

Abstract
RXRα represents an intriguing and unique target for pharmacologic interventions. We recently showed that Sulindac and a designed analog could bind to RXRα and modulate its biological activity, including inhibition of the interaction of an N-terminally truncated RXRα (tRXRα) with the p85α regulatory subunit of phosphatidylinositol-3-OH kinase (PI3K). Here we report the synthesis, testing and SAR of a series of novel analogs of Sulindac as potential modulators for inhibiting tRXRα-dependent AKT activation. A new compound 30 was identified to have improved biological activity.
Keywords: RXRα, αtRXR modulator, AKT activation, Sulindac analogs
1. Introduction
Retinoid X receptor-α (RXRα) is a unique member of the nuclear receptor (NR) superfamily, playing an important role in many biological processes ranging from apoptosis, cell differentiation and growth to lipid metabolism [1–3]. RXRα acts primarily as a ligand-dependent transcription factor through forming homodimer with itself or heterodimer with other members of the NR family. Structurally RXRα shares a modular organization with other nuclear receptors, consisting of three main functional domains: an N-terminal region where the ligand-independent transcriptional activation function (AF-1) is located, a DNA-binding domain and a ligand-binding domain (LBD) [2]. The transcriptional activity is directly mediated by the LBD and thus the LBD has been the most studied domain. The LBD possesses a ligand-binding pocket (LBP) for the binding of small molecule ligands, a transactivation function domain termed AF-2 composed of Helix 12 (H12) of the LBD, a coregulator binding surface, and a dimerization surface. Numerous ligands targeting the LBP have been designed and reported [4, 5]. Natural RXRα ligand 9-cis-Retinoic Acid (9-cis-RA) and synthetic RXR ligands (rexinoids) have been effective in preventing tumorigenesis in animals [6] and RXRα has been a drug target for therapeutic applications, especially in the treatment of cancer [7]. Targretin, a synthetic RXR-selective retinoid, was approved for treating cutaneous T-cell lymphoma [8, 9], and it has also been explored for the treatment of other form of cancer such as lung cancer, breast cancer, and prostate cancer [10–12].
Sulindac, a nonsteroidal antiinflammatory drug (NSAID) drug, has been investigated as a cancer chemopreventive agent, because of its potent induction of apoptosis and inhibition of cancer cell growth [13–16]. It has been documented that the anti-cancer effect of Sulindac can be mediated through COX-2-independent mechanisms [14, 15, 17]. We recently reported that Sulindac induces apoptosis in several cancer cell lines and primary tumors by binding to an N-terminally-truncated RXRα (tRXRα) [18]. Tumor necrosis factor-α (TNFα) promoted tRXRα interaction with the p85α subunit of phosphatidylinositol-3-OH kinase (PI3K), activating PI3K/AKT signaling. When combined with TNFα, Sulindac inhibited TNFα-induced tRXRα/p85α interaction, leading to activation of the death receptor-mediated apoptotic pathway [18]. Furthermore, we showed, a designed Sulindac analog K-80003 (2) (Fig. 1) exhibits increased affinity to RXRα without COX inhibitory activity, and displays enhanced efficacy in inhibiting tRXRα-dependent AKT activation and tRXRα tumor growth in animals, demonstrating the feasibility of developing a new generation of RXRα-specific molecules for therapeutic application or mechanistic studies of RXRα. Here we present the synthesis, SAR studies and biological evaluation of a series of K-80003 derivatives and the discovery of a new scaffold 30.
Figure 1.
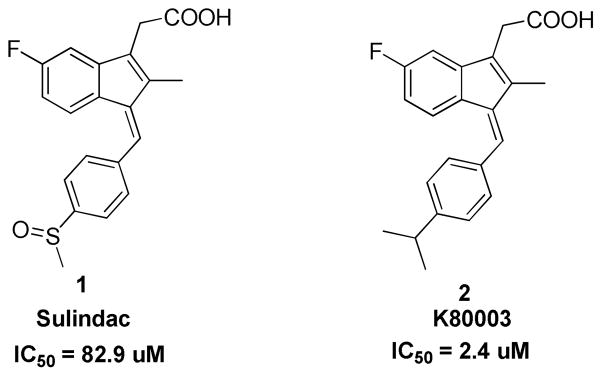
Structure and RXRα binding activity of Sulindac and K-80003
2. Results and discussion
Compared to Sulindac (1, Fig. 1), compound 2 displays an increased binding to RXRα and potency in inhibiting tRXRα-dependent AKT activation [18]. 1 and 2 differ in the replacement of the sulfide group in 1 by an isopropyl group in 2 at R1 (Fig. 2). Thus, for the first round of SAR study we investigated the effects of various substituents of R1 (Fig. 2) on the binding affinity to the RXRα LBD. Scheme 1 outlines the synthetic chemistry used for the preparation of this group of compounds (3–14) and the testing results are listed in Table 1. The designed compounds 3–14 provide an opportunity to study the effect of the size of the group and the influence of electron-deficient and electron-rich groups. It seems that the binding capability is sensitive to the size of R1 group at position 4. Compound 3 with R1 = H displayed weaker binding whereas compound 4 with R1= CF3 exhibited slightly weaker binding and when R1= C(CH3)3 the compound 5 showed no binding. Replacing R1 of –CH(CH3)2 in 2 with either electron-donating (compounds 7 and 8, 10, 14) diminished the compounds’ binding. Electron-withdrawing group of -CN (compounds 9) also abolished the binding. Similar trend was observed when the substitution was moved from position 4 to 3 (11–13, table 1).
Figure 2.
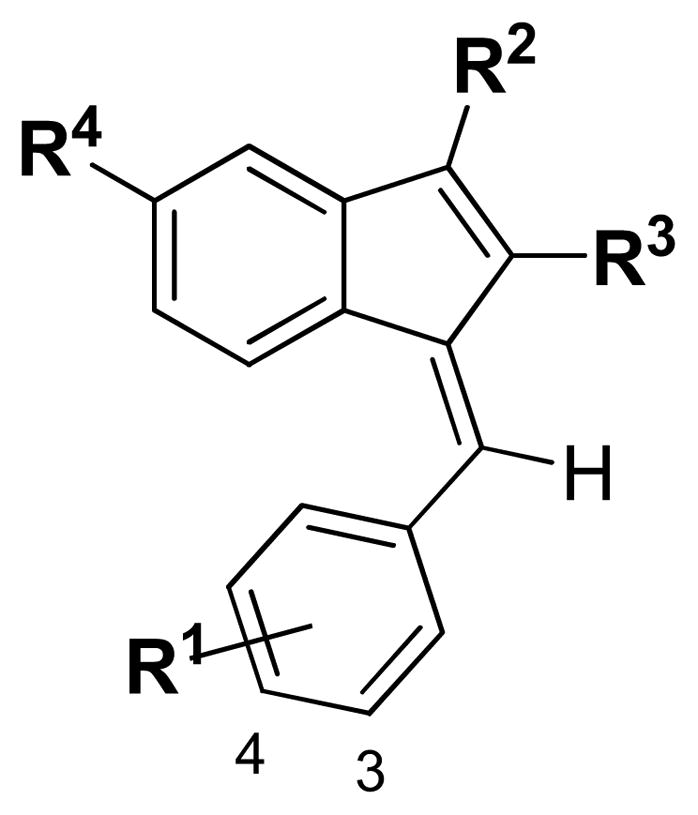
Scaffold for the SAR study
Scheme 1.

Synthesis of compounds 3~14
Table 1.
Summary of the binding data for the R1 derivatives
| Compound | R1 (R2= CH2COOH; R3= CH3; R4= F) | RXRα Binding IC50 (μM) |
|---|---|---|
| 3 | H | 10.65 |
| 4 | 4-CF3 | 4.73 |
| 5 | 4-C(CH3)3 | ≥100 |
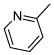
| 6 |
|
≥100 |
| 7 | 4-OCH3 | ≥100 |
| 8 | 4-OCH2CH3 | ≥100 |
| 9 | 4-CN | ≥100 |
| 10 | 4-N(CH3)2 | ≥100 |
| 11 | 3-CF3 | 8.27 |
| 12 | 3-OCH3 | ≥100 |
| 13 | 3-CN | ≥100 |
| 14 | 4-NHC(O)CH3 | ≥100 |
Our previous molecular docking study showed that the carboxylate group of 2 formed charge-charge interaction with Arg316 in the ligand-binding pocket of RXRα in a similar fashion to the carboxylate group found in other RXRα ligands [18]. This binding model is consistent with the SAR study of R2 group as shown in Table 2. Although extending the carboxylate group by one carbon (15) weakens the binding, replacing the carboxylate group with non-charged groups (16, 17 and 19) resulted in the loss of the binding activity. Compounds 15–17 and 19 were synthesized according to Scheme 2.
Table 2.
Summary of the binding data for the R2 derivatives
| Compound | R2 (R1= CH(CH3)2; R3= CH3; R4= F) | RXRα Binding IC50 (μM) |
|---|---|---|
| 15 | CH2CH2COOH | 5.71 |
| 16 | CH2CH2CN | ≥100 |
| 17 | CH2CONH2 | ≥100 |
| 18 | CH2C(O)NHCH3 | ≥100 |
Scheme 2.

Synthesis of compounds 15~20
A few substituents at R3 and R4 were also examined and the binding results are outlined in Table 3. It shows that R3 can tolerate bulkier groups. For example, replacing methyl at R3 with ethyl (21) or isobutyl group (22) did not affect the binding dramatically. However, R4 is sensitive to different substituents. Except for the ethoxyl group, replacing floride by other groups including hydrogen, chloride, methyl (23–26, 28, 29) caused steep drop in binding. Compounds 21–29 were prepared according to the procedure outlined in Scheme 3.
