caerulein. Animals were euthanized by isoflurane at one hour after the last caerulein injection for collection of serum and pancreatic tissue.
Pancreatic Trypsin Activity and Serum Amylase and Alanine Aminotransferase (ALT) Assays
Pancreatic tissue was flash frozen in liquid nitrogen and then homogenized with a rotor stator homogenizer in 5 mM MOPS, 250 mM sucrose, 1 mM MgSO4, pH 7.00. To 50 µL of pancreatic homogenate is added 175 µL of 50 mM Tris HCl, 150 mM NaCl, 1.3 mM CaCl2, pH 8.10 and 25 µL of 10 mM fluorescent trypsin substrate Boc-QAR-MCA (Peptides International, Osaka, Japan). The samples were then incubated for 5 minutes in a Synergy II fluorescence plate reader with excitation and emission wavelengths of 380 nm and 460 nm (Biotek, Winooski, VT). Mean velocity of emission fluorescence was calculated from 10 readings over 5 minutes per sample. This was normalized to total amylase content as determined using a Phadebas Kit (Amersham Pharmacia, Rochester, NY). Heparinized mouse blood was centrifuged at 10,000 × g at 4 °C and assessed for serum amylase activity using this assay kit as well. Serum alanine aminotransferase levels were determined in the Yale Clinical Chemistry Laboratory (New Haven, CT).
Quantitation of Liver and Pancreas Infiltrating Neutrophils
Neutrophil quantitation was performed in paraffin embedded liver sections after immunolabelling with Ly-6B.2 monoclonal antibody (AbD Serotec,Raleigh, NC) by scoring for positive cells in five high power fields (40×).
Statistics
Pharmacokinetic data was analyzed by noncompartmental analysis using Pharsight WinNonLin software. Statistical analysis were performed using Microsoft Excel 2007. Unpaired 2-tailed Student’s t test was used to compare groups. A P value of less than 0.05 was considered significant. Percent bioavailability for SC and IP doses for COV08-0064 were calculated as the ratio relative to IV dosing of area under the curve for the respective route divided by dose administered.
Results
COV08-0064 is specific small molecule antagonist of TLR9 signaling
A library of small molecules structurally related to traditional opioids, but having opposite stereochemistry and novel functional group substitutions, were screened for antagonism of TLR9 using the HEK-Blue TLR9 cell line. Several compounds were thereby identified as TLR9 antagonists by antagonism to dose-dependent inhibition of ODN2006 TLR9 ligand stimulation of an NF-κB inducible transgene for secreted placental alkaline phosphatase (SEAP) (Fig. 1A). In particular, the two most efficacious compounds, COV08-0064 and COV08-0093, were noted to have similar 5-membered saturated heterocyclic rings containing oxygen (Fig. 1B). COV08-0064 was identified as the more efficacious antagonist of TLR9 for further study with an IC50 of ~100 nM.
Figure 1.
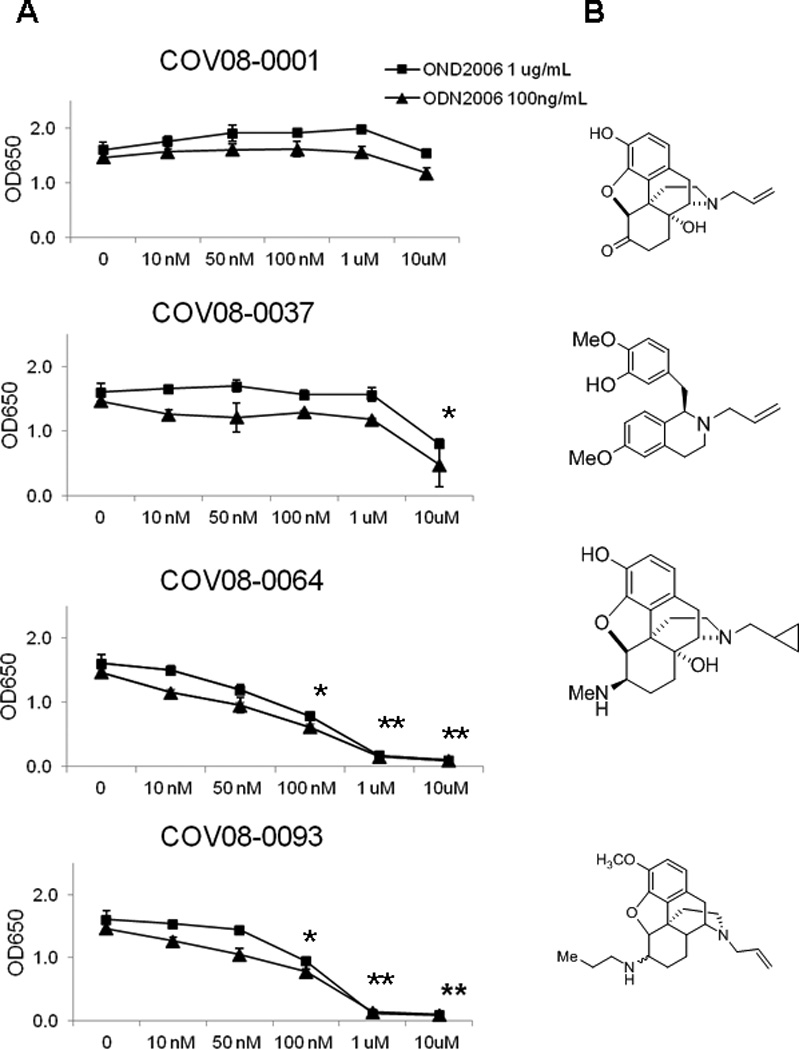
COV08-0064 is a small molecule antagonist of TLR9 signaling. Small molecules based on the (+)-morphinan structures were assessed for dose dependent antagonism of TLR9 using a HEK-Blue TLR9 cell line expressing a TLR9 responsive NF-κB inducible secreted placental alkaline phosphatase (SEAP). (A) NF-κB induced SEAP was quantitated in culture supernatant using a colorimetric assay as described in Materials and Methods. TLR9 agonist ODN2006 was used at 1 ug per mL (closed boxes) and 100 ng per mL (closed triangles). Each data point represents the mean of three independent experiments with standard deviations shown in the brackets. Asterisks denote significant differences relative to groups with no compound added (P<0.01) and two askerisks denote significant differences relative to 100 nM of compound added using an unpaired 2-tailed Student’s t test. (B) Chemical structures of four of the small molecules tested, specifically COV08-001 which was (+)-naloxone as well as COV08-0037, COV08-0064, and COV08-0093.
