

Abstract
Background
Previous studies show that the rise in skin blood flow and cutaneous vascular conductance (CVC) during heat stress is substantially attenuated in chronic heart failure (CHF) patients. The mechanism(s) responsible for this finding is not clear. In particular, little is known regarding the responses of skin sympathetic nerve activity (SSNA) that control the skin blood flow during heat stress in CHF patients. We examined the effects of a modest heat stress to test the hypothesis that SSNA responses could be attenuated in CHF.
Methods and Results
We assessed SSNA (microneurography) from the peroneal nerve and skin blood flow (forearm laser Doppler) in 9 patients with stable class II-III CHF and in matched healthy subjects during passive whole body heating with a water perfused suit. Whole body heating induced similar increases in internal temperature (~0.6 °C) in both groups. Whole body heat stress evoked similar SSNA activation in CHF patients (Δ891±110 units/min) and the control subjects (Δ787±84 units/min, P=0.66), while the elevation in forearm CVC in patients with CHF was significantly lower than that in healthy control subjects (Δ131±29 vs. Δ623±131%, P=0.001).
Conclusions
The present data show that SSNA activation during a modest whole body heat stress is not attenuated in CHF. Thus, the attenuated skin vasodilator response in CHF patients is not due to a reduction in total activity of sympathetic outflow to skin.
Keywords: autonomic, regional blood flow, vasodilation, heart failure
Patients with heart disease are particularly vulnerable to injury from heat stress.1 For example, “excess deaths” were noted during a “heat wave”2, 3, and cardiovascular diseases are a major contributor to the excess mortality noted during a “heat wave”.2, 3 Moreover, adverse cardiac events occur at higher frequency during summer months than spring and autumn.1 The mechanisms causing this phenomenon are not understood thoroughly.
We4 and others5 have demonstrated that the rise in skin blood flow (SkBF) and cutaneous vascular conductance (CVC) evoked by whole body heating is less in chronic heart failure (CHF) patients than that in healthy subjects. Although it may be one of the ways the body adapts to heart failure, the attenuated rise in CVC may be a key factor contributing to the heat related injury in those with cardiovascular disease. The mechanism(s) contributing to the impaired rise in cutaneous conductance are not thoroughly understood.
Both cutaneous vascular beds and sweat glands are innervated by the skin sympathetic nerves.6 Upon exposure to a warm/hot environment, the initial increase in SkBF occurs via withdrawal of the cutaneous vasoconstrictor activity.6-8 As internal temperature continues to increase, the cutaneous active vasodilator system is engaged9, and sudomotor activity is activated (i.e. sweating).6 Thus, whole body heat stress increases the total activity of skin sympathetic nerve activity (SSNA)6, 8 and SkBF8 in young healthy individuals. Our previous studies in normal subjects8, 10, 11 demonstrated that both total activity of SSNA and SkBF rose dramatically when whole body heating increased core temperature by more than 0.5 °C. A 0.5 °C increase in core temperature is considered a modest stressor.
SSNA responses to whole body heat stress in CHF patients have not been reported. It is well known that autonomic control is impaired in CHF patients. For example, baseline muscle sympathetic nerve activity is increased, and baroreflex function is impaired.12 Although no significant difference has been found in normothermic baseline SSNA in CHF and control subjects13-15, one might speculate that autonomic adjustment to thermal challenge could be impaired in CHF. Specifically, since the cutaneous vasodilator response to heat stress in CHF is attenuated, it is reasonable to postulate that cutaneous sympathetic vasodilator nerve activity (as well as total SSNA) also is attenuated in CHF. Therefore, the aim of the present study was to examine the SSNA response to whole body heating in CHF. We hypothesized that the SSNA increase with a modest heat stress would be attenuated in CHF patients as compared with control subjects.
Methods
Subjects
Nine patients with CHF (male, age, 61±2 years; height, 180±3 cm; weight, 101±7 kg), and nine age, gender and race matched healthy control subjects (male; age, 62±2 years; height, 177±1 cm; weight, 84±3 kg) participated in this study.
Patients with CHF were eligible on the basis of the following inclusion criteria: 1) New York Heart Association class II–III after stabilization;2) ejection fraction <40% determined by 2-dimensional echocardiography; and 3) no underlying aortic outflow obstruction as assessed by echocardiography. The mean ejection fraction of the CHF patients was 28±3%. Patients were recruited from the Penn State Hershey Heart and Vascular Institute, Hershey, PA. Patients with ischemic and nonischemic pathology were considered eligible for the study. Patients were excluded if they had a recent myocardial infarction, unstable angina, or any angina without dyspnea or exertional fatigue. Patients with serious arrhythmias and other systemic diseases such as liver disease, renal failure, diabetes, lung diseases including asthma and chronic obstructive pulmonary disease were also excluded. Table 1 lists the patients’ conditions and the medication class. All matched control subjects were healthy, and none were taking any medication.
Table 1.
Clinical Findings and Medications for the Patients with CHF
| N | % | |
|---|---|---|
| NYHA Class | ||
| II | 6 | 67 |
| III | 3 | 33 |
| Comorbidities | ||
| Hypertension | 8 | 89 |
| Atrial Fibrillation | 3 | 33 |
| Hyperlipidemia | 9 | 100 |
| Medications | ||
| Beta-Blocker | 9 | 100 |
| ACEI | 8 | 89 |
| ARB | 1 | 11 |
| Digoxin | 2 | 22 |
| Loop Diuretic | 9 | 100 |
| Spironolactone | 4 | 44 |
| Aspirin | 8 | 89 |
| Statin | 9 | 100 |
Medications: medications or class of medications patients were taking; ACEI: angiotensin-converting enzyme inhibitor; ARB:angiotensin receptor blocker. N: subject number. %: percentage in all patients.