Table 3.
Summary of the binding data for the R3/R4 derivatives
| Compound | R3 (R1=CH(CH3)2 R2= CH2COOH) |
R4 (R1= CH(CH3)2 R2= CH2COOH) |
RXRα Binding IC50 (μM) |
|---|---|---|---|
| 21 | CH2CH3 | F | 6.29 |
| 22 | CH2CH(CH3)2 | F | 3.15 |
| 23 | CH3 | H | ≥100 |
| 24 | CH3 | CH3 | ≥100 |
| 25 | CH3 | OCH3 | ≥100 |
| 26 | CH3 | CH2CH3 | ≥100 |
| 27 | CH3 | OCH2CH3 | 3.08 |
| 28 | CH3 | CH(CH3)2 | ≥100 |
| 29 | CH3 | Cl | ≥100 |
Scheme 3.


Synthesis of compounds 21~29
Overall, the analogs synthesized for this SAR study didn’t improve the binding activity compared to the original lead 2. Therefore we decided to explore the E-isomer of 2. Compound 30 (Figure 3) was prepared according to Scheme 4 and was found to exhibit slightly tighter binding with an IC50 of 1.6 μM. Compound 30 was then evaluated for the effect on RXRα transactivation activity by employing the Gal4 reporter assay. The LBD of RXRα was cloned as a Gal4 fusion and the resulting Gal4-RXRα/LBD chimera and Gal4 reporter system were used to evaluate the effect of compound 30. Gal4-RXRα/LBD strongly activated the Gal4 reporter in the presence of 9-cis-RA, which was inhibited by BI-1003, a RXRα antagonist [19]. As shown in Fig. 4, treatment of cells with 2 or 30 resulted in inhibition of 9-cis-RA-induced reporter activity in a dose dependent manner. So, Like 2, 30 acts as a RXRα antagonist, however 30 showed stronger antagonism activity.
Figure 3.
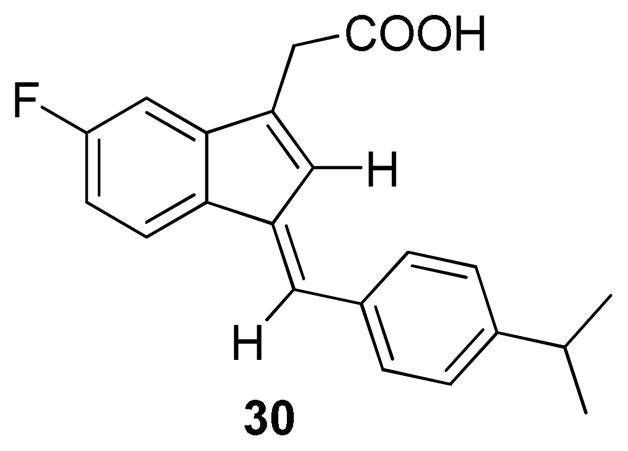
Structure of E-isomer 30
Scheme 4.
Synthesis of compounds 30
Figure 4.
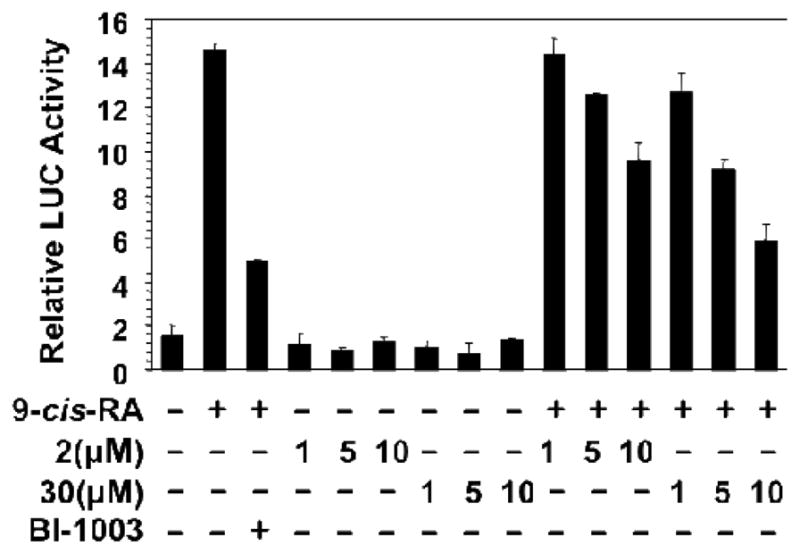
Inhibition of Gal4-RXRα-LBD activit by 30. HCT116 cells transfected with pG5 luciferase reporter vector and pGAL4-RXRα-LBD expression vector (50ng/well) were incubated with or without 9-cis-RA (10−7 M) in the presence or absence of the indicated concentrations of 2 or 30 for 12 hr. Luciferase activities were measured using the Dual-Luciferase Assay System Kit. For comparison, the effect of BI-1003 (1 μM) was shown.
30, being an E-isomer of 2, displays a different shape from 2 due to the different orientation of the isopropyl benzene motif. With such a difference in shape, it would be expected intuitively that 30 would not be tolerated in the same pocket where 2 binds. Thus we were intrigued to understand how 2 and 30 bind to the same LBP. Docking study was performed to explore the potential binding modes of 2 and 30. RXRα/antagonist complex structure 3A9E [20] from Protein Data Bank (PDB) was used and Glide docking program [21] from Schrodinger was applied. The docked binding mode of 2 suggested that 2 bound to the LBP of RXRα in a similar mode as previously proposed for Sulindac [18], in which the carboxylate interacts with Arg316 of RXRα and the isopropyl benzene portion of the compound interacts with hydrophobic side chains residing in H3, H5 and H7 (Fig. 5A). For 30, it was found that 30 could be tolerated and docked into the same pocket (Fig. 5B). This is most likely explained by the large size and the hydrophobic nature of the LBP. However, the docked 30 adopts a different orientation from 2 and forms different interactions with the protein (Fig. 5C). In the docked mode, the carboxylate group of 30 is not close to Arg316 to make the same interaction as seen in 2. Instead, 30 makes more extensive hydrophobic interactions with hydrophobic side chains in H3, H5, H7 and H11 (Fig. 5B). Recent crystal structures of RXRα in complex with antagonists have demonstrated the significance of the hydrophobic interactions that were found dominant in the ligand binding [22, 23]. Therefore in the case of 30, it is conceivable that even though the acid group of 30 may not contribute as much to the binding as that of 2, the hydrophobic interactions play a key role in the binding.
Figure 5.
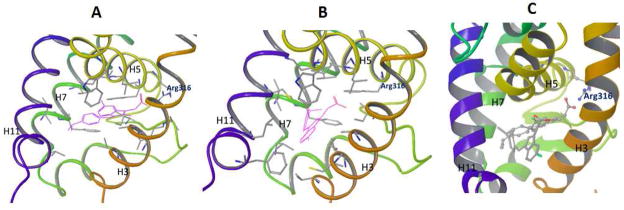
Docking study of 2 and 30. (A) Proposed binding mode of 2 by docking. 2 is shown in magenta and the side chains in the LBP that could interact favorably with the ligand are displayed. (B) Proposed binding mode of 30 by docking. 30 is shown in magenta and the side chains in the LBP that could interact favorably with the ligand are displayed. (C) Relative orientation of the docked 2 (in ball and stick) and 30 (in tube) in the LBP.
We further tested 30 in other biological assays for its effect on the apoptosis of cancer cells and its ability to inhibit the PI3K/AKT activation. In the MTT assay, 30 could dose-dependently induce growth inhibition in some cancer cell lines such as PC3 prostate cancer cells and ZR75-1 breast cancer cells (Figure 6A). In the induction of PARP cleavage, 30 was more effective than 1 or 2 (Figure 6B).
Figure 6.
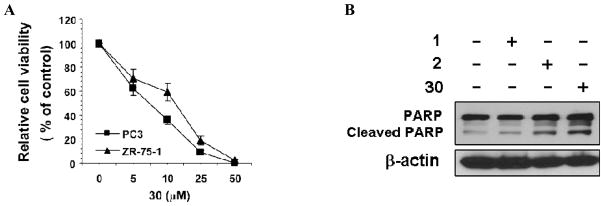
Compound 30 induces cancer cell apoptosis. (A) Growth inhibition by 30. PC3 and ZR-75-1 breast cancer cells were treated with the indicated concentration of 30 for 24 hr. Cell viability was determined by the MTT colorimetric assay. (B) Induction of PARP cleavage by 1, 2 or 30. ZR-75-1 cells were treated with vehicle or 30 μM compound as indicated for 6 hr. PARP cleavage was analyzed.
We previously demonstrated that inhibition of AKT activation by the Sulindac/TNFα combination was closely associated with its apoptotic effect [18]. We then investigated whether compound 30 could inhibit TNFα-induced AKT activation. In agreement with previous studies, treatment of A549 lung cancer cells with TNFα led to a strong AKT activation [18], which was inhibited by 1, 2 or 30 (Figure 7A). Such effects were also observed in HCT-116 colon cancer cells and HepG2 liver cancer cells (data not shown). Consistently, compound 30 showed a better effect on the inhibition of TNFα-induced AKT activation. Knocking down tRXRα by siRNA significantly impaired the inhibitory effect of 30 on AKT activation (Figure 7B). These results indicated that inactivation of AKT by 30 was tRXRα-dependent.
Figure 7.