COV08-0064 did not demonstrate antagonist activity at TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, or the Null-1 NF-kB reporter cell line (Fig. 2A). COV08-0064 had no noted functionality as an agonist of TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, TLR9, or the Null-1 NF-kB reporter cell line using standard agonists as positive controls (Fig. 2B).
Figure 2.
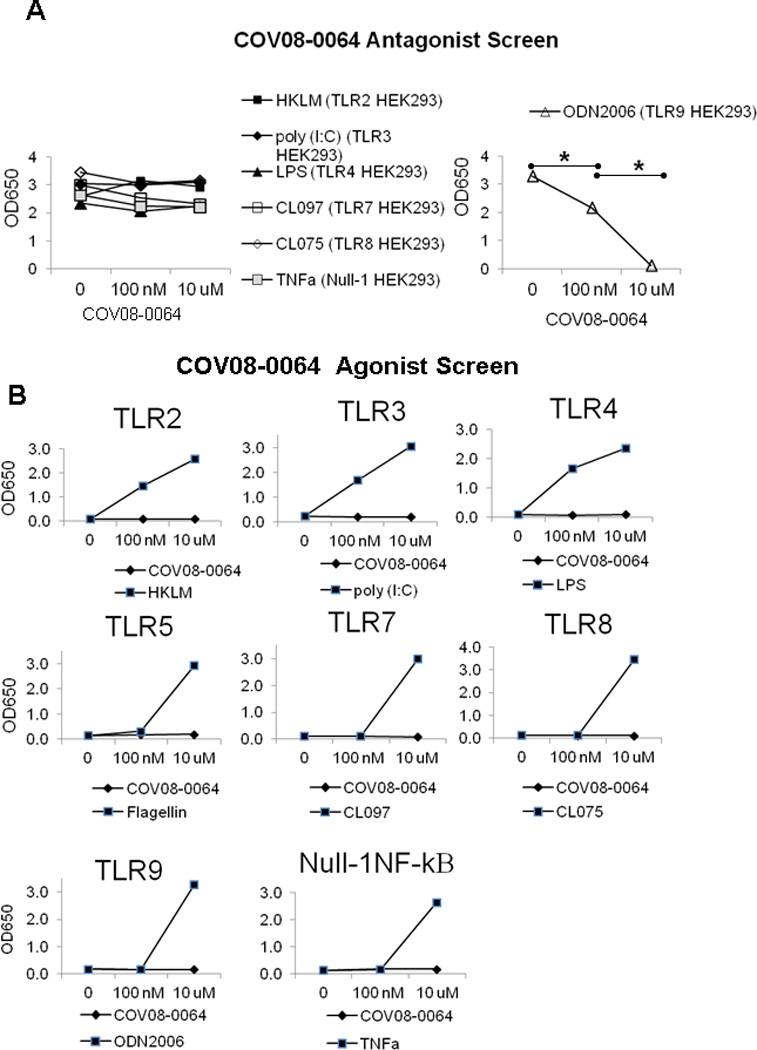
COV08-0064 is a specific antagonist of TLR9 with no detected antagonism or agonism at other TOLL-like receptors assayed, specifically TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, and the parent cell line Null-1 HEK293,which is endogenously responsive to TNF-α induction of NF-κB. (A) NF-κB induced SEAP was quantitated in culture supernatant using a colorimetric assay as described in Materials and Methods. COV08-0064 was co-incubated at 1, 100 nM, and 10 uM with 10 uM of each TLR agonist heat killed Listeria monocytogenes (HKLM) (closed box), poly I:C (closed rhombus), LPS (closed triangle), CLO97 (open box), CLO75 (open rhombus), ODN2006 (open triangle) or TNF-α (gray box) in the respective HEK-Blue TLR reporter cell lines. Each data point represents the mean of three independent experiments with standard deviations shown in the brackets. (B) COV08-0064 (closed box) or the respective TLR ligand (closed rhombus) were separately incubated with of each of HEK-Blue TLR reporter cell lines noted. SEAP activity was quantitated. Each data point represents the mean of three independent experiments with standard deviations shown in the brackets. Asterisks denote significant differences between bracketed groups (P<0.01) using an unpaired 2-tailed Student’s t test.
COV08-0064 was further screened for alterations of binding of radioligand agonist and antagonists of many receptor families as well as inhibition or activation of enzymes in cell culture at Cerep (Redmond, WA) (data not shown). COV08-0064 modestly inhibited yohimbine binding to alpha-2B adrenergic receptors and protryptiline binding to norepinephrine transporters. Additionally, COV08-0064 was assessed for aqueous solubility, cell culture cytotoxicity, as well as CYP metabolism and interaction at Cerep and was not cytotoxic (data not shown).
COV08-0064 is widely bio-available with a long serum half-life
COV08-0064 dosing by intravenous and subcutaneous dosing resulted in similar peak serum concentrations (Fig. 3A, 3B). Peak serum concentrations were significantly and markedly reduced by intraperitoneal administration by comparison (Fig. 3C). Additionally, per oral administration at ten times the intravenous dose resulted in similar peak serum concentrations (Fig. 3D). The half life of intravenous, intraperitoneal, and subcutaneous administration were similar at 116-122 minutes. Per oral administration resulted in longer serum half-life at 149 ± minutes (Fig. 3E). Additionally, subcutaneous and oral bioavailability were high at 92. ± 5 6.6% and 55.1 ± 7.1% of the intravenous dose, respectively.