Due to safety concerns for the CHF patients, beta-blockers were not withheld. All other medications, including angiotensin-converting enzyme (ACE) inhibitors and diuretics were held after midnight before the study. All patients and the control subjects refrained from caffeine, alcohol, and exercise 24 hours prior to the study. The experimental protocol was approved by the Institutional Review Board of the Milton S. Hershey Medical Center and conformed with the Declaration of Helsinki. Each subject had the purposes and risks of the protocol explained to them before written informed consent was obtained.
Measurements
Internal (i.e., core) temperature (Tcore) was indexed from an ingestible pill telemetry system (HTI Technologies, Palmetto, FL) that was swallowed by volunteers (n=12) approximately 1.5-2 hours prior to data collection, or from a thermocouple placed in the sublingual sulcus (n=6, 4 patients and 2 controls). Telemetry pill measurements correlate well with other internal temperature measurements such as esophageal temperature.16 Mean skin temperature (Tsk) was measured via the weighted average of six thermocouples attached to the skin.17 To indicate the increase in both skin and internal temperatures, mean body temperature (Tbody) was used in this report, and was calculated as follows: 0.9×Tcore + 0.1×Tsk.8, 18 Each volunteer was dressed in a tube-lined suit that permitted the control of Tsk by changing the temperature of the water perfusing the suit. SkBF was indexed from dorsal forearm skin using the mean values of 2 integrating flow probes of laser-Doppler flowmetry (MoorLab, Moor Instruments Ltd, Devon, UK). CVC was calculated from the ratio of the SkBF to mean arterial blood pressure (MAP). The final CVC was expressed as percentage of the normothermic baseline. Forearm sweat rate was measured from forearm skin via capacitance hygrometry (Vaisala, Woburn, MA) using the ventilated capsule method (surface area=2.0 cm2). The areas from which SkBF and sweat rate were measured was not covered by the suit, and the local temperature of these areas were not controlled.
Arterial blood pressure was measured by auscultation of the brachial artery (SureSigns VS3, Philips, Philip Medical System). Heart rate was monitored from the electrocardiogram (Cardicap®5, Datex-Ohmeda, GE Healthcare, NJ). Respiratory frequency was monitored using piezoelectric pneumography. Multifiber recordings of SSNA were obtained with a tungsten microelectrode inserted into the common peroneal nerve. A reference electrode was placed subcutaneously 2-3 cm from the recording electrode. The recording electrode was adjusted until a site was found in which SSNA bursts were clearly identified using previously established criteria19, 20: 1) integrated nerve activity nonsynchronous with the heartbeat; 2) irregular burst activity; 3) generation of reflex bursts during mental or somatosensory stimuli (e.g., loud sound and light stroking of the innervated region); and 4) absence of an increase in activity during inspiratory apnea. The nerve signal was amplified, passed through a band-pass filter with a bandwidth of 500-5,000 Hz, and integrated with a time constant of 0.1 s (Iowa Bioengineering, Iowa City, IA). The nerve signal was routed to a computer screen and a loudspeaker for monitoring throughout the study.
Protocols
The study was conducted in a temperature-controlled room (~23 °C) in the morning (~8:30 to 12:00). After the instrumentation, the tube-lined suit worn by the subject was perfused with 34 °C water. After 5-min of rest, 6-min of data were collected as normothermic baseline. Thereafter, whole-body heating was initiated by elevating Tsk to ~38 °C by perfusing warm (46 °C) water through the water-perfused suit. Whole-body heating continued until Tcore increased a minimum of 0.5 °C; in healthy individuals this is sufficient to cause SSNA activation and pronounced cutaneous vasodilation and sweating.8, 10, 11 Once Tcore was elevated ~0.5 °C, the temperature of the water perfusing the suit was slightly reduced to attenuate the rate of increase in Tcore. Six minutes of data were then obtained as whole body heat stress (WBH) data (see Figure 1).
Figure 1.
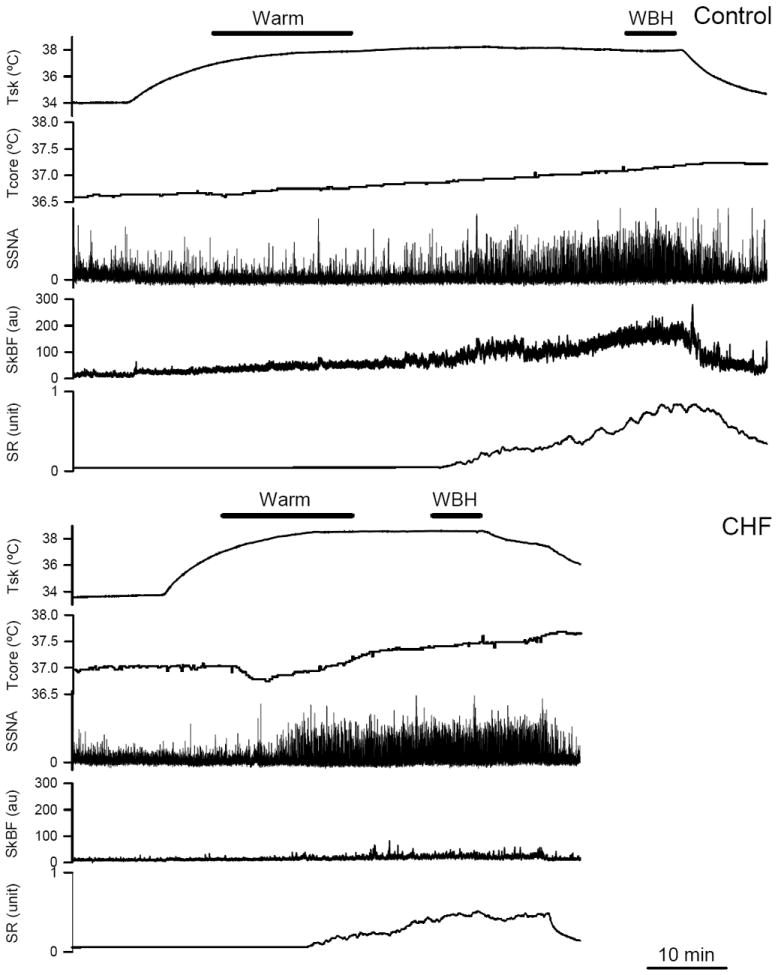
Representative tracings of mean skin temperature (Tsk), internal temperature (Tcore), skin sympathetic nerve activity (SSNA), skin blood flow (SkBF) and sweat rate (SR) during whole body heating in a control subject (upper panel) and a CHF patient (lower panel). SkBF unit: Flux unit. SR unit: mg/cm2/min. Warm: Tsk >37 °C while the increase in Tcore <0.2 °C. WBH: whole body heat stress, the increase in Tcore was greater than 0.5 °C when Tsk was clamped at ~38 °C. Notice the clear SSNA activation in both control subject (upper panels) and CHF patient (lower panels) during WBH.