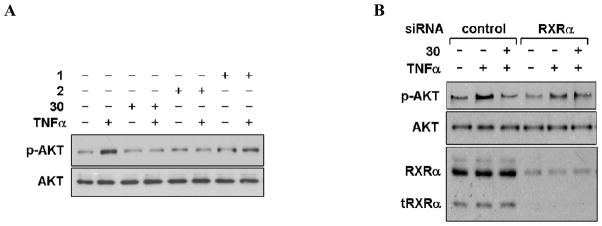
Inhibition of tRXRα-dependent AKT activation by compound 30. (A) Synergistic inhibition of AKT activation by Sulindac and its analogs. A549 cells were pretreated with 30 μM, 1, 2 or 30 for 1 hr before exposed to TNFα (10 ng/mL) for 30 min. Phosphorylated AKT and total AKT were analyzed by immunoblotting. (B) 30 inhibits AKT activation dependent on RXRα expression. HeLa cells transfected with scramble or RXRα siRNA were pretreated with 20 μM 30 for 1 hr before exposed to TNFα (10 ng/mL) for 30 min. The effect of RXRα siRNA and 30 on the inhibition of TNFα-induced AKT activation was analyzed by immunoblotting.
We also examined whether 30 could enhance the TNFα-induced apoptosis. Figure 8 showed that 30 could significantly enhance the PARP cleavage in combination with TNFα, suggesting that 30 could activate TNFα-dependent apoptotic pathway. The observed synergistic effect of 30/TNFα on the TNFα-induced apoptosis was stronger than that of 2/TNFα or 1/TNFα.
Figure 8.
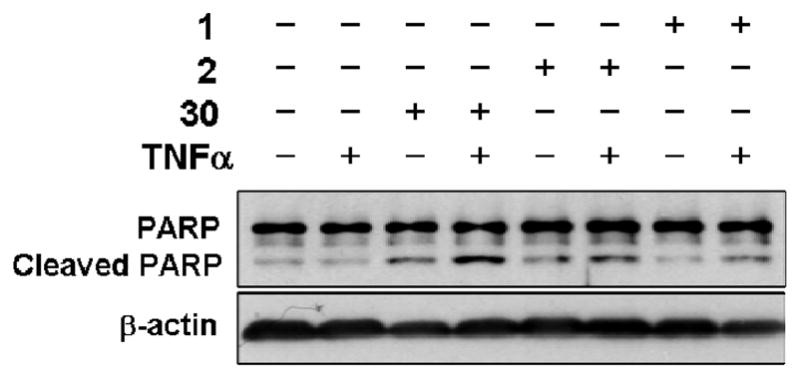
Synergistic induction of apoptosis by compound/TNFα combination. HCT116 cells treated with 40 μM, 1, 2 or 30 in the presence or absence of TNFα for 4 hr were analyzed by immunoblotting.
3. Chemistry
The synthesis started from the Perkin reaction of 4-fluorobenzaldehyde 31 with propionate anhydride [24], in which K2CO3 was used as a base to substitute hygroscopic sodium propionate, providing the desired product 32 in 83% yield (Scheme 1). Catalytic hydrogenation in the presence of Pd/C and under 10 atm of hydrogen gave carboxylic acid 33 in 90% yield. Polyphosphoric acid (PPA)-promoted intermolecular Friedel-Crafts acylation reaction produced indenone 34 in 74% yield. Treatment of indenone 34 with the enolate generated from ethyl acetate and LDA gave the corresponding β-hydroxy ester, which was treated with a mixture of HOAc and concentrated H2SO4 (v/v 10:1) to yield the indene 35 in 80% yield. Finally, two methods were used for the Claisen-Schmidt condensation reactions of compound 35 with differently substituted benzaldehydes to give compounds 3~14, respectively. The electronic properties of the substituents on the aromatic aldehydes were found to have an impact on the reaction, and slightly different conditions should be used for the synthesis of a specific compound. The results of the reactions are summarized in Table 4.
Table 4.
Results of the Claisen-Schmidt reactions
| Compound | R1 | Yield (%) | Compound | R1 | Yield (%) |
|---|---|---|---|---|---|
| 3 | H | 77 | 4 | 4-CF3 | 62 |
| 5 | 4-(CH3)3C | 72 | 6 |
|
33 |
| 7 | 4-CH3O | 78 | 8 | 4-CH3CH2O | 66 |
| 9i | 4-CN | 73 | 10 | 4-(CH3)2N | 20 |
| 11 | 3-CF3 | 71 | 12 | 3-CH3O | 70 |
| 13 | 3-CN | 60 | 14 | 4-CH3CONH | 22 |
4. Conclusion
In conclusion, we have described the synthesis and SAR studies on a series of novel analogs of Sulindac as potential modulators for inhibiting tRXRα-dependent AKT activation. Compound 30, a geometric isomer of the original lead 2 and with better binding activity and improved biological effects, could bind to the LBP of RXRα in a different mode from 2, which offers a new design strategy. 30 is a promising lead for further optimization studies and may find application as a small molecule probe in studying the mechanism of the tRXRα-dependent AKT signaling.
5. Experimental section
5.1. Chemistry
5.1.1. General methods
Melting points (M.p.) were determined on a Yanaco MP-500 micro melting point apparatus and were uncorrected. Infrared spectra were measured with a Nicolet Avatar 360 FT-IR spectrometer using film KBr pellet techniques. 1H and 13C NMR spectra were recorded in CDCl3 or CD3OD on a Bruker 400 spectrometer with tetramethylsilane as an internal standard. Chemical shifts are expressed in δ (ppm) units downfield from TMS. Mass spectra were recorded by a Bruker Dalton ESquire 3000 plus liquid chromatography-mass spectrum (direct injection). Optical rotations were measured with a Perkin-Elmer 341 automatic polarimeter. Diastereoselectivities and enantioselectivities were determined by chiral HPLC analysis using a Shimadzu LC-10AT VP series and a Shimadzu SPD-M10Avp photo diode array detector (190–370 nm) with a Chiralcel OJ-H column using n-hexane/i-PrOH (98:2, v/v) as a mobile phase. Flash column chromatography was carried out with silica gel (300–400 mesh). THF was distilled over sodium benzophenone ketyl under N2.
5.1.2. General Procedure A: the synthesis of acrylic acid from aromatic aldehyde
Appropriate anhydride (300 mmol, 1.6 equiv.) was added to potassium carbonate (224 mmol, 1.2 equiv.) at 0 °C. After stirring for 5 min to mix up, appropriate aromatic aldehyde (186 mmol, 1.0 equiv.) was added. The mixture was heated to reflux for 12 h. After cooling with an ice bath, to the reaction mixture was added water and solid Na2CO3 (30 g). After the resultant yellow precipitate was filtered, the reaction mixture was acidified to pH 6.0 using concentrated HCl to afford acrylic acid as a solid.
5.1.3. 3-(4-Fluorophenyl)-2-methylacrylic acid (32)
Compound 32 [24] was synthesized according to the general procedure A. Pale yellow crystals, yield: 83%. M.p. 155–158 °C (MeOH); IR (film): νmax 3429, 3076, 2972, 1665, 1596, 1508, 1425, 1313, 1298, 1224 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 2.00 (d, J = 1.2H Hz, 3H, C 3), 7.19–7.25 (m, 2H, Ar-H), 7.46–7.52 (m, 2H, 2H, Ar-H), 7.58 (s, 1H, vinyl-H), 12.50 (br s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ 14.2, 115.8 (d, JC-F = 21.0 Hz), 129.0, 132.2 (d, JC-F = 9.0 Hz), 132.46 (d, JC-F = 3.0 Hz), 136.9, 162.3 (d, JC-F = 245.0 Hz), 169.7 ppm; MS (ESI) m/z 179 (M+H+).
5.1.4. General Procedure B: the synthesis of propanoic acid derivative from acrylic acid by Pd/C-catalyzed reduction
A mixture of acrylic acid (55 mmol, 1.0 equiv.) and Pd/C (10%) in methanol (70 mL) was hydrogenated under 10 atm of hydrogen for 24 h. The catalyst was filtered off and the filtrate concentrated to afford propanoic acid, which was used in the next step as it was. An analytical sample of compound was obtained by flash column chromato- graphy on silica gel.
5.1.5. 3-(4-Fluorophenyl)-2-methylpropanoic acid (33)
Compound 33 [24] was synthesized according to the general procedure B. Colorless oil, yield: 90%. IR (film): νmax 3406, 2972, 2933, 1701, 1560, 1509, 1460, 1406, 1223 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.12 (d, J = 6.7 Hz, 3H, CH3), 2.60 (dd, J = 13.0, 7.9 Hz, 1H, CH2CH), 2.66 (ddq, J = 7.9, 6.0, 6.7 Hz, CHCH3), 2.99 (dd, J = 13.0, 6.0 Hz, 1H, CH2CH), 6.90–7.00 (m, 2H, Ar-H), 7.06–7.14 (m, 2H, Ar-H), 9.80 (br s, 1H, COOH) ppm; 13C NMR (100 MHz, CDCl3) δ 16.5, 38.6, 41.8, 115.1 (d, JC-F = 21.0 Hz), 130.35 (d, JC-F = 8.0 Hz), 134.87 (d, JC-F = 3.0 Hz), 161.6 (d, JC-F = 243.0 Hz), 182.3 ppm; MS (ESI) m/z 181 (M+H+).