Figure 3.
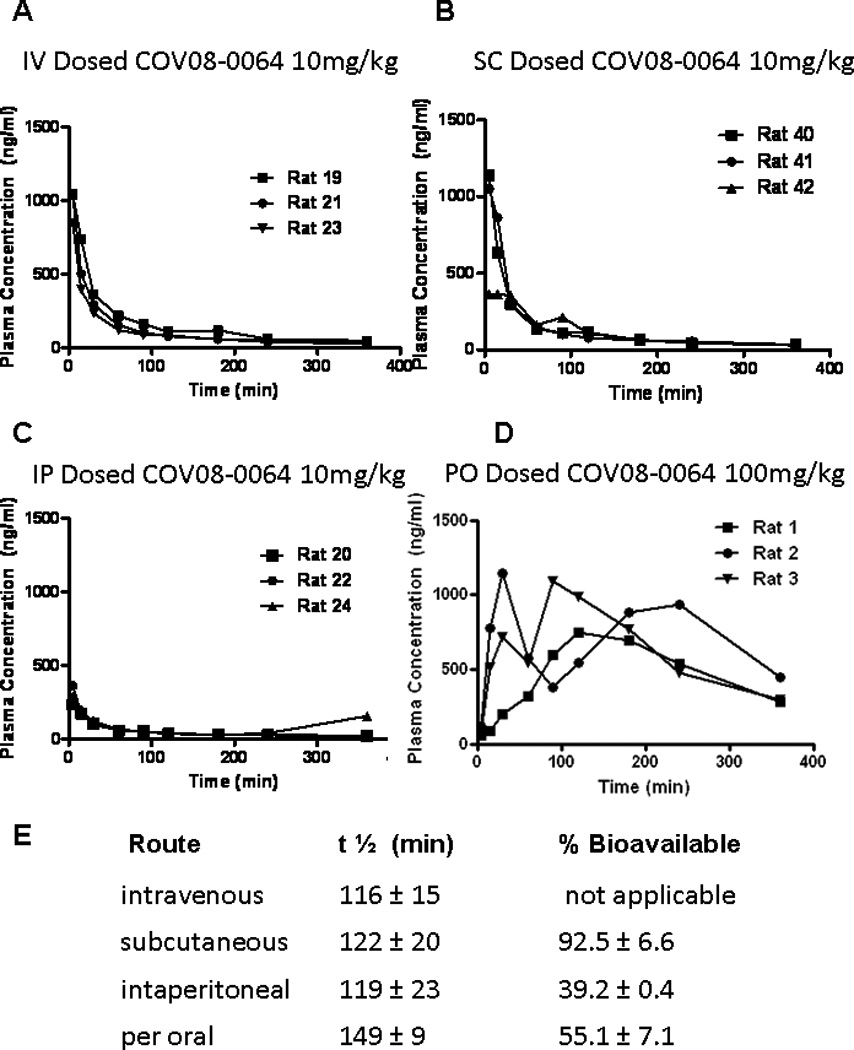
COV08-0064 is widely bio-available with a long serum half-life. Sprague-Dawley rats were administered COV08-0064 at the doses noted (n=3). Serum levels of COV08-0064 were sampled by jugular venous cannulation at the time pints indicated for (A) intravenous tail vein injection, (B) subcutaneous injection, (C) intraperitoneal injection, and (D) per oral administration. Each data point represents one rat. (E) Serum half-life and percent bioavailabilty in comparison to intravenous administration were determined as noted in Materials and Methods. Percent bioavailability for SC and IP doses for COV08-0064 were calculated as the ratio relative to IV dosing of area under the curve for the respective route divided by dose administered. Data are presented as mean and standard deviation from 3 animals.
COV08-00064 dose dependently inhibits TLR9 but not TLR7 effects in peritoneal elicited cells
COV08-0064 significantly decreases TLR9 type B ligand ODN1826 stimulated induction of pro-IL-1β and Nlrp3 expression in peritoneal elicited cells in a dose-dependent manner (Figs. 4A). COV08-0064 at the same doses does not inhibit imiquimod (TLR7 ligand) stimulated induction of pro-IL-1β and Nlrp3 expression in peritoneal elicited cells (Fig. 4B). COV08-0064 also significantly inhibits TLR9 type B ligand ODN1826 stimulated induction of IL12p40 in peritoneal elicited cells from 342 ± 6 to 108 ± 27 pg/mL (Figure 4C).
Figure 4.