Data Analysis
Data were sampled at 200 Hz via a data acquisition system (MacLab, AD Instruments, Castle Hill, Australia). SSNA bursts were first identified in real time by visual inspection of the trace, coupled with the burst sound from the audio amplifier. SSNA bursts were further analyzed offline with visual determination. In the case of complex multi-peaked bursts of SSNA, peaks that were separated by a decrease in voltage to less than one-half of the smaller peak were counted as separate bursts.14 Integrated SSNA was normalized by assigning a value of 100 to the mean amplitude of the largest sympathetic bursts (top 10% of identified bursts) during normothermic baseline.8, 10 Subsequent bursts in the neurogram were normalized against that value. To assess total SSNA activity, the baseline was carefully identified and the area of the integrated neurogram above this baseline was measured from the digitized record.8
The mean values of SSNA, thermal and hemodynamic variables during the 6-min normothermic baseline and the 6-min WBH data were calculated. In addition, the mean values of the variables over an early period of heating (“Warm”), in which Tsk was greater that 37 °C while the increase in Tcore was less than 0.2 °C, also were calculated (see Figure 1). These data were analyzed because differences in SSNA responses over this period were observed between the groups. Statistical analyses were performed with the use of SigmaStat software (SPSS Science). SSNA, thermoregulatory and hemodynamic variables were used to examine two main effects: 1) the effect of the heating (i.e. baseline, Warm and WBH); 2) the effects of group (CHF or control); and the interaction between the two factors via repeated measures (one factor repetition) two-way ANOVA. When appropriate, Tukey’s post-hoc method to adjust for multiple comparisons was employed. The differences in the changes (delta) from the baseline to WBH response between the groups were evaluated via unpaired t-test to further examine the effects of heating. Since our sample size was limited, we examined residual diagnostics to ensure that parametric modeling assumptions were met. Moreover, the changes from baseline to heat stress condition between the 2 groups were also evaluated using the non-parametric Mann-Whitney Rank Sum test. This was done to determine if similar results would be obtained. A post-hoc power calculation was performed via an unpaired t-test. For this analysis we assumed our study sample size, a type I error of 5%, and the observed SSNA standard deviation in the present study. Values are reported as mean ± SEM. Probability values <0.05 were considered statistically significant.
Results
Temperatures Change During Whole Body Heating
Baseline temperatures were not different between groups (Table 2). Figure 1 shows examples of Tsk, Tcore, SSNA, SkBF and sweat rate during whole body heating in a CHF patient and the matched healthy control subject. Interestingly, during the initial period of heating, Tsk increased while Tcore did not change or slightly decreased in some subjects. As heating continued, Tcore began to rise. During the later period of whole body heating, Tsk was clamped at ~38 °C with the suit, while Tcore rose continually. As a result of controlling the Tsk and the increase in Tcore, the elevation in Tcore for the CHF patients (Δ0.62 ± 0.03 °C) for the WBH was not different from the control subjects (Δ0.60 ± 0.06 °C, P=0.38). Combined with the similar increases in Tsk in CHF patients (Δ4.3 ± 0.3 °C) and control subjects (Δ4.2 ± 0.1°C, P=0.26), the increase in Tbody during WBH was not different between CHF patients (Δ1.0 ± 0.1 °C) and control subjects (Δ1.0 ± 0.1 °C, P=0.16). Thus, the heat stress challenge was similar in the two groups.
Table 2.
Thermal and Hemodynamic Variables during Whole Body Heating
| Baseline CHF | Control | Warm CHF | Control | WBH CHF | Control | P for Heating | P for Groups | P for Interaction | |
|---|---|---|---|---|---|---|---|---|---|
| SBP (mmHg) | 116±5 | 123±4 | 109±3* | 117±3* | 110±3* | 118±3 | 0.001 | 0.152 | 0.761 |
| DBP (mmHg) | 68±3 | 74±2 | 62±2* | 69±2* | 59±3*† | 68±2* | 0.001 | 0.049 | 0.443 |
| MAP (mmHg) | 84±4 | 90±3 | 78±2* | 85±2* | 76±3*† | 85±2* | 0.001 | 0.057 | 0.547 |
| HR (beats/min) | 63±2 | 58±3 | 67±3 | 68±4* | 73±4* | 77±4* | 0.001 | 0.972 | 0.016 |
| Tsk (C°) | 33.7±0.2 | 34.0±0.1 | 37.6±0.2*† | 38.0±0.1* | 38.0±0.1* | 38.2±0.1* | 0.001 | 0.005 | 0.206 |
| Tcore (C°) | 36.6±0.1 | 36.8±0.1 | 36.8±0.1* | 36.9±0.1* | 37.3±0.1* | 37.4±0.1* | 0.001 | 0.109 | 0.925 |
| Tbody (C°) | 36.3±0.1 | 36.5±0.1 | 36.8±0.1* | 37.0±0.1* | 37.4±0.1* | 37.5±0.1* | 0.001 | 0.506 | 0.478 |
Mean arterial blood pressure (MAP) was calculated as two-third diastolic blood pressure (DBP) plus one-third systolic blood pressure (SBP), measured by auscultation of the brachial artery. HR: heart rate. Tsk: Mean skin temperature. Tcore: Internal temperature. Mean body temperature (Tbody) was calculated as follows: 0.9×Tcore + 0.1×Tsk. Warm: an early period of heating. WBH: the last 5-min of whole body heating
: significantly different from normothermic Baseline (P<0.05).