5.1.6. General Procedure C: the synthesis of indenone from propanoic acid derivative by F-C acylation
A mixture of the crude propanoic acid derivative (42.0 mmol, 1.0 equiv.) and polyphosphoric acid (400 mmol, 9.5 equiv.) was stirred at 80 °C for 12 hours. The resulting mixture was poured into ice water and extracted with EtOAc (30 mL × 3). The combined extracts were washed with a saturated aqueous NaHCO3 (10 mL × 3) to remove the starting acids, and then washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography to afford indenone.
5.1.7. 6-Fluoro-2-methyl-2,3-dihydroinden-1-one (34)
Compound 34 [24] was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). Pale yellow oil, yield: 74%. IR (film) νmax 3064, 2968, 2932, 2873, 1716, 1611, 1509, 1486, 1444, 1264, 1158 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.32 (d, J = 7.4 Hz, 3H, CH3), 2.70 (dd, J = 16.7, 3.9 Hz, 1H, CH2CH), 2.74–2.82 (m, 1H, CHCH3), 3.37 (dd, J = 16.7, 7.6 Hz, 1H, CH2CH), 7.26–7.33 (m, 1H, Ar-H), 7.36–7.44 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.2, 34.4, 42.9, 109.7 (d, JC-F = 22.0 Hz), 122.3 (d, JC-F = 24.0 Hz), 127.85 (d, JC-F = 8.0 Hz), 138.1, 148.8, 162.3 (d, JC-F = 247.0 Hz), 208.4 ppm; MS (ESI) m/z 187 (M+Na+).
5.1.8. General Procedure D: the synthesis of inden-3-yl acetate from indenone
To a solution of LDA or LHMDS (48.0 mmol, 2.0 equiv.) in anhydrous THF (100 mL) was added EtOAc (61.0 mmol, 2.5 equiv.) at −78 °C. The mixture was stirred at −78 °C for 30 min. To the resulting mixture was added dropwise a solution of indenone (24.0 mmol, 1.0 equiv.) in anhydrous THF (20 mL). The mixture was stirred at −78 °C for another 4 hr and then quenched with a saturated aqueous NH4Cl. The mixture was extracted with EtOAc (20 mL × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. To the residue was added HOAc/H2SO4 (10/1, 40 mL). After stirring for 4 hr at room temperature, the mixture was extracted with EtOAc (15 mL × 3). The combined extracts were washed successively with water, saturated NaHCO3, and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel to afford inden-3-yl acetate.
5.1.9. Ethyl 2-(5-fluoro-2-methyl-1H-inden-3-yl)acetate (35)
Compound 35 [24] was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Colorless oil, yield: 80%. IR (film) νmax 2981, 2911, 1736, 1614, 1590, 1473, 1368, 1329, 1308, 1256, 1154, 1034 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.12 (s, 3H, C=CCH3), 3.29 (s, 2H, ArCH2C=C), 3.48 (s, 2H, CH2COOEt), 4.14 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.77–6.83 (m, 1H, Ar-H), 6.94–6.99 (m, 1H, Ar-H), 7.23–7.27 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.15, 14.26, 31.5, 42.1, 60.9, 105.8 (d, JC-F = 23.0 Hz), 110.3 (d, JC-F = 23.0 Hz), 123.7 (d, JC-F = 9.0 Hz), 129.6, 137.19 (d, JC-F = 2.0 Hz) 144.5, 147.87 (d, JC-F = 9.0 Hz), 162.4 (d, JC-F = 239.0 Hz), 170.7 ppm; MS (ESI) m/z 257 (M+Na+).
5.1.10. General Procedure E: the synthesis of indene derivative from appropriate inden-3-yl acetate
To a solution of indene-3-yl acetate 35 (1.3 mmol, 1.0 equiv.) in MeOH (4.0 mL) was added 2.5 N NaOMe (4.0 mmol, 3.0 equiv.) at room temperature to get an orange mixture. After stirring for 30 min, to the mixture was added appropriate aromatic aldehyde (1.3~2.0 mmol, 1.0~1.5 equiv.). The resulting mixture was refluxed at 80 °C for 4 h. After concentrated under reduced pressure, the residue was acidified with a 1N HCl solution to pH 4.0~6.0. After stirring for another 0.5 hr at room temperature, the mixture was extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography to afford indene derivative. An analytical sample of compound was obtained by recrystallization.
5.1.11. (Z)-2-(1-Benzylidene-5-fluoro-2-methyl-1H-inden-3-yl) acetic acid (3)
Compound 3 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:3). Yellow solid, yield: 77%. M.p. 175–176 °C (hexane/EtOAc); IR (film): νmax 3430, 3021, 2918, 1705, 1604, 1467, 1415, 1302, 1168 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.10 (s, 3H, C=CCH3), 3.50 (s, 2H, CH2COOH), 6.43–6.50 (m, 1H, Ar-H), 6.75–6.84 (m, 1H, Ar-H), 7.13 (s, 1H, vinyl-H), 7.25–7.43 (m, 6H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 31.4, 105.7 (d, JC-F = 24.0 Hz), 110.6 (d, JC-F = 22.0 Hz), 123.8 (d, JC-F = 9.0 Hz), 128.0, 128.2, 128.5 (2C), 129.2 (2C), 129.4, 129.8, 130.1, 130.7, 136.5, 138.8, 140.2, 146.2 (d, JC-F = 8.0 Hz), 163.1 (d, JC-F = 245.0 Hz), 176.6 ppm; MS (ESI) m/z 317.1 (M+Na+, 100%); HRMS (ESI) calcd for C19H15FNaO2+ [M+Na+]: 317.0948; found: 317.0951.
5.1.12. (Z)-2-(5-Fluoro-2-methyl-1-(4-trifluoromethylbenzylidene)-1H-inden-3-yl)acetic acid (4)
Compound 4 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:3). Yellow solid, yield: 62%. M.p. 188–189 °C (hexane/EtOAc); IR (film): νmax 3435, 2918, 1708, 1604, 1467, 1321, 1165, 1122, 1065, 1016 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.21 (s, 3H, C=CCH3), 3.60 (s, 2H, CH2COOH), 6.54–6.61 (m, 1H, Ar-H), 6.86–6.91 (m, 1H, Ar-H), 7.08–7.14 (m, 1H, Ar-H), 7.18 (s, 1H, vinyl-H), 7.57–7.63 (m, 2H, Ar-H), 7.67–7.74 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 31.3, 106.1 (d, JC-F = 23.0 Hz), 110.9 (d, JC-F = 22.0 Hz), 122.7, 123.7 (d, JC-F = 9.0 Hz), 125.46, 125.49, 128.3, 129.4, 129.5, 131.1, 138.6, 140.3, 141.6, 146.4 (d, JC-F = 8.0 Hz), 163.4 (d, JC-F = 245.0 Hz), 176.0 ppm; MS (ESI) m/z 385.1 (M+Na+, 100%); HRMS (ESI) calcd for C20H14F4NaO2+ [M+Na+]: 385.0822; found: 385.0819.
5.1.13. (Z)-2-(1-(4-tert-Butylbenzylidene)-5-fluoro-2-methyl-1H-inden-3-yl)acetic acid (5)
Compound 5 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:3). Yellow solid, yield: 72%. M.p. 187–188 °C (hexane/EtOAc); IR (film): νmax 3420, 2964, 1708, 1604, 1503, 1464, 1412, 1363, 1266, 1168 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 9H, C(CH3)3), 2.20 (s, 3H, C=CCH3), 3.60 (s, 2H, CH2COOH), 6.56–6.64 (m, 1H, Ar-H), 6.86–6.93 (m, 1H, Ar-H), 7.20 (s, 1H, vinyl-H), 7.38–7.52 (m, 5H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 31.3, 31.4, 34.8, 105.6 (d, JC-F = 24.0 Hz), 110.6 (d, JC-F = 22.0 Hz), 123.8 (d, JC-F = 9.0 Hz), 125.4 (2C), 129.2 (2C), 129.7, 129.8, 130.9, 133.4, 139.0, 139.7, 146.2 (d, JC-F = 9.0 Hz), 151.6, 163.1 (d, JC-F = 244.0 Hz), 176.3 ppm; MS (ESI) m/z 373.2 (M+Na+, 100%); HRMS (ESI) calcd for C23H23FNaO2+ [M+Na+]: 373.1574; found: 373.1571.
5.1.14. (Z)-2-(5-Fluoro-2-methyl-1-(4-(pyridin-2-yl)benzylidene)-1H-inden-3-yl)acetic acid (6)
To a solution of indene 35 (150 mg, 0.64 mmol) in toluene (4.0 mL) was added DBU (0.9 mL, 6.4 mmol) at room temperature. After stirring for 30 min at 80 °C, to the mixture was added the solution of aromatic aldehyde (168 mg, 0.96 mmol) in toluene (2.0 mL). The resulting mixture was heated at 80 °C for 36 h, then quenched with a saturated aqueous NH4Cl. The mixture was extracted with EtOAc (20 mL × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Then to the residue in MeOH (3 mL) was added 2N NaOH (2 mL). The mixture was stirring for 2 hr at 65 °C. After concentrated under reduced pressure, the residue was acidified with a 1N HCl solution to pH 6.0~7.0. The residue was purified by flash chromatography on silica gel (eluent: 1H NMR (400 MHz, DMSO-d6) δ 2.18 (s, 3H, C=CCH3), 3.59 (s, 2H, CH2COOH), 6.70–6.77 (m, 1H, Ar-H), 7.00–7.05 (m, 1H, Ar-H), 7.31–7.36 (m, 1H, Ar-H), 7.36–7.40 (m, 1H, Ar-H), 7.40 (s, 1H, vinyl-H), 7.64–7.70 (m, 2H, Ar-H), 7.89–7.95 (m, 1H, Ar-H), 8.04–8.08 (m, 1H, Ar-H), 8.20–8.25 (m, 2H, Ar-H), 8.68–8.72 (m, 1H, Ar-H), 12.45 (br s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ 10.3, 31.1, 105.9 (d, JC-F = 24.0 Hz), 110.3 (d, JC-F = 23.0 Hz), 120.3, 122.9, 123.2 (d, JC-F = 9.0 Hz), 126.6 (2C), 129.6, 129.7, 130.4 (2C), 132.1, 136.8, 137.3, 138.0, 138.4, 139.8, 146.96 (d, JC-F = 8.0 Hz), 149.6, 155.3, 162.42 (d, JC-F = 242.0 Hz), 171.6 ppm; MS (ESI) m/z 372.1 (M+H+, 100%); HRMS (ESI) calcd for C24H19FNO2+ [M+H+]: 372.1394; found: 372.1395.