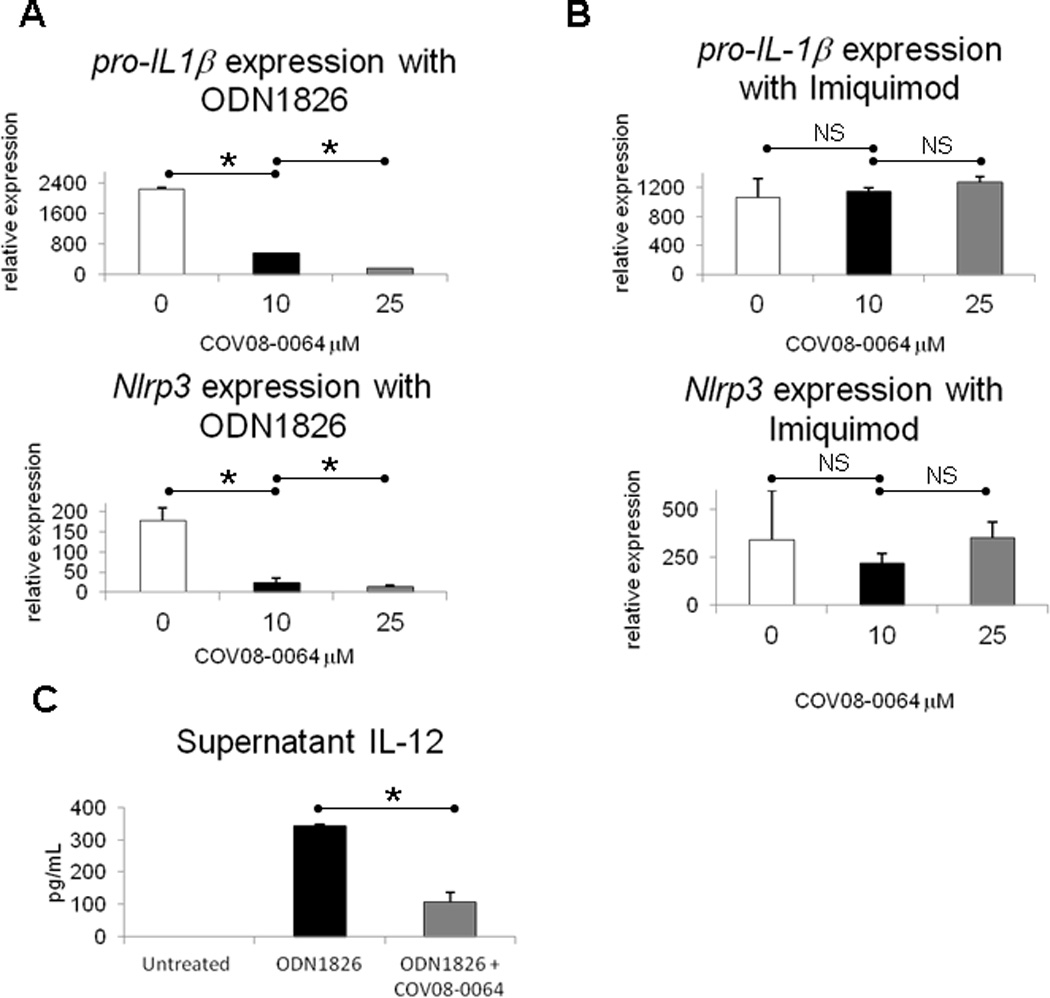
COV08-0064 dose dependently suppresses TLR9 but not TLR7 mediated proinflammatory gene expression and inflammasome priming in murine peritoneal elicited cells (PECs). (A) Transcript for pro-IL-1β was quantitated by quantitative PCR in PECs treated with increasing doses of COV-08-0064 as detailed in Materials and Methods. (B) Transcript for pro-IL-1β was quantitated by quantitative PCR in PECs co-incubated with imiquimod at 5 uM and increasing doses of COV-08-0064. (A) Transcript for pro-IL-1β and Nlrp3 were quantitated by quantitative PCR in PECs treated ODN1826 at 1uM with increasing doses of COV-08-0064 as detailed in Materials and Methods. (B) Transcript for pro-IL-1β and Nlrp3 were quantitated by quantitative PCR in PECs co-incubated with imiquimod at 5 uM and increasing doses of COV-08-0064. (C) Supernatant protein levels of IL-12p40 were quantitated in PECs co-incubated with ODN182 at 1uM and COV08-0064 at 10 uM. Each data point represents the mean of three independent experiments with standard deviations shown in the brackets. Asterisks denote significant differences between bracketed groups (P<0.01) and NS denotes no significant differences using an unpaired 2-tailed Student’s t test.
COV08-0064 protects from acetaminophen (APAP) induced acute liver injury and inflammation
COV08-0064 pretreatment at 60 mg per kilogram body weight protects wild-type mice from APAP induced acute liver injury and inflammation. Liver necrosis was significantly reduced with COV08-0064 pre-treatment compared to saline pre-treatment as noted on hematoxylin and eosin-stained liver tissue histology and on blinded histology scoring with necrosis scores of 1.2 ± 0.4 versus 2.1 ± 0.4, respectively (Fig. 5A and 5B). Liver hemorrhage was also significantly reduced with COV08-0064 pre-treatment compared to saline pre-treatment with hemorrhage scores of 1.1 ± 0.6 versus 2.2 ± 0.4, respectively (Fig. 5A and 5B). Neutrophil infiltration into the liver parenchyma was significantly reduced with COV08-0064 pre-treatment compared to saline pre-treatment with neutrophil counts per 40× magnified high powered field of 15 ± 2 versus 36 ± 3, respectively (Fig. 5C and 5D). Serum ALT levels were significantly reduced with COV08-0064 pre-treatment from 10,375 ± 1990 to 3010 ± 549 units per mL (Fig. 5E). Liver caspase-1 activity was significantly reduced with COV08-0064 pre-treatment from 93 ± 32 to 14 ± 21 activity units per gram liver tissue (Fig. 5F). Liver IL-12p40 levels were significantly reduced with COV08-0064 pre-treatment from 125 ± 13 to 76 ± 17 pg per gram liver tissue (Fig. 5G). Liver IL-6 levels were significantly reduced with COV08-0064 pre-treatment from 292 ± 17 to 238 ± 18 pg per gram liver tissue (Fig. 5H).
Figure 5.