: significantly different from Control subjects (P<0.05).
Blood Pressure and Heart Rate Responses to Whole Body Heating
Baseline hemodynamic variables were not different between groups (Table 2). During heating, blood pressures dropped and heart rate rose in both CHF patients and the control subjects (Table 2). However, the increase in heart rate during WBH was significantly less in CHF patients (Δ10±3 beats/min) than in the control subjects (Δ19±3 beats/min, P=0.02), while the MAP decrease in CHF patients (Δ8±2 mmHg) was not significantly different from that in the control subjects (Δ6±1 mmHg, P=0.16).
SSNA, CVC and Sweating Responses to Whole Body Heating
SSNA did not rise during the initial period of heating. As heating continued, SSNA progressively increased (Figure 1). The mean values of SSNA, CVC and sweat rate responses as Tbody increases are illustrated in Figure 2. During the “Warm” period (i.e. Tsk >37 °C and increase in Tcore <0.2 °C), SSNA and sweat rate rose from baseline in CHF patients, but not in the control groups (Figure 3). During WBH, SSNA was significantly greater than the respective baseline in both groups, and there was no difference in the SSNA responses in the two groups (Figure 3). Moreover, the SSNA increase (i.e. the delta from the baseline) in CHF patients (Δ29.6±1.9 bursts/min; Δ891±110 units/min) was not significantly different from the control groups (Δ27.1±2.2 bursts/min, P=0.20; Δ787±84 units/min, P=0.23).
Figure 2.
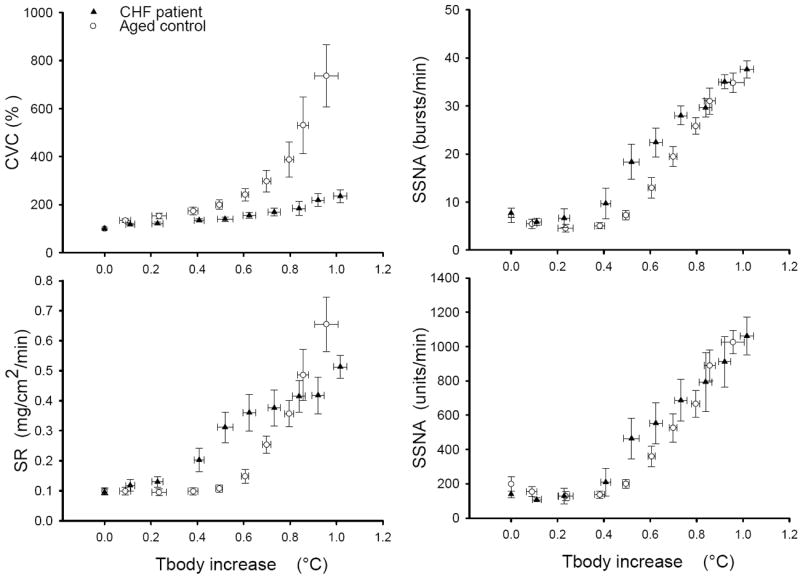
Mean SSNA, cutaneous vascular conductance (CVC) and SR responses to whole body heating. During the initial period of heating, Tsk increased, but the Tcore did not rise. In the later period of whole body heating, Tsk was clamped at ~38 °C, while the Tcore increased. Mean body temperature (Tbody) was calculated as follows: 0.9×Tcore + 0.1×Tsk.
Figure 3.
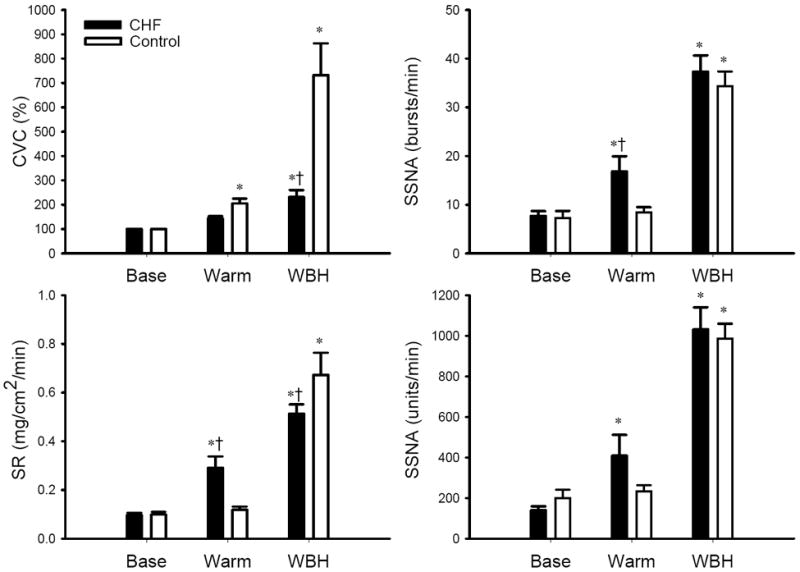
SSNA, CVC and SR responses to Warm and WBH. CVC is expressed as percent of normothermic baseline (Base). Warm and WBH: see definition in Figure 1. *: significantly different from Baseline (P<0.05). †: significantly different from Control subjects (P<0.05). For CVC (left upper panel), P=0.002 for subject groups; P<0.001 for heating, P<0.001 for the interaction. For SR (left lower panel), P=0.931 for subject groups; P<0.001 for heating, P=0.003 for the interaction. For SSNA burst rate change (right upper panel), P=0.04 for subject groups; P<0.001 for heating, P=0.049 for the interaction. For SSNA total activity change (right lower panel), P=0.342 for subject groups, P<0.001 for heating, P=0.238 for the interaction.