5.1.15. (Z)-2-(5-Fluoro-1-(4-methoxybenzylidene)- 2-methyl-1H-inden-3-yl)acetic acid (7)
Compound 7 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 78%. M.p. 182–183 °C (hexane/EtOAc); IR (film): νmax 3418, 2927, 2833, 1708, 1601, 1507, 1464, 1296, 1250, 1171, 1028 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.20 (s, 3H, C=CCH3), 3.60 (s, 2H, CH2COOH), 3.89 (s, 3H, OCH3), 6.55–6.63 (m, 1H, Ar-H), 6.85–6.91 (m, 1H, Ar-H), 6.92–6.99 (m, 2H, Ar-H), 7.18 (s, 1H, vinyl-H), 7.36–7.43 (m, 1H, Ar-H), 7.44–7.50 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.6, 31.3, 55.3, 105.6 (d, JC-F = 24.0 Hz), 110.5 (d, JC-F = 22.0 Hz), 113.9 (2C), 123.6 (d, JC-F = 9.0 Hz), 128.7, 129.5, 129.8, 130.8, 130.9 (2C), 139.0, 139.2, 146.1 (d, JC-F = 9.0 Hz), 159.7, 163.0 (d, JC-F = 245.0 Hz), 175.6 ppm; MS (ESI) m/z 347.1 (M+Na+, 100%); HRMS (ESI) calcd for C20H17FNaO3+ [M+Na+]: 347.1054; found: 347.1060.
5.1.16. (Z)-2-(1-(4-Ethoxybenzylidene)-5-fluoro-2-methyl-1H-inden-3-yl)acetic acid (8)
Compound 8 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 66%. M.p. 186–187 °C (hexane/EtOAc); IR (film): νmax 3410, 2976, 2921, 1705, 1601, 1507, 1464, 1296, 1247, 1168, 1043 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.47 (t, J = 7.0 Hz, 3H, OCH 2CH3), 2.21 (s, 3H, C=CCH3), 3.59 (s, 2H, CH2COOH), 4.11 (q, J = 7.0 Hz, 2H, OCH2CH3), 6.56–6.63 (m, 1H, Ar-H), 6.86–6.99 (m, 3H, Ar-H), 7.18 (s, 1H, vinyl-H), 7.40–7.49 (m, 3H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.6, 14.8, 31.4, 63.5, 105.5 (d, JC-F = 23.0 Hz), 110.5 (d, JC-F = 22.0 Hz), 114.4 (2C), 123.5 (d, JC-F = 9.0 Hz), 128.5, 129.4, 129.8, 130.88, 130.94 (2C), 139.0, 139.1, 146.1 (d, JC-F = 9.0 Hz), 159.1, 163.0 (d, JC-F = 244.0 Hz), 176.9 ppm; MS (ESI) m/z 361.1 (M+Na+, 100%); HRMS (ESI) calcd for C21H19FNaO3+ [M+Na+]: 361.1210; found: 361.1212.
5.1.17. (Z)-2-(1-(4-Cyanobenzylidene)-5-fluoro-2-methyl-1H-inden-3-yl)acetic acid (9)
Compound 9 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 73%. M.p. 199–201 °C (hexane/EtOAc); IR (film): νmax 3434, 2915, 2223, 1705, 1601, 1467, 1496, 1314, 1266, 1226, 1165, 1131, 1113, 1016 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.20 (s, 3H, C=CCH3), 3.60 (s, 2H, CH2COOH), 6.53–6.60 (m, 1H, Ar-H), 6.85–6.90 (m, 1H, Ar-H), 7.04–7.10 (m, 1H, Ar-H), 7.12 (s, 1H, vinyl-H), 7.59–7.62 (m, 2H, Ar-H), 7.70–7.75 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 31.3, 106.3 (d, JC-F = 24.0 Hz), 111.0 (d, JC-F = 22.0 Hz), 111.7, 118.6, 123.7 (d, JC-F = 9.0 Hz), 127.6, 129.2, 129.9 (2C), 131.5, 132.3 (2C), 138.5, 141.4, 142.1, 146.5 (d, JC-F = 8.0 Hz), 163.4 (d, JC-F = 246.0 Hz), 176.0 ppm; MS (ESI) m/z 342.1 (M+Na+, 100%); HRMS (ESI) calcd for C20H14FNNaO2+ [M+Na+]: 342.0901; found: 342.0902.
5.1.18. (Z)-2-(1-(4-(Dimethylamino)benzylidene)-5-fluoro-2-methyl-1H-inden-3-yl)acetic acid (10)
Compound 10 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 20%. M.p. 174–175 °C (hexane/EtOAc); IR (film): νmax 3415, 2911, 1708, 1594, 1522, 1464, 1363, 1189, 1162, 1135, 1061 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.21 (s, 3H, C=CCH3), 3.04 (s, 6H, N(CH3)2), 3.60 (s, 2H, CH2COOH), 6.58–6.65 (m, 1H, Ar-H), 6.72–6.77 (m, 2H, Ar-H), 6.87–6.92 (m, 1H, Ar-H), 7.17 (s, 1H, vinyl-H), 7.45–7.51 (d, 2H, Ar-H), 7.64–7.70 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.7, 31.3, 40.3, 105.3 (d, JC-F = 23.0 Hz), 110.3 (d, JC-F = 23.0 Hz), 111.7 (2C), 123.4 (d, JC-F = 9.0 Hz), 123.8, 128.3, 130.0, 131.3 (2C), 132.1, 137.2, 139.2, 145.8 (d, JC-F = 10.0 Hz), 150.4, 162.8 (d, JC-F = 247 Hz), 175.8 ppm; MS (ESI) m/z 338.2 (M+H+, 100%); HRMS (ESI) calcd for C21H21FNO2+ [M+H+]: 338.1551; found: 338.1549.
5.1.19. (Z)-2-(5-Fluoro-2-methyl-1-(3-trifluoromethylbenzylidene)-1H-inden-3-yl)acetic acid (11)
Compound 11 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 71%. M.p. 188–190 °C (hexane/EtOAc); IR (film): νmax 3433, 2921, 1708, 1604, 1464, 1409, 1330, 1165, 1263, 1122, 1208, 1068 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.21 (s, 3H, C=CCH3), 3.60 (s, 2H, CH2COOH), 6.53–6.61 (m, 1H, Ar-H), 6.86–6.92 (m, 1H, Ar-H), 7.07–7.13 (m, 1H, Ar-H), 7.18 (s, 1H, vinyl-H), 7.53–7.59 (m, 1H, Ar-H), 7.62–7.72 (m, 2H, Ar-H), 7.74–7.79 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 31.4, 106.1 (d, JC-F = 24.0 Hz), 110.9 (d, JC-F = 23.0 Hz), 122.6, 123.6 (d, JC-F = 9.0 Hz), 124.8, 125.3, 126.12, 128.3, 129.0, 129.4, 131.0, 132.5, 137.3, 138.6, 141.5, 146.4 (d, JC-F = 9.0 Hz), 163.3 (d, JC-F = 245.0 Hz), 176.5 ppm; MS (ESI) m/z 385.1 (M+Na+, 100%); HRMS (ESI) calcd for C20H14F4NaO2+ [M+Na+]: 385.0822; found: 385.0825.
5.1.20. (Z)-2-(5-Fluoro-1-(3-methoxybenzylidene)- 2-methyl-1H-inden-3-yl)acetic acid (12)
Compound 12 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 70%. M.p. 132–133 °C (hexane/EtOAc); IR (film): νmax 3418, 2936, 2833, 1708, 1598, 1464, 1424, 1275, 1159, 1049 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.21 (s, 3H, C=CCH3), 3.59 (s, 2H, CH2COOH), 3.82 (s, 3H, OCH3), 6.53–6.61 (m, 1H, Ar-H), 6.86–6.91 (m, 1H, Ar-H), 6.91–6.96 (m, 1H, Ar-H), 7.01–7.10 (m, 2H, Ar-H), 7.20 (s, 1H, vinyl-H), 7.27–7.37 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 31.3, 55.3, 105.7 (d, JC-F = 23.0 Hz), 110.7 (d, JC-F = 23.0 Hz), 114.27, 114.30, 121.6, 124.0 (d, JC-F = 9.0 Hz), 129.6, 129.8, 130.2, 130.4, 137.9, 138.8, 140.4, 146.3 (d, JC-F = 9.0 Hz), 159.7, 163.2 (d, JC-F = 245.0 Hz), 175.9 ppm; MS (ESI) m/z 347.1 (M+Na+, 100%); HRMS (ESI) calcd for C20H17FNaO3+ [M+Na+]: 347.1054; found: 347.1054.