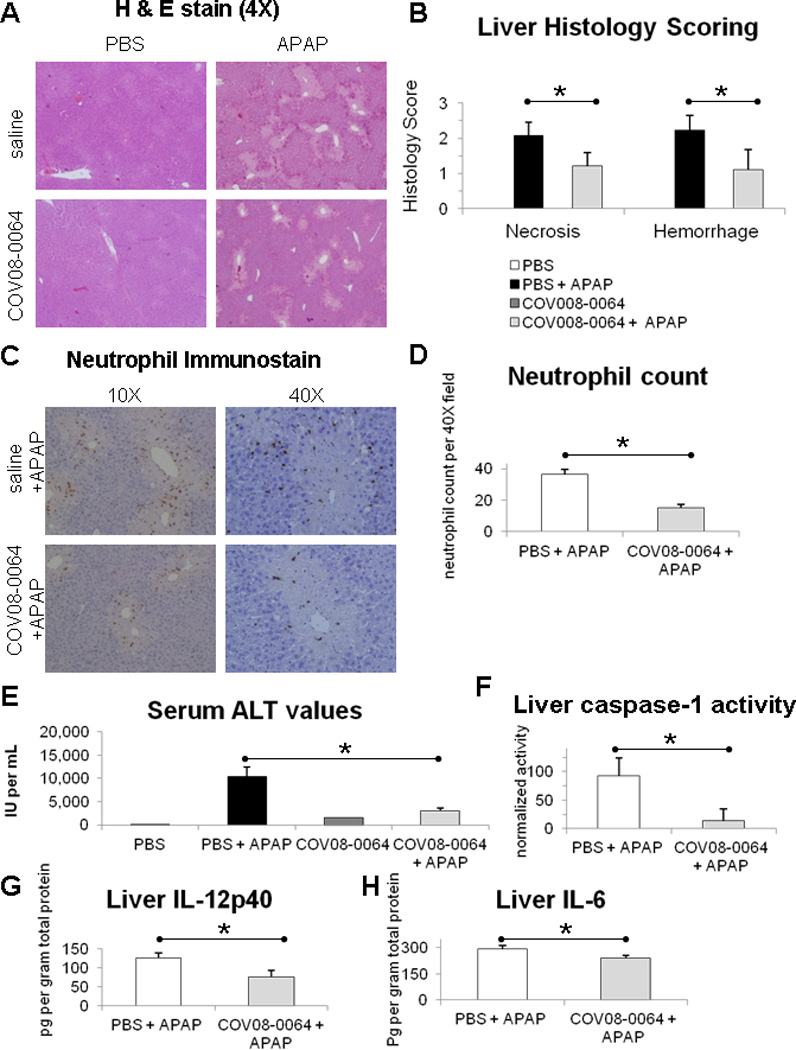
COV08-0064 pre-treatment decreases liver inflammation and injury in acetaminophen hepatotoxicity in mice. C57BL/6 mice were administered COV08-0064 at 60 mg per kilogram body weight or saline by intraperitoneal injection one hour before administration of acetaminophen (APAP) at 500 mg per kilogram body weight or PBS. Mice were sacrificed at 12 hours post-APAP treatment for assessment detailed herein. (A) Representative histology of hematoxylin and eosin (H & E) stained formalin fixed liver sections at 10 × magnification. (B) Blinded histology scoring of liver H & E stained sections for hemorrhage and necrosis as described in Materials and Methods. (C) Representative immunostaining for Ly-6B.2 in formalin fixed liver sections at 10 × magnification as described in Materials and Methods. (D) Blinded scoring for neutrophils per 40 × field in liver sections immunostained for Ly-6B.2. (E) Serum ALT values. (F) Liver caspase-1 activity normalized to total protein level. (G) Liver IL-12p40 levels normalized to total protein level. (H) Liver IL-6 levels normalized to total protein level. Saline and PBS group (n=2) (white bars), PBS and APAP group (n=8) (black bars), COV08-0064 and PBS group (n=2) (dark gray bars), and COV08-0064 and APAP (n=8) (light gray bars). Each data point represents the mean with standard deviations shown in the brackets of these respective groups. Asterisks denote significant differences between the PBS and APAP group and the COV08-0064 and APAP group (P<0.01) using an unpaired 2-tailed Student’s t test.
COV08-0055, a morphinan structurally related to COV08-0064, has no TLR9 antagonist activity and does not protect from acetaminophen induced acute liver injury when administered at the same dose, timing, and route of administration as COV08-0064 (Supplementary Figure 1).
COV08-0064 protects from caerulein hyperstimulation induced acute pancreatic injury and inflammation
COV08-0064 treatment at 30 mg per kilogram body weight administered for three subcutaneous injections concurrent with caerulein treatment protects wild-type mice from acute pancreatic induced acute liver injury and inflammation. Pancreatic edema is markedly reduced with COV08-0064 co-treatment as noted in tissue sections (Fig. 6A). Neutrophil infiltration into the pancreas was significantly reduced with COV08-0064 co-treatment compared to saline co-treatment with neutrophil counts per 40× magnified high powered field of 10 ± 1 versus 24 ± 4, respectively (Fig. 6A and 6C). Serum amylase levels were significantly reduced with COV08-0064 co-treatment from 0.8 ± 0.1 to 0.5 ± 0.1 units per mL (Fig. 6B). Pancreatic trypsin activity was significantly reduced with COV08-0064 co-treatment from 13.1 ± 4.5 to 2.4 ± 0.5 normalized units (Fig. 6D). Pancreatic caspase-1 activity was significantly reduced with COV08-0064 co-treatment from 5.8 ± 0.9 to 2.3 ± 0.4 normalized units (Fig. 6E). Pancreatic IL-12p40 levels were significantly reduced with COV08-0064 co-treatment from 101 ± 17 to 40 ± 11 pg per gram pancreatic tissue (Fig. 6F). Finally, pancreatic IL-6 levels were also significantly reduced with COV08-0064 co-treatment from 43 ± 7 to 25 ± 3 pg per gram pancreatic tissue (Fig. 6G).
Figure 6.