During the “Warm” period, the CVC in the control subjects rose, but the CVC in CHF patients was not significantly different from baseline (Figure 3). WBH induced a significant increase in CVC in both groups, but the CVC in CHF patients was significantly lower than that noted in the control subjects (Figure 3). Moreover, the elevation (the delta from the baseline) in CVC in CHF patients (Δ131±29%) was significantly attenuated when compared with control subjects (Δ632±131%, P=0.001). WBH induced sweating in both groups, but the sweat rate in CHF patients was lower than the control subjects (Figure 3).
When a Mann-Whitney Rank Sum was employed, similar results (i.e. P> or <0.05 for the comparisons between the 2 groups) were found for ΔTsk, ΔTcore, ΔTbody, Δheart rate, ΔMAP, ΔSSNA and ΔCVC.
Discussion
The major finding of the present study is that despite the fact that modest whole body heat stress evokes similar increases in SSNA in CHF and control subjects, the cutaneous vasodilator response to the heat stimulus is significantly attenuated in CHF patents. These observations do not support our hypothesis that the attenuated cutaneous vasodilator responses to heat in CHF would be associated with reduced sympathetic activation.
Consistent with previous reports4, 5, the present data show that the cutaneous vasodilator responses to heat in CHF are significantly attenuated. The forearm skin, in which the SkBF was measured, was not directly heated by the suits. Thus, the observed cutaneous vasodilation was likely due to indirect heating via neural control mechanisms.
Classic early studies21, 22 demonstrate that human skin is innervated by sympathetic vasodilator nerves, since nerve blockade prevented the large increases in SkBF that were seen with core hyperthermia in the absence of nerve blockade. Activation of the active vasodilator system is responsible for ~80-90% of the elevation in SkBF to indirect heat stress.23 Thus, the cutaneous vasodilation during body heating is primarily neutrally mediated events.9 The precise systems engaged to evoke neural vasodilator activity are not well understood.24 It has been hypothesized that cholinergic sudomotor nerve activity 25 or a co-transmitter system24, 26 are involved in the cutaneous active vasodilation. Based on these observations we were surprised that vasodilation was attenuated in CHF, whereas SSNA responses to heat were preserved.
We cannot exclude that cutaneous vasodilator activity might be decreased while the discharges of the constrictor and/or sudomotor nerve might be enhanced. However, cutaneous constrictor tone falls in normal subjects with heat stress27, and the sweat rate was low in CHF patients in the present study. Thus, we doubt that our results could be explained by combination of attenuated vasodilator and enhanced vasoconstrictor or sudomotor activity in CHF patients during heat stress.
The maximal peripheral vasodilator response to stimuli such as forearm circulatory arrest (i.e. reactive hyperemia response) is impaired in CHF.28 Previous work suggests that maximal cutaneous vasodilation evoked by local heating is also reduced in CHF patients.4 However, the impaired cutaneous vasodilator response to whole body heat stress was not due to a ceiling effect.4 Thus, we speculate other factors such as cutaneous vascular dysfunction are likely to contribute to the attenuated cutaneous vasodilator response seen in CHF. In an additional set of analyses, we compared CVC in the CHF and the control subjects at the same sweat rate. To perform these analyses, the end heat stress CVC values in CHF were compared to CVC at the same sweat rate in the control subjects (i.e. prior to the end of heating). When the sweat rates were matched in this way (CHF vs. Control: 0.513±0.038 vs. 0.512±0.041 mg/cm2/min, P=0.98), CVC were dramatically lower in the CHF patients (Δ131±29% of baseline) than that in control subjects (Δ532±101% of baseline, P=0.002). These data support the concept that for a given level cholinergic nerve activity25, cutaneous vasodilator responses are attenuated in CHF.
Alone this line, it is known that CHF mediates structural changes29 in the cutaneous vasculature, impairs endothelial function30-33 and nitric oxide production, and reduces the vascular responsiveness to nitric oxide.30, 34, 35 Green and colleagues have shown a significant nitric oxide contribution to heat-induced skin vasodilation in control subjects, but not in CHF patients.5 Thus, the impaired SkBF response to heating observed in CHF patients may be in part explained by impaired nitric oxide function.31, 36 On the other hand, it is well known that plasma norepinephrine levels are much higher in CHF patients in normothermic37 and whole body heating38 conditions. The increased plasma norepinephrine concentration could limit the vasodilation. However, this effect would be in part counterbalanced by an impairment in cutaneous adrenergic receptors that is seen with heating.39
Beta-blockers were taken by all CHF patients on the day of study. This could have restrained the rise in heart rate and cardiac output, and have limited the increase in SkBF. It is known that beta-blockers attenuate cutaneous vasodilation during exercise in healthy individuals.40, 41 However, it should be noted that cutaneous vasodilation is attenuated in CHF patients even when not taking beta-blockers in the previous report.4 Thus, beta-blockers should not be the only factor for the attenuated vasodilation.
We withheld other medications on the study day. However, this approach might not have entirely removed untoward effects of these agents. For example, Holowatz et al showed that chronic low-dose aspirin therapy attenuates reflex cutaneous vasodilation in middle-aged patients.42
The WBH induced significant sweating responses in CHF patients, but the sweat rate in CHF patients in WBH condition was significantly lower than the control subjects. Since the total SSNA activation to WBH (sudomotor + vasodilator activity) is similar to healthy controls, the attenuated sweat rate in CHF patients could be caused by altered responsiveness of sweat glands. There is no report regarding whether the sweat gland function is altered in CHF patients. Beta-blockers might alter the sweat rate43, however, oral propranolol administration in healthy individuals has been shown to increase40, decrease43, or have no effect44 on whole body sweat rate during exercise in healthy individuals. Thus, function of sweat glands in CHF patients should be examined in further studies.