5.1.21. (Z)-2-(1-(3-Cyanobenzylidene)-5-fluoro-2-methyl-1H-inden-3-yl)acetic acid (13)
Compound 13 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 60%. M.p. 187–189 °C (hexane/EtOAc); IR (film): νmax 3433, 2917, 2226, 1711, 1601, 1464, 1409, 1311, 1271, 1168, 1131, 1037 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.20 (s, 3H, C=CCH3), 3.59 (s, 2H, CH2COOH), 6.54–6.61 (m, 1H, Ar-H), 6.85–6.91 (m, 1H, Ar-H), 6.97–7.02 (m,1H, Ar-H), 7.09 (s, 1H, vinyl-H), 7.52–7.58 (m, 1H, Ar-H), 7.65–7.70 (m, 1H, Ar-H), 7.71–7.78 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 31.3, 106.3 (d, JC-F = 23.0 Hz), 111.0 (d, JC-F = 23.0 Hz), 112.9, 118.3, 123.5 (d, JC-F = 9.0 Hz), 127.0, 129.2, 129.4, 131.4, 131.6, 132.6, 133.5, 138.0, 138.4, 142.0, 146.5 (d, JC-F = 8.0 Hz), 163.4 (d, JC-F = 246.0 Hz), 175.6 ppm; MS (ESI) m/z 342.1 (M+Na+, 100%); HRMS (ESI) calcd for C20H14FNNaO2+ [M+Na+]: 342.0901; found: 342.0899.
5.1.22. (Z)-2-(1-(4-Acetamidobenzylidene)-5-fluoro-2-methyl-1H-inden-3-yl)acetic acid (14)
Compound 14 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 22%. M.p. 240–242 °C (hexane/EtOAc); IR (film): νmax 3411, 3296, 2921, 1662, 1595, 1476, 1406, 1381, 1318, 1220, 1171, 1122, 1037 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 2.08 (s, 3H, COCH3), 2.14 (s, 3H, C=CCH3), 3.57 (s, 2H, CH2COOH), 6.70–6.79 (m, 1H, Ar-H), 6.97–7.04 (m, 1H, Ar-H), 7.28 (s, 1H, vinyl-H), 7.32–7.37 (m, 1H, Ar-H), 7.45–7.53 (m, 2H, Ar-H), 7.65–7.73 (m, 2H, Ar-H), 10.13 (s, 1H, NHCOCH3), 12.40 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ 10.2, 24.0, 31.1, 105.7 (d, JC-F = 23.0 Hz), 110.1 (d, JC-F = 22.0 Hz), 118.6 (2C), 122.9 (d, JC-F = 9.0 Hz), 129.6, 130.0 (2C), 130.4, 130.9, 131.5, 137.9, 138.7, 139.5, 146.7 (d, JC-F = 9.0 Hz), 162.2 (d, JC-F = 241.0 Hz), 168.5, 171.6 ppm; MS (ESI) m/z 374.1 (M+Na+, 100%); HRMS (ESI) calcd for C21H18FNNaO3+ [M+Na+]: 374.1163; found: 374.1162.
5.1.23. Methyl 3-(5-fluoro-2-methyl-1H-inden-3-yl)propanoate (38)
A solution of compound 34 (164.0 mg, 1.0 mmol), iso-propanol (0.38 mL, 5.0 mmol), and methyl acrylate (0.9 mL, 10 mmol) in THF (4 mL) was purged with argon for 20 min and cooled to 0 °C. A SmI2 (3.0 mmol) solution in THF (30 mL) was added through transfer needle. After 5 min, the reaction was quenched with saturated Na2CO3 (3 mL). The resulting mixture was extracted with Et2O (5 mL × 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. To a solution of the residue in CH3OH (4.0 mL) was added p-TsOH (cat.), then the mixture was refluxed for 3 h. The reaction was quenched with a saturated aqueous NaHCO3 (2.0 mL). The resulting mixture was extracted with EtOAc (5 mL × 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10) to afford compound 38 (171 mg, 73%) as a colorless oil. IR (film): νmax 2948, 1735, 1610, 1589, 1473, 1430, 1281, 1171, 1046 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.09 (s, 3H, C=CCH3), 2.53 (t, J = 7.8 Hz, 2H, CH2CH2COOMe), 2.82 (t, J = 7.8 Hz, 2H, CH2CH2COOMe), 3.23 (s, 2H, ArCH2C=C), 3.68 (s, 3H, COOCH3), 6.76–6.82 (m, 1H, Ar-H), 6.89–6.93 (m, 1H, Ar-H), 7.23–7.28 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.0, 20.6, 32.8, 42.0, 51.6, 105.2 (d, JC-F = 23.0 Hz), 110.0 (d, JC-F = 23.0 Hz), 123.75 (d, JC-F = 9.0 Hz), 134.85 (d, JC-F = 3.0 Hz), 137.56 (d, JC-F = 2.0 Hz), 142.3, 147.9 (d, JC-F = 9.0 Hz), 162.4 (d, JC-F = 240.0 Hz), 173.4 ppm; MS (ESI) m/z 257.1 (M+Na+, 100%); HRMS (ESI) calcd for C14H15FNaO2+ [M+Na+]: 257.0948; found: 257.0946.
5.1.24. (Z)-3-(5-Fluoro-1-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl)propanoic acid (15)
Compound 15 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 86%. M.p. 130–131 °C (hexane/EtOAc); IR (film): νmax 3426, 2961, 1711, 1601, 1464, 1412, 1290, 1193, 1138 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.18 (s, 3H, C=CCH3), 2.62 (t, J = 7.8 Hz, 2H, CH2CH2COOH), 2.90 (t, J = 7.8 Hz, 2H, CH2CH2COOH), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 6.55–6.62 (m, 1H, Ar-H), 6.82–6.87 (m, 1H, Ar-H), 7.14 (s, 1H, vinyl-H), 7.27–7.31 (m, 2H, Ar-H), 7.35–7.40 (m, 1H, Ar-H), 7.42–7.46 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.3, 20.7, 23.9, 32.7, 34.0, 105.3 (d, JC-F = 23.0 Hz), 110.3 (d, JC-F = 23.0 Hz), 123.8 (d, JC-F = 9.0 Hz), 126.5, 129.4, 129.9, 130.24 (d, JC-F = 3.0 Hz), 134.0, 136.06 (d, JC-F = 2.0 Hz), 136.7, 140.0, 146.36 (d, JC-F = 8.0 Hz), 149.1, 163.1 (d, JC-F = 244.0 Hz), 178.3 ppm; MS (ESI) m/z 373.2 (M+Na+, 100%); HRMS (ESI) calcd for C23H23FNaO2+ [M+Na+]: 373.1574; found: 373.1572.
5.1.25. 3-(5-Fluoro-2-methyl-1H-inden-3-yl)propanenitrile (39)
A solution of compound 34 (300.0 mg, 1.8 mmol), and iso-propanol (0.7 mL, 9.0 mmol), and acrylonitrile (1.2 mL, 18.0 mmol) in THF (4 mL) was purged with argon for 20 min and cooled to 0 °C. A SmI2 (5.4 mmol) solution in THF (54 mL) was added through transfer needle. After 5 min, the reaction was quenched with saturated aqueous Na2CO3 (10 mL). The resulting mixture was extracted with Et2O (15 mL × 3). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. To the residue was added HOAc/H2SO4 (10/1, 3.0 mL). After stirring for 4 hr at room temperature, the mixture was extracted with EtOAc (15 mL × 3). The combined extracts were washed successively with saturated NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4) to afford compound 39 as a white solid (108 mg, 30%). M.p. 91–92 °C (hexane/EtOAc); IR (film): νmax 2915, 2247, 1610, 1592, 1476, 1275, 1190, 1165 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.16 (s, 3H, C=CCH3), 2.57 (t, J = 7.3 Hz, 2H, CH2CH2CN), 2.86 (t, J = 7.3 Hz, 2H, CH2CH2CN), 3.31 (s, 2H, CH2C=C), 6.79–6.88 (m, 2H, Ar-H), 7.27–7.32 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.3, 16.6, 21.3, 41.2, 104.8 (d, JC-F = 24.0 Hz), 110.5 (d, JC-F = 23.0 Hz), 119.2, 124.17 (d, JC-F = 9.0 Hz), 132.8, 137.5, 144.6, 146.9 (d, JC-F = 9.0 Hz), 162.4 (d, JC-F = 241.0 Hz) ppm; MS (ESI) m/z 224.1 (M+Na+, 100%); HRMS (ESI) calcd for C13H12FNNa+ [M+Na+]: 224.0846; found: 224.0848.
5.1.26. (Z)-3-(5-Fluoro-1-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl)propanenitrile (16)
Compound 16 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:20). Yellow solid, yield: 53%. M.p. 108–109 °C (hexane/EtOAc); IR (film): νmax 2957, 2927, 2866, 2247, 1598, 1464, 1199, 1162, 1138, 1055, 1016 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.32 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.24 (s, 3H, C=CCH3), 2.60 (t, J = 7.4 Hz, 2H, CH2CH2CN), 2.93 (t, J = 7.4 Hz, 2H, CH2CH2CN), 2.98 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 6.58–6.65 (m, 1H, Ar-H), 6.75–6.80 (m, 1H, Ar-H), 7.21 (s, 1H, vinyl-H), 7.28–7.33 (m, 2H, Ar-H), 7.39–7.48 (m, 3H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 16.6, 21.6, 23.9, 34.0, 104.7 (d, JC-F = 23.0 Hz), 110.6 (d, JC-F = 22.0 Hz), 119.1, 124.0 (d, JC-F = 8.0 Hz), 126.5 (2C), 129.4 (2C), 130.2, 131.1, 133.68, 133.78, 138.2, 139.6, 145.47 (d, JC-F = 8.0 Hz), 149.3, 163.0 (d, JC-F = 244.0 Hz) ppm; MS (ESI) m/z 354.2 (M+Na+, 100%); HRMS (ESI) calcd for C23H22FNNa+ [M+Na+]: 354.1628; found: 354.1625.