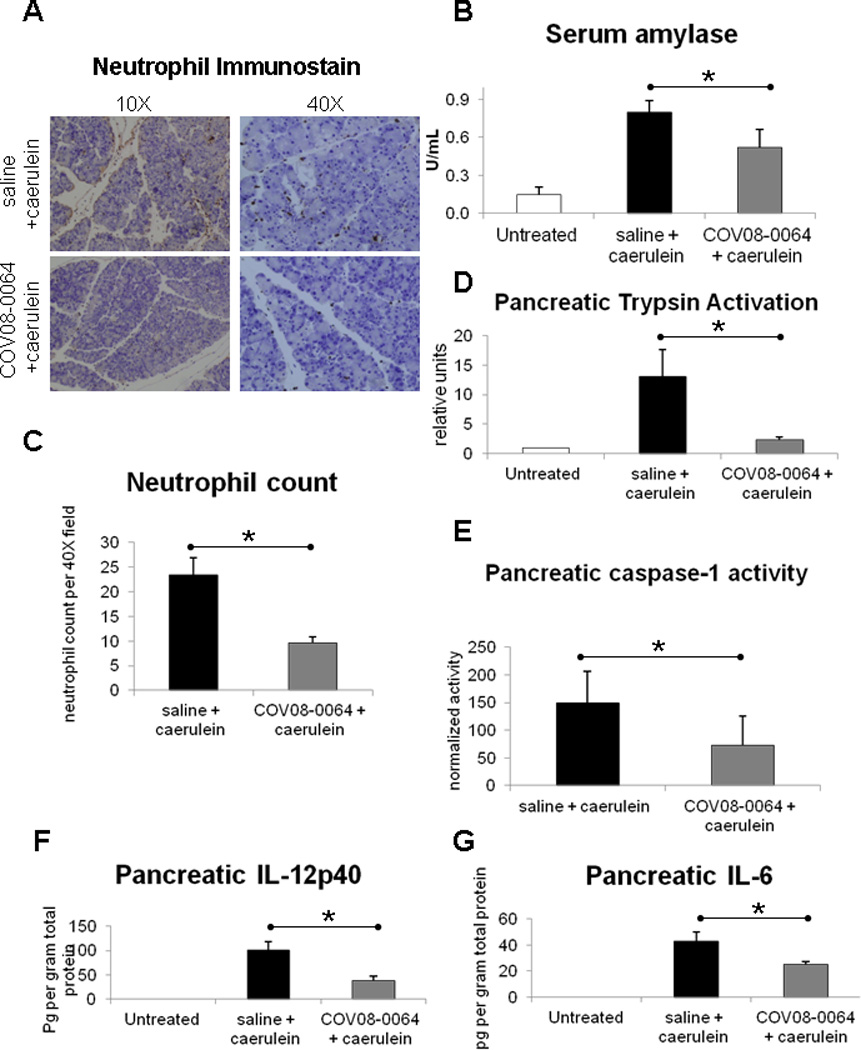
COV08-0064 co-treatment decreases pancreatic inflammation and injury in caerulein hyperstimulation induced acutre pancreatitis in mice. C57BL/6 mice were administered COV08-0064 at 30 mg per kilogram body weight or saline by subcutaneous injection concurrent with doses one, four, and seven of 10 sequential hourly doses of caerulein administered at 50 ug per kilogram body weight by intraperitoneal injection. Mice were sacrificed at 1 hour after the last dose of caerulein. (A) Representative immunostaining for Ly-6B.2 in formalin fixed pancreatic sections at 10 × and 40 × magnification as described in Materials and Methods. (B) Serum amylase values determined by commercial colorimetric assay, Phadebas kit. (C) Blinded scoring for neutrophils per 40 × field in pancreatic sections immunostained for Ly-6B.2. (D) Pancreatic trypsin activation as determined by cleavage of fluorescent trypsin substrate Boc-QAR-MCA normalized to total amylase content and trypsin activity in untreated pancreas. (E) Pancreatic caspase-1 activation as determined by cleavage of fluorescent caspase-1 substrate Ac-YVAD-MCA normalized to total amylase content. (F) Pancreatic IL-12p40 levels normalized to total protein content. (G) Pancreatic IL-6 levels normalized to total protein content. For all experiments, untreated animals (n=2) (white bars), saline and caerulein group (n=7) (black bars), and COV08-0064 and caerulein group (n=7) (dark gray bars). Each data point represents the mean with standard deviations shown in the brackets of these respective groups. Asterisks denote significant differences between the saline plus caerulein group and COV08-0064 plus caerulein group (P<0.01) using an unpaired 2-tailed Student’s t test.
Discussion
Pattern conserved molecules in bacteria and viruses were initially identified as ligands for TLRs. Activation of TLRs in-vivo results in a range of biological responses with initiation of innate and adaptive immunity being dominant. Subsequently it was identified that a wide range of self-molecules can also activate TLRs and result in sterile inflammatory responses which are responsible for a wide range of pathologies from acute liver failure, pancreatitis, to end-organ damage after crush injuries(22). This has identified specific TLRs as candidates for antagonism, with TLR9 being a required receptor for the development of many of these pathologies (2, 3, 10,23–27).
Nuclear and mitochondrial DNA are known to be endogenous ligands for TLR9, and synthetic antagonists have the structure of nucleic acids and inhibitory oligonucleotides (ODN) or immunoregulatory DNA sequences (IRS), with chemical modification of the oligophosphate backbone to prevent degradation (11, 28). These are a rational choice but have a number of limitations. Firstly they have antagonist activity against TLR7 which is activated by single stranded RNA molecules. This may be of significance as TLR7 and TLR9 have complex interactions which at times are opposing (13, 14). In addition to a lack of specificity nucleic acid based antagonists have immunoregulatory consequences independent of TLR9 which may become an issue with repeated administrations (17, 18).
To identify novel molecules with TLR9 antagonism we screened over 200 compounds using a TLR9 reporter cell line and identified four with some antagonist activity. Of these COV08-0064 had the best antagonist profile, with almost complete inhibition of TLR9 response at a concentration of 1 microM (Fig. 1A). These compounds are unique TLR9 antagonists as their structure is based on the (+)-morphinan scaffold. The (+)-morphinan class of compounds refers to the enantiomeric analogues of traditional (−)-morphinans such as (−)-Naltrexone and (−)-Naloxone. These compounds are predicted to have no activity on any of the morphine receptors and this was the case when tested by us (data not shown).