In the present study, the time for heating to increase Tcore by 0.5 °C (i.e. the target) in CHF (2571±131 sec) was significantly shorter than that in control subjects (3318±177 sec, P=0.002), while the water temperature and flow in the suit were well controlled. The body mass index (BMI) of the patients was greater than the controls (31±1 vs. 27±1, P=0.007), which might affect the Tcore increase rate. To our knowledge, there are no reports regarding the internal temperature increase rate during passive heating in obese subjects. Theoretically, compared to lean subjects, larger BMI (i.e. body mass/surface) subjects have more mass relative to skin surface. This could conceivably slow the increase in core temperature in obese subjects. Thus, the greater Tcore increase rate in CHF likely indicates an impairment in thermoregulatory responses. For example, the lower sweat rate in CHF would act to attenuate heat dissipation during heating, and this could contribute to the greater increase rate in Tcore.
Thus during exposure to heat, CHF patients have attenuated cutaneous vasodilator and sweating responses, and a more rapid increase in body temperature. These data help explain prior clinical observations of cardiac decompensation during “heat waves”.1, 2 Clearly these responses need to be contrasted with the effects of chronic exposure to warm, not extreme, weather which may be associated with a reduced incidence of heart failure decompensation.45 The mechanisms for this beneficial effect are not clear but may include the improvement in cardiac function.38 It is clear that further studies are necessary to systemically examine the range of cardiovascualr responses to thermoregulatory challenge seen in CHF patients.
Additionally, CVC rises during the “Warm” period in the controls, where cutaneous vasodilation was not seen in CHF. It is interesting that in CHF patients both SSNA and sweat rate increased during the “Warm” period of heating, while neither increased in healthy controls during this period. It should be noted that this is a very mild heating condition. No patients or control subjects reported any discomfort during this very mild heating stage. Thus, the SSNA increase in CHF is likely not due to mental stress. The mechanism(s) for the SSNA and sweat rate increases in CHF during this stage is unclear and can only be postulated. It should be noted that a prior clinical report has suggested that an increased sweating rate might be an important symptom in CHF.46 Our data can support the clinical observations of excessive sweating in CHF during simple activities of daily living in patients with CHF.
Study Limitations
Multiunit SSNA recordings do not afford the opportunity to discriminate between sympathetic influences to blood vessels and sweat glands.19 Thus, the present experiment does not afford the opportunity to directly evaluate the various components of SSNA response in CHF. More work using local pharmacological approaches will be needed.27 However, the present observations clearly show that the attenuated cutaneous vasodilation seen in CHF is not caused by an attenuated total SSNA response to whole body heat stress.
We recognize that the variability between subjects and the relatively small subject number increase the likelihood of committing a type II error for the result of no significant difference in SSNA responses between the controls and CHF subjects. Increasing the number of subjects would increase the statistical power. However, it is quite doubtful that the observed ~4.6% differences in SSNA between the groups in heat stress condition in this study are biologically meaningful. Previous studies showed no significant difference in SSNA between study groups when mean differences between groups were in the range of ~9-40%13-15, 47. Grassi et al found SSNA differences between study groups when there was a 51% difference 7. Based on prior literture we assume that a minimal 40-50% mean difference is needed to realize a biologically meaningful group difference in SSNA. Based on the SSNA standard deviation of 30% (CHF group) in this study, our post-hoc power calculation suggests that our sample size of 9 subjects for each group would provide 76-91% power to detect such biologically meaningful difference in SSNA (i.e. Δ40-50%). Thus, we believe our interpretation of the data suggesting that SSNA responses to heat stress were not different in controls and CHF is valid.
In conclusion, our findings provide the first direct evidence that SSNA activation to a modest whole body heat stress in CHF patients is not attenuated. Thus, the attenuated cutaneous vasodilator response to heat in CHF patients is not induced by a reduction in total activity of sympathetic outflow to skin. We speculate that impaired intrinsic vasodilator pathways contribute to this response.
Supplementary Material
Acknowledgments
We are grateful to Jennifer L. Stoner for secretarial help in preparing this manuscript.
Sources of Funding
This work was supported by the American Heart Association Grant 0565399U (Cui), 0635245 N (Cui), NIH P01 HL096570 (Sinoway), M01 RR010732 (GCRC Grant), C06 RR016499 (Construction Grant) and UL1 RR033184 (Sinoway).
Footnotes
Disclosures
None.
References
- 1.Depasquale NP, Burch GE. The seasonal incidence of myocardial infarction in new orleans. Am J Med Sci. 1961;242:468–474. doi: 10.1097/00000441-196110000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Semenza JC, Rubin CH, Falter KH, Selanikio JD, Flanders WD, Howe HL, Wilhelm JL. Heat-related deaths during the july 1995 heat wave in chicago. N Engl J Med. 1996;335:84–90. doi: 10.1056/NEJM199607113350203. [DOI] [PubMed] [Google Scholar]