5.1.27. 2-(5-Fluoro-1-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl)acetamide (17 and 18)
A solution of compound 3 (Z : E = 2.5 : 1) (140.0 mg, 0.42 mmol), HOBt (72 mL, 0.53 mmol), and EDCI (101 mg, 0.53 mmol) in CH2Cl2 (4 mL) was stirred at room temperature under argon for 1 hr and cooled to 0 °C. Then to the solution was added NH3·H2O (0.1 mL), and stirred for 12 hr at room temperature. The reaction was quenched with 1.0 N citric acid, extracted with CH2Cl2 (5 mL × 3). The combined organic layers were washed with brine, saturated NHCO3, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:1) to give compound (Z)-17 as a yellow solid (82 mg, 58%), and compound (E)-18 as a yellow solid (26 mg, 19%). The data for (Z)-17: M.p. 138–139 °C (hexane/EtOAc); IR (film) νmax 3393, 3195, 2960, 2860, 1659, 1601, 1464, 1409, 1272, 1165, 1131, 1052, 1016 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.32 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.21 (s, 3H, C=CCH3), 2.98 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.51 (s, 2H, CH2CO), 5.75 (s, 1H, CONH2), 6.28 (s, 1H, CONH2), 6.57–6.64 (m, 1H, Ar-H), 6.85–6.90 (m, 1H, Ar-H), 7.22 (s, 1H, vinyl-H), 7.28–7.32 (m, 2H, Ar-H), 7.40–7.48 (m, 3H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 23.8, 33.2, 33.9, 105.5 (d, JC-F = 24.0 Hz), 110.8 (d, JC-F = 23.0 Hz), 123.79 (d, JC-F = 9.0 Hz), 126.5 (2C), 129.3 (2C), 129.85, 129.87, 131.2, 133.5, 138.8, 139.5, 145.9 (d, JC-F = 8.0 Hz), 149.3, 163.0 (d, JC-F = 244.0 Hz), 172.4 ppm; MS (ESI) m/z 358.2 (M+Na+, 100%); HRMS (ESI) calcd for C22H22FNNaO+ [M+Na+]: 358.1578; found: 358.1572. The data for (E)-18: M.p. 169–170 °C (hexane/EtOAc); IR (film): νmax 3393, 3192, 2961, 2918, 2872, 1656, 1607, 1461, 1397, 1257, 1147, 1113 cm−1; 1H NMR (400 MHz, CDCl3) δ: 1.29 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.90 (s, 3H, C=CCH3), 2.96 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.50 (s, 2H, CH2CO), 5.60 (s, 1H, CONH2), 6.87 (s, 1H, CONH2), 6.83–6.90 (m, 1H, Ar-H), 6.90–6.95 (m, 1H, Ar-H), 7.23–7.33 (m, 4H, Ar-H), 7.49–7.54 (m, 1H, Ar-H), 7.63 (s, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.2, 23.9, 33.2, 33.9, 105.4 (d, JC-F = 24.0 Hz), 111.3 (d, JC-F = 23.0 Hz), 119.5 (d, JC-F = 9.0 Hz), 126.2 (2C), 129.6 (2C), 130.3, 133.3, 133.5, 135.3, 136.2, 138.8, 143.07 (d, JC-F = 9.0 Hz), 149.2, 163.16 (d, JC-F = 243.0 Hz), 171.9 ppm; MS (ESI) m/z 358.2 (M+Na+, 100%); HRMS (ESI) calcd for C22H22FNNaO+ [M+Na+]: 358.1578; found: 358.1581.
5.1.28. 2-(5-Fluoro-1-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl)-N-methylacetamide (19 and 20)
Following the procedure described for 17 and 18, compounds 19 and 20 were synthesized respectively, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:2). (Z)-19 is a yellow solid (63%). The data for (Z)-19: M.p. 199–200 °C (hexane/EtOAc); IR (film): νmax 3289, 2957, 2869, 1647, 1601, 1467, 1409, 1262, 1162, 1055 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.20 (s, 3H, C=CCH3), 2.76 (d, J = 4.8 Hz, 3H, NCH3), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.51 (s, 2H, CH2CO), 5.85 (q, J = 4.8 Hz, 1H, CONH), 6.57–6.64 (m, 1H, Ar-H), 6.83–6.88 (m, 1H, Ar-H), 7.22 (s, 1H, vinyl-H), 7.27–7.32 (m, 2H, Ar-H), 7.40–7.48 (m, 3H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ: 10.5, 23.8, 26.4, 33.4, 33.9, 105.5 (d, JC-F = 23.0 Hz), 110.7 (d, JC-F = 22.0 Hz), 123.76 (d, JC-F = 9.0 Hz), 126.5 (2C), 129.3 (2C), 129.85, 129.87, 131.1 (2C), 133.5, 139.0, 139.5, 146.0 (d, JC-F = 9.0 Hz), 149.4, 163.0 (d, JC-F = 244.0 Hz), 169.9 ppm; MS (ESI) m/z 372.2 (M+Na+, 100%); HRMS (ESI) calcd for C23H24FNNaO+ [M+Na+]: 372.1734; found: 372.1733. (E)-20 is a yellow solid (23%). The data for (E)-20: M.p. 135–136 °C (hexane/EtOAc); IR (film): νmax 3296, 2957, 1644, 1598, 1470, 1409, 1202, 1150, 1049, 1013 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.29 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.89 (s, 3H, C=CCH3), 2.75 (d, J = 4.8 Hz, 3H, NCH3), 2.95 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.51 (s, 2H, CH2CO), 5.60 (q, J = 4.8 Hz, 1H, CONH), 6.84–6.92 (m, 2H, Ar-H), 7.23–7.33 (m, 4H, Ar-H), 7.50–7.56 (m, 1H, Ar-H), 7.63 (s, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.2, 23.9, 26.5, 33.5, 33.9, 105.5 (d, JC-F = 23.0 Hz), 111.35 (d, JC-F = 23.0 Hz), 119.5 (d, JC-F = 9.0 Hz), 126.2 (2C), 129.6 (2C), 130.2, 133.3, 133.5, 135.3, 136.4, 138.8, 143.18 (d, JC-F = 9.0 Hz), 149.2, 163.2 (d, JC-F = 244.0 Hz), 169.7 ppm; MS (ESI) m/z 372.2 (M+Na+, 100%). HRMS (ESI) calcd for C23H24FNNaO+ [M+Na+]: 372.1734; found: 372.1732.
5.1.29. 2-(4-Fluorlbenzylidene)butanoic acid (40)
Compound 40 was synthesized according to the general procedure A. White solid, yield: 50%. M.p. 113–114 °C (hexane/EtOAc); IR (film): νmax 3425, 2957, 1668, 1595, 1506, 1424, 1308, 1257, 1223, 1162, 1141 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.22 (t, J = 7.4 Hz, 3H, CH2CH3), 2.56 (q, J = 7.4 Hz, 2H, CH2CH3), 7.06–7.15 (m, 2H, Ar-H), 7.35–7.45 (m, 2H, Ar-H), 7.76 (s, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.6, 20.5, 115.66 (d, JC-F = 22.0 Hz), 131.3 (d, JC-F = 8.0 Hz), 131.5 (d, JC-F = 4.0 Hz), 133.7, 139.6, 162.8 (d, JC-F = 249.0 Hz), 174.0 ppm; MS (ESI) m/z 193.1 (M−H+); HRMS (ESI) calcd for C11H10FO2− [M−H+]: 193.0670; found: 193.0667.
5.1.30. 2-(4-Fluorlbenzyl)butanoic acid (41)
Compound 41 was synthesized according to the general procedure B. Colorless oil, yield: 90%. IR (film): νmax 3415, 2969, 1705, 1598, 1509, 1458, 1415, 1229, 1149 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.96 (t, J = 7.4 Hz, 3H, CH2CH3), 1.52–1.72 (m, 2H, CH2CH3), 2.52–2.63 (m, 1H, CH2CH), 2.73 (dd, J = 13.8, 6.6 Hz, 1H, CH2CH), 2.93 (dd, J = 13.8, 8.2 Hz, 1H, CH2CH), 6.92–7.00 (m, 2H, Ar-H), 6.09–7.17 (m, 2H, Ar-H), 10.40 (br s, 1H, COOH) ppm; 13C NMR (100 MHz, CDCl3) δ 11.6, 24.8, 36.9, 49.1, 115.2 (d, JC-F = 21.0 Hz) (2C), 130.26 (d, JC-F = 8.0 Hz) (2C), 134.78 (d, JC-F = 3.0 Hz), 162.6 (d, JC-F = 243.0 Hz), 181.7 ppm; MS (ESI) m/z 195.1 (M−H+); HRMS (ESI) calcd for C11H12FO2− [M−H+]: 195.0827; found: 195.0824.