In contrast to DNA based TLR9 antagonists, COV08-0064 had a high degree of specificity for TLR9, with no antagonist activity against TLRs 2, 3, 4, 7, or 8 at a concentration ten times greater than required to inhibit most TLR9 response (Fig. 2A). These results from the TLR reporter cell lines are confirmed by in-vitro antagonism of primary peritoneal macrophages where COV08-0064 inhibited responses from a TLR9 but not a TLR7 agonist (Fig. 4A and B). Tissue macrophages including Kupffer cells have been implicated as major innate immune effector cells of sterile inflammation in the liver (29) and, more recently, in the pancreas (30, 31). Our laboratory has found that TLR9 contributes to tissue injury in acetaminophen hepatotoxicity (2) and multiple mouse models of acute pancreatitis (32). We have also established that TLR9 induces pro-inflammatory signals in Kupffer cells (2), is expressed in the immune cell compartment of the pancreas, which is predominantly tissue macrophages, and is required for full inflammatory signaling in this compartment in the pancreas (32). As our investigation is on early acute inflammation and tissue injury, the TLR9 contribution to innate immune responses through tissue macrophages was our focus for in vitro investigation.
In addition to the therapeutic implications of TLR9 selective antagonism, the fact that TLR7 and 9 share many aspects of their biology including compartmentalization and downstream signaling suggests that COV08-0064 is functioning very proximally. To further obtain information on unpredicted off-target effects we performed a large screen of the effect of COV08-0064 on receptor binding and enzyme activity in many pathways. This screen demonstrated that COV08-0064 did not have a significant effect on major pathways (data not shown). COV08-0064 modestly inhibited yohimbine binding to alpha-2B adrenergic receptors, protryptiline binding to norepinephrine transporters and inhibition of COX-1 by diclofenac. Additionally, COV08-0064 was assessed for aqueous solubility, cell culture cytotoxicity, as well as CYP metabolism, all of which were negative. Collectively this entirely novel structure of molecules has potent TLR9 antagonist activity with a high degree of selectivity. In addition to selectivity, DNA based antagonists have poor oral bioavailability. COV08-0064 had 55% oral bioavailability and a half-life of 149 minutes. Intravenous and subcutaneous routes had slightly shorter half-lives (116 +/− 15 and 122 +/− 20), but still significantly longer than the half-lives of DNA based antagonists which have half-lives in the range of 20-25 minutes.
Since TLR receptors were initially identified as sensors of molecules on infectious agents the use of TLR antagonists raises questions regarding increased susceptibility to pathogens (11). This is however unlikely to be the case as there is now a significant amount of data on the infectious consequences of the loss of these pathways in mice and in humans. Mice deficient in individual TLRs or in common signaling pathways are viable and healthy without special care. Humans with loss of analogous pathways or individual TLRs also have a phenotype limited to childhood (33). Direct interventional information is available from a phase one trial of a combined TLR9 and 7 antagonists (DV1179) which did not produce any adverse effects(34).
The ability of this novel structure of molecules to inhibit TLR9 activation is unexpected and many of the above characteristics of specificity, delivery and pharmokinetics makes them attractive for therapeutic antagonism. The range of non-infectious pathologies in which TLR9 has been demonstrated to play a role has been rapidly expanding. Currently these include acute liver failure, pancreatitis, liver and lung fibrosis, ischemia reperfusion injury, graft versus host disease after bone marrow transplantation as well as the systemic inflammatory response after a crush injury (2, 23, 24, 32, 35). We tested the ability of COV08-0064 in two models, one of acute liver injury by acetaminophen and one of acute pancreatitis. In both conditions COV08-0064 significantly reduced serum markers of tissue injury, and reduced tissue inflammation and damage as assessed by histology, neutrophil migration, caspase-1 activity, and TLR9 induced cytokine production (Figures 5 and 6). These data have a number of important implications. The first is that COV08-0064 in addition to having the desired TLR9 antagonist activities in in-vitro tests is also able to reduce a full in-vivo disease response. The additional point is that COV08-0064 was protective in disease models of two different organs, in which injury was initiated by very different mechanisms. To confirm that off-target effects were not responsible for COV08-0064 mediated protection, we assessed COV08-0055, a structurally similar analog with no TLR9 antagonist function, and found the latter to be without hepatoprotective effects in vivo (Supplementary Figure 1). The liver injury was initiated by acetaminophen which is responsible for the majority of cases of acute liver failure in the United States. Toxic metabolites of acetaminophen induce a necrotic hepatocyte death which results in the release of DAMPs including nuclear and mitochondrial DNA further amplifying the initial injury (2, 36). Caerulein induced acute pancreatitis is not due to the production of toxic metabolites of the drug, but by hyperstimulation of the exocrine pancreas which results in the excess production of digestive enzymes. This is a mechanism that is known to be active in many forms of acute pancreatitis. The ability of COV08-0064 to protect in two models demonstrates the importance of downstream activation of TLR9 after initial tissue injury, and also the therapeutic opportunities of TLR9 antagonism.
Supplementary Material
Acknowledgments
Financial support
This work was supported by Covidien Mallinckrodt, NIH R01DK076674-01A2 (WM), VA Merit Award (WM), NIH T32 DK7356 (RH), and NIH K08DK092281 (RH), and the resources of the Yale Liver Center supported by NIH P30DK34989.