- 3.Fouillet A, Rey G, Laurent F, Pavillon G, Bellec S, Guihenneuc-Jouyaux C, Clavel J, Jougla E, Hemon D. Excess mortality related to the august 2003 heat wave in france. Int Arch Occup Environ Health. 2006;80:16–24. doi: 10.1007/s00420-006-0089-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui J, Arbab-Zadeh A, Prasad A, Durand S, Levine BD, Crandall CG. Effects of heat stress on thermoregulatory responses in congestive heart failure patients. Circulation. 2005;112:2286–2292. doi: 10.1161/CIRCULATIONAHA.105.540773. [DOI] [PubMed] [Google Scholar]
- 5.Green DJ, Maiorana AJ, Siong JH, Burke V, Erickson M, Minson CT, Bilsborough W, O’Driscoll G. Impaired skin blood flow response to environmental heating in chronic heart failure. Eur Heart J. 2006;27:338–343. doi: 10.1093/eurheartj/ehi655. [DOI] [PubMed] [Google Scholar]
- 6.Bini G, Hagbarth K-E, Hynninen P, Wallin BG. Thermoregulatory and rhythm-generating mechanisms governing the sudomotor and vasoconstrictor outflow in human cutaneous nerves. J Physiol (London) 1980;306:537–552. doi: 10.1113/jphysiol.1980.sp013413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grassi G, Seravalle G, Turri C, Bertinieri G, Dell’Oro R, Mancia G. Impairment of thermoregulatory control of skin sympathetic nerve traffic in the elderly. Circulation. 2003;108:729–735. doi: 10.1161/01.CIR.0000081769.02847.A1. [DOI] [PubMed] [Google Scholar]
- 8.Cui J, Sathishkumar M, Wilson TE, Shibasaki M, Davis SL, Crandall CG. Spectral characteristics of skin sympathetic nerve activity in heat-stressed humans. Am J Physiol Heart Circ Physiol. 2006;290:H1601–H1609. doi: 10.1152/ajpheart.00025.2005. [DOI] [PubMed] [Google Scholar]
- 9.Johnson JM, Proppe DW. Cardiovascular adjustments to heat stress. In: Fregly MJ, Blatteis CM, editors. Handbook of physiology - environmental physiology. New York: Oxford University Press; 1996. pp. 215–243. [Google Scholar]
- 10.Cui J, Wilson TE, Crandall CG. Orthostatic challenge does not alter skin sympathetic nerve activity in heat-stressed humans. Auton Neurosci. 2004;116:54–61. doi: 10.1016/j.autneu.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Wilson TE, Cui J, Crandall CG. Absence of arterial baroreflex modulation of skin sympathetic activity and sweat rate during whole-body heating in humans. J Physiol. 2001;536:615–623. doi: 10.1111/j.1469-7793.2001.0615c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grassi G, Seravalle G, Cattaneo BM, Lanfranchi A, Vailati S, Giannattasio C, Del Bo A, Sala C, Bolla GB, Pozzi M. Sympathetic activation and loss of reflex sympathetic control in mild congestive heart failure. Circulation. 1995;92:3206–3211. doi: 10.1161/01.cir.92.11.3206. [DOI] [PubMed] [Google Scholar]
- 13.Grassi G, Colombo M, Seravalle G, Spaziani D, Mancia G. Dissociation between muscle and skin sympathetic nerve activity in essential hypertension, obesity, and congestive heart failure. Hypertension. 1998;31:64–67. doi: 10.1161/01.hyp.31.1.64. [DOI] [PubMed] [Google Scholar]
- 14.Silber DH, Sutliff G, Yang QX, Smith MB, Sinoway LI, Leuenberger UA. Altered mechanisms of sympathetic activation during rhythmic forearm exercise in heart failure. J Appl Physiol. 1998;84:1551–1559. doi: 10.1152/jappl.1998.84.5.1551. [DOI] [PubMed] [Google Scholar]
- 15.Middlekauff HR, Hamilton MA, Stevenson LW, Mark AL. Independent control of skin and muscle sympathetic nerve activity in patients with heart failure. Circulation. 1994;90:1794–1798. doi: 10.1161/01.cir.90.4.1794. [DOI] [PubMed] [Google Scholar]
- 16.O’Brien C, Hoyt RW, Buller MJ, Castellani JW, Young AJ. Telemetry pill measurement of core temperature in humans during active heating and cooling. Med Sci Sports Exerc. 1998;30:468–472. doi: 10.1097/00005768-199803000-00020. [DOI] [PubMed] [Google Scholar]
- 17.Taylor WF, Johnson JM, Kosiba WA, Kwan CM. Cutaneous vascular responses to isometric handgrip exercise. J Appl Physiol. 1989;66:1586–1592. doi: 10.1152/jappl.1989.66.4.1586. [DOI] [PubMed] [Google Scholar]
- 18.Wenger CB, Roberts MF, Stolwijk JAJ, Nadel ER. Forearm blood flow during body temperature transients produced by leg exercise. J Appl Physiol. 1975;38:58–63. doi: 10.1152/jappl.1975.38.1.58. [DOI] [PubMed] [Google Scholar]
- 19.Hagbarth K-E, Hallin RG, Hongell A, Torebjörk HE, Wallin BG. General characteristics of sympatheric activity in human skin nerves. Acta Physiol Scand. 1972;84:164–176. doi: 10.1111/j.1748-1716.1972.tb05167.x. [DOI] [PubMed] [Google Scholar]
- 20.Vallbo AB, Hagbarth K-E, Torebjörk HE, Wallin BG. Somatosensory, proprioceptive and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- 21.Roddie IC, Shepherd JT, Whelan RF. The contribution of constrictor and dilator nerves to the skin vasodilatation during body heating. J Physiol. 1957;136:489–497. doi: 10.1113/jphysiol.1957.sp005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edholm OG, Fox RH, Macpherson RK. Vasomotor control of the cutaneous blood vessels in the human forearm. J Physiol. 1957;139:455–465. doi: 10.1113/jphysiol.1957.sp005904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowell LB. Reflex control of the cutaneous vasculature. J Invest Dermatol. 1977;69:154–166. doi: 10.1111/1523-1747.ep12497938. [DOI] [PubMed] [Google Scholar]
- 24.Kellogg DL, Jr, Pergola PE, Piest KL, Kosiba WA, Crandall CG, Grossmann M, Johnson JM. Cutaneous active vasodilation in humans is mediated by cholinergic nerve cotransmission. Circ Res. 1995;77:1222–1228. doi: 10.1161/01.res.77.6.1222. [DOI] [PubMed] [Google Scholar]