References
- 1.Plitas G, Burt BM, Nguyen HM, Bamboat ZM, DeMatteo RP. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J Exp Med. 2008;205:1277–1283. doi: 10.1084/jem.20080162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011;141:358–369. doi: 10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Jong SD, Basha G, Wilson KD, Kazem M, Cullis P, Jefferies W, Tam Y. The immunostimulatory activity of unmethylated and methylated CpG oligodeoxynucleotide is dependent on their ability to colocalize with TLR9 in late endosomes. J Immunol. 184:6092–6102. doi: 10.4049/jimmunol.0802442. [DOI] [PubMed] [Google Scholar]
- 5.Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75:99–1000. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- 6.Wei T, Gong J, Jamitzky F, Heckl WM, Stark RW, Rossle SC. Homology modeling of human Toll-like receptors TLR7, 8, and 9 ligand-binding domains. Protein Sci. 2009;18:1684–1691. doi: 10.1002/pro.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peter ME, Kubarenko AV, Weber AN, Dalpke AH. Identification of an N-terminal recognition site in TLR9 that contributes to CpG-DNA-mediated receptor activation. J Immunol. 2009;182:7690–7697. doi: 10.4049/jimmunol.0900819. [DOI] [PubMed] [Google Scholar]
- 8.Gursel I, Gursel M, Yamada H, Ishii KJ, Takeshita F, Klinman DM. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J Immunol. 2003;171:1393–1400. doi: 10.4049/jimmunol.171.3.1393. [DOI] [PubMed] [Google Scholar]
- 9.Yu D, Wang D, Zhu FG, Bhagat L, Dai M, Kandimalla ER, Agrawal S. Modifications incorporated in CpG motifs of oligodeoxynucleotides lead to antagonist activity of toll-like receptors 7 and 9. J Med Chem. 2009;52:5108–5114. doi: 10.1021/jm900730r. [DOI] [PubMed] [Google Scholar]
- 10.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrat FJ, Coffman RL. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev. 2008;223:271–283. doi: 10.1111/j.1600-065X.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 12.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, Reith W, Mohan C, Kotzin BL, Izui S. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34:339–348. doi: 10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Bhagat L, Yu D, Zhu FG, Tang JX, Kandimalla ER, Agrawal S. Oligodeoxyribonucleotide-based antagonists for Toll-like receptors 7 and 9. J Med Chem. 2009;52:551–558. doi: 10.1021/jm8014316. [DOI] [PubMed] [Google Scholar]
- 16.Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, Zheng W, Ataide MA, de Oliveira RB, Golenbock DT, Gazzinelli RT. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci U S A. 2011;108:3689–3694. doi: 10.1073/pnas.1015406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trieu A, Bokil N, Dunn JA, Roberts TL, Xu D, Liew FY, Hume DA, Stacey KJ, Sweet MJ. TLR9-independent effects of inhibitory oligonucleotides on macrophage responses to S. typhimurium. Immunol Cell Biol. 2009;87:218–225. doi: 10.1038/icb.2008.95. [DOI] [PubMed] [Google Scholar]
- 18.Babiuk S, Mookherjee N, Pontarollo R, Griebel P, van Drunen Littel-van den Hurk S, Hecker R, Babiuk L. TLR9−/− and TLR9+/+ mice display similar immune responses to a DNA vaccine. Immunology. 2004;113:114–120. doi: 10.1111/j.1365-2567.2004.01938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol. 23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2010;51:621–623. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, Mehal WZ. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 25.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–334. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meneghin A, Choi ES, Evanoff HL, Kunkel SL, Martinez FJ, Flaherty KR, Toews GB, Hogaboam CM. TLR9 is expressed in idiopathic interstitial pneumonia and its activation promotes in vitro myofibroblast differentiation. Histochem Cell Biol. 2008;130:979–992. doi: 10.1007/s00418-008-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, Strutz F, Bauer S, Rutz M, Wagner H, Grone HJ, Schlondorff D. Activation of toll-like receptor 9 induces progression of renal disease in MRL-Fas(lpr) mice. Faseb J. 2004;18:534–536. doi: 10.1096/fj.03-0646fje. [DOI] [PubMed] [Google Scholar]
- 28.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 29.Goldin RD, Ratnayaka ID, Breach CS, Brown IN, Wickramasinghe SN. Role of macrophages in acetaminophen (paracetamol)-induced hepatotoxicity. J Pathol. 1996;179:432–435. doi: 10.1002/(SICI)1096-9896(199608)179:4<432::AID-PATH609>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 30.Dang SC, Jiang DL, Chen M, Li D, Zhang JX. Clodronate-containing liposomes attenuate lung injury in rats with severe acute pancreatitis. J Zhejiang Univ Sci B. 11:828–835. doi: 10.1631/jzus.B1000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamichi I, Habtezion A, Zhong B, Contag CH, Butcher EC, Omary MB. Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase 1 induction. J Clin Invest. 2005;115:3007–3114. doi: 10.1172/JCI24912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 141:358–369. doi: 10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang K, Puel A, Zhang S, Eidenschenk C, Ku CL, Casrouge A, Picard C, von Bernuth H, Senechal B, Plancoulaine S, Al-Hajjar S, Al-Ghonaium A, Marodi L, Davidson D, Speert D, Roifman C, Garty BZ, Ozinsky A, Barrat FJ, Coffman RL, Miller RL, Li X, Lebon P, Rodriguez-Gallego C, Chapel H, Geissmann F, Jouanguy E, Casanova JL. Human TLR-7-, 8-, and 9-mediated induction of IFN-alpha/beta and -lambda Is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity. 2005;23:465–478. doi: 10.1016/j.immuni.2005.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Connolly DJ, O'Neill LA. New developments in Toll-like receptor targeted therapeutics. Curr Opin Pharmacol. 2012;12:510–518. doi: 10.1016/j.coph.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Calcaterra C, Sfondrini L, Rossini A, Sommariva M, Rumio C, Menard S, Balsari A. Critical role of TLR9 in acute graft-versus-host disease. J Immunol. 2008;181:6132–6139. doi: 10.4049/jimmunol.181.9.6132. [DOI] [PubMed] [Google Scholar]
- 36.McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.