- 25.Brengelmann GL, Freund PR, Rowell LB, Olerud JE, Kraning KK. Absence of active cutaneous vasodilation associated with congenital absence of sweat glands in humans. Am J Physiol Heart Circ Physiol. 1981;240:H571–H575. doi: 10.1152/ajpheart.1981.240.4.H571. [DOI] [PubMed] [Google Scholar]
- 26.Hokfelt T, Johansson O, Ljungdahl A, Lundberg JM, Schultzberg M. Peptidergic neurones. Nature. 1980;284:515–521. doi: 10.1038/284515a0. [DOI] [PubMed] [Google Scholar]
- 27.Kellogg DL, Jr, Johnson JM, Kosiba WA. Selective abolition of adrenergic vasoconstrictor responses in skin by local iontophoresis of bretylium. Am J Physiol Heart Circ Physiol. 1989;257:H1599–H1606. doi: 10.1152/ajpheart.1989.257.5.H1599. [DOI] [PubMed] [Google Scholar]
- 28.Zelis R, Mason DT, Braunwald E. A comparison of the effects of vasodilator stimuli on peripheral resistance vessels in normal subjects and in patients with congestive heart failure. J Clin Invest. 1968;47:960–970. doi: 10.1172/JCI105788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wroblewski H, Norgaard T, Haunso S, Kastrup J. Microvascular distensibility in two different vascular beds in idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 1995;269:H1973–H1980. doi: 10.1152/ajpheart.1995.269.6.H1973. [DOI] [PubMed] [Google Scholar]
- 30.Drexler H, Hayoz D, Munzel T. Endothelium function in chronic congestive heart failure. Am J Cardiol. 1992;69:1596–1601. doi: 10.1016/0002-9149(92)90710-g. [DOI] [PubMed] [Google Scholar]
- 31.Kubo SH, Rector TS, Bank AJ, Williams RE, Heifetz SM. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation. 1991;84:1589–1596. doi: 10.1161/01.cir.84.4.1589. [DOI] [PubMed] [Google Scholar]
- 32.Andersson SE, Edvinsson ML, Edvinsson L. Cutaneous vascular reactivity is reduced in aging and in heart failure: Association with inflammation. Clin Sci (Lond) 2003;105:699–707. doi: 10.1042/CS20030037. [DOI] [PubMed] [Google Scholar]
- 33.Andreassen AK, Gullestad L, Holm T, Simonsen S, Kvernebo K. Endothelium-dependent vasodilation of the skin microcirculation in heart transplant recipients. Clin Transplant. 1998;12:324–332. [PubMed] [Google Scholar]
- 34.Ramsey MW, Goodfellow J, Jones CJ, Luddington LA, Lewis MJ, Henderson AH. Endothelial control of arterial distensibility is impaired in chronic heart failure. Circulation. 1995;92:3212–3219. doi: 10.1161/01.cir.92.11.3212. [DOI] [PubMed] [Google Scholar]
- 35.Hornig B, Maier V, Drexler H. Physical training improves endothelial function in patients with chronic heart failure. Circulation. 1996;93:210–214. doi: 10.1161/01.cir.93.2.210. [DOI] [PubMed] [Google Scholar]
- 36.Katz SD, Biasucci L, Sabba C, Strom JA, Jondeau G, Galvao M, Solomon S, Nikolic SD, Forman R, LeJemtel TH. Impaired endothelium-mediated vasodilation in the peripheral vasculature of patients with congestive heart failure. J Am Coll Cardiol. 1992;19:918–925. doi: 10.1016/0735-1097(92)90271-n. [DOI] [PubMed] [Google Scholar]
- 37.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 38.Tei C, Horikiri Y, Park JC, Jeong JW, Chang KS, Toyama Y, Tanaka N. Acute hemodynamic improvement by thermal vasodilation in congestive heart failure. Circulation. 1995;91:2582–2590. doi: 10.1161/01.cir.91.10.2582. [DOI] [PubMed] [Google Scholar]
- 39.Wilson TE, Cui J, Crandall CG. Effect of whole-body and local heating on cutaneous vasoconstrictor responses in humans. Auton Neurosci. 2002;97:122–128. doi: 10.1016/s1566-0702(02)00046-2. [DOI] [PubMed] [Google Scholar]
- 40.Freund BJ, Joyner MJ, Jilka SM, Kalis J, Nittolo JM, Taylor JA, Peters H, Feese G, Wilmore JH. Thermoregulation during prolonged exercise in heat: Alterations with beta-adrenergic blockade. J Appl Physiol. 1987;63:930–936. doi: 10.1152/jappl.1987.63.3.930. [DOI] [PubMed] [Google Scholar]
- 41.Pescatello LS, Mack GW, Leach CN, Jr, Nadel ER. Effect of beta-adrenergic blockade on thermoregulation during exercise. J Appl Physiol. 1987;62:1448–1452. doi: 10.1152/jappl.1987.62.4.1448. [DOI] [PubMed] [Google Scholar]
- 42.Holowatz LA, Jennings JD, Lang JA, Kenney WL. Systemic low-dose aspirin and clopidogrel independently attenuate reflex cutaneous vasodilation in middle-aged humans. J Appl Physiol. 2010;108:1575–1581. doi: 10.1152/japplphysiol.01362.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mack GW, Shannon LM, Nadel ER. Influence of beta-adrenergic blockade on the control of sweating in humans. J Appl Physiol. 1986;61:1701–1705. doi: 10.1152/jappl.1986.61.5.1701. [DOI] [PubMed] [Google Scholar]
- 44.Pescatello LS, Mack GW, Leach CN, Jr, Nadel ER. Thermoregulation in mildly hypertensive men during beta-adrenergic blockade. Med Sci Sports Exerc. 1990;22:222–228. [PubMed] [Google Scholar]
- 45.Martinez-Selles M, Garcia Robles JA, Prieto L, Serrano JA, Munoz R, Frades E, Almendral J. Annual rates of admission and seasonal variations in hospitalizations for heart failure. European journal of heart failure. 2002;4:779–786. doi: 10.1016/s1388-9842(02)00116-2. [DOI] [PubMed] [Google Scholar]
- 46.Morgan CL, Nadas AS. Sweating and congestive heart failure. N Engl J Med. 1963;268:580–585. [Google Scholar]
- 47.Park J, Campese VM, Nobakht N, Middlekauff HR. Differential distribution of muscle and skin sympathetic nerve activity in patients with end-stage renal disease. J Appl Physiol. 2008;105:1873–1876. doi: 10.1152/japplphysiol.90849.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.