5.1.31. 2-Ethyl-6-fluoro-2,3-dihydro-1H-inden-1-one (42)
Compound 42 was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). Colorless oil, yield: 85%. IR (film): νmax 2967, 2930, 2872, 1717, 1613, 1482, 1439, 1287, 1263, 1229, 1162 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.00 (t, J = 7.4 Hz, 3H, CH2CH3), 1.48–1.61 (m, 1H, CH2CH3), 1.90–2.01 (m, 1H, CH2CH3), 2.62–2.70 (m, 1H, CH2CH), 2.78 (dd, J = 17.0, 3.7 Hz, 1H, CH2CH), 3.28 (dd, J = 17.0, 7.8 Hz, 1H, CH2CH), 7.25–7.32 (m, 1H, Ar-H), 7.34–7.38 (m, 1H, Ar-H), 7.39–7.44 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 11.5, 24.4, 31.7, 49.6, 109.5 (d, JC-F = 22.0 Hz), 122.2 (d, JC-F = 23.0 Hz), 127.85 (d, JC-F = 8.0 Hz), 138.6 (d, JC-F = 7.0 Hz), 149.1, 162.25 (d, JC-F = 246.0 Hz), 207.9 ppm; MS (ESI) m/z 201.1 (M+Na+, 100%); HRMS (ESI) calcd for C11H11FNaO+ [M+Na+]: 201.0686; found: 201.0688.
5.1.32. Ethyl-2-(2-ethyl-5-fluoro-1H-inden-3-yl)acetate (43)
Compound 43 was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Colorless oil, yield: 67%. IR (film): νmax 2970, 2930, 1735, 1613, 1592, 1473, 1372, 1321, 1260, 1150, 1089, 1043 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.18 (t, J = 7.6 Hz, 3H, CH2CH3), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.53 (q, J = 7.6 Hz, 2H, CH2CH3), 3.32 (s, 2H, ArCH2C=C), 3.50 (s, 2H, CH2CO), 4.14 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.78–6.85 (m, 1H, Ar-H), 6.97–7.04 (m, 1H, Ar-H), 7.26–7.31 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.0, 14.1, 21.9, 31.5, 39.3, 60.9, 105.9 (d, JC-F = 24.0 Hz), 110.4 (d, JC-F = 23.0 Hz), 123.84 (d, JC-F = 9.0 Hz), 128.8, 137.24 (d, JC-F = 2.0 Hz), 147.86 (d, JC-F = 9.0 Hz), 150.5, 162.4 (d, JC-F = 240.0 Hz), 170.7 ppm; MS (ESI) m/z 271.1 (M+Na+, 100%); HRMS (ESI) calcd for C15H17FNaO2+ [M+Na+]: 271.1105; found: 271.1104.
5.1.33. (Z)-2-(2-Ethyl-5-fluoro-1-(4-isopropylbenzylidene)-1H-inden-3-yl)acetic acid (21)
Compound 21 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:4). Yellow solid, yield: 45%. M.p. 160–161 °C (hexane/EtOAc); IR (film): νmax 3426, 2967, 2939, 2866, 1707, 1598, 1464, 1412, 1299, 1174, 1055 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.22 (t, J = 7.6 Hz, 3H, CH2CH3), 1.32 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.65 (q, J = 7.6 Hz, 2H, CH2CH3), 2.98 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.60 (s, 2H, CH2CO), 6.56–6.63 (m, 1H, Ar-H), 6.89–6.94 (m, 1H, Ar-H), 7.22 (s, 1H, vinyl-H), 7.27–7.32 (m, 2H, Ar-H), 7.38–7.43 (m, 1H, Ar-H), 7.44–7.49 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 15.6, 18.3, 23.9, 31.3, 34.0, 105.8 (d, JC-F = 23.0 Hz), 110.7 (d, JC-F = 22.0 Hz), 123.96 (d, JC-F = 9.0 Hz), 126.5 (2C), 129.0, 129.5 (2C), 129.9, 130.9, 133.8, 138.1, 145.0, 146.1 (d, JC-F = 9.0 Hz), 149.3, 163.1 (d, JC-F = 244.0 Hz), 176.4 ppm; MS (ESI) m/z 373.2 (M+Na+, 100%); HRMS (ESI) calcd for C23H23FNaO2+ [M+Na+]: 373.1574; found: 373.1574.
5.1.34. 6-Fluoro-2-isobutyl-2,3-dihydro-1H-inden-1-one (44)
To the solution of 6-fluoro-1-indanone 34 (600 mg, 4.0 mmol), KOH (336 mg, 6.0 mmol), and Pd/C (60 mg, 10 %) in EtOH (40 mL), was added i-PrCHO (0.55 mL, 6.0 mmol) at 0 °C. After stirring for 1 hr at room temperature, the catalyst was filtered off and the mixture was acidified with a 6N HCl solution to pH 7.0, concentrated, extracted with EtOAc (20 mL × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentration under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent: petroleum ether) to afford compound 44 as white solid (511 mg, 62%). M.p. 42–43 °C (hexane/EtOAc); IR (film): νmax 2961, 2930, 2872, 1717, 1607, 1488, 1436, 1281, 1260, 1162, 1034 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.97 (dd, J = 6.2, 1.3 Hz, 6H, CH(CH3)2), 1.24–1.37 (m, 1H, CH2CH(CH3)2), 1.70–1.87 (m, 2H, CH(CH3)2 and CH2CH(CH3)2), 2.71–2.80 (m, 2H, ArCH2CH and ArCH2CH), 3.30 (dd, J = 17.6, 8.6 Hz, 1H, ArCH2CH), 7.26–7.32 (m, 1H, Ar-H), 7.36–7.38 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 21.7, 23.4, 26.5, 32.8, 40.6, 46.9, 109.6 (d, JC-F = 22.0 Hz), 122.2 (d, JC-F = 24.0 Hz), 127.8 (d, JC-F = 7.0 Hz), 138.4 (d, JC-F = 7.0 Hz), 148.93 (d, JC-F = 2.0 Hz), 162.29 (d, JC-F = 247.0 Hz), 208.26 (d, JC-F = 3.0 Hz) ppm; MS (ESI) m/z 229.1 (M+Na+, 100%); HRMS (ESI) calcd for C13H15FNaO+ [M+Na+]: 229.0999; found: 229.0996.
5.1.35. Ethyl-2-(5-fluoro-2-isobutyl-1H-inden-3-yl) acetate (45)
Compound 45 was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Yellow oil, yield: 57%. IR (film): νmax 2957, 1738, 1610, 1586, 1476, 1366, 1263, 1153, 1031 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.94 (d, J = 6.6 Hz, 6H, CH(CH3)2), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.85–2.00 (m, 1H, CH(CH3)2), 2.38 (d, J = 7.4 Hz, 2H, CH2CH), 3.31 (s, 2H, ArCH2C=C), 3.51 (s, 2H, CH2COOEt), 4.14 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.78–6.85 (m, 1H, Ar-H), 6.98–7.04 (m, 1H, Ar-H), 7.25–7.30 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 28.9, 31.6, 38.1, 40.2, 60.8, 106.0 (d, JC-F = 23.0 Hz), 110.4 (d, JC-F = 22.0 Hz), 123.7 (d, JC-F = 9.0 Hz), 130.32, 130.35, 137.3, 147.79 (d, JC-F = 8.0 Hz), 148.3, 162.4 (d, JC-F = 240.0 Hz), 170.7 ppm; MS (ESI) m/z 299.1 (M+Na+, 100%); HRMS (ESI) calcd for C17H21FNaO2+ [M+Na+]: 299.1418; found: 299.1419.
5.1.36. (Z)-2-(5-Fluoro-2-isobutyl-1-(4-isopropylbenzylidene)-1H-inden-3-yl) acetic acid (22)
Compound 22 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 75%. M.p. 135–136 °C (hexane/EtOAc); IR (film): νmax 3425, 2957, 2927, 2866, 1714, 1604, 1467, 1412, 1278, 1165, 1049 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.80 (d, J = 6.6 Hz, 6H, CH(CH3)2), 1.32 (d, J = 6.9 Hz, 6H, PhCH(CH3)2), 1.88–2.01 (m, 1H, CH(CH3)2), 2.51 (d, J = 7.3 Hz, 2H, CH2CH), 2.98 (sept, J = 6.9 Hz, 1H, PhCH(CH3)2), 3.63 (s, 2H, CH2CO), 6.57–6.64 (m, 1H, Ar-H), 6.88–6.94 (m, 1H, Ar-H), 7.20 (s, 1H, vinyl-H), 7.27–7.33 (m, 2H, Ar-H), 7.36–7.42 (m, 1H, Ar-H), 7.43–7.48 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 22.8 (2C), 23.9 (2C), 30.4, 31.6, 34.0, 34.2, 105.85 (d, JC-F = 23.0 Hz), 110.66 (d, JC-F = 23.0 Hz), 123.85 (d, JC-F = 9.0 Hz), 126.5 (2C), 129.4 (2C), 129.9, 130.4, 131.4, 133.8, 139.0, 142.4, 145.97 (d, JC-F = 9.0 Hz), 149.3, 163.0 (d, JC-F = 244.0 Hz), 176.5 ppm; MS (ESI) m/z 401.2 (M+Na+, 100%); HRMS (ESI) calcd for C25H27FNaO2+ [M+Na+]: 401.1887; found: 401.1887.
5.1.37. 12-Methyl-3-phenylacrylic acid (47a)
Compound 47a [25] was synthesized according to the general procedure A. Yellow solid, yield: 40%. M.p. 80–82 °C (hexane/EtOAc); IR (film): νmax 3415, 3425, 3040, 1662 (CO), 1609, 1443, 1407,1260 cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.15 (s, 3H, CH=CC H3), 7.30–7.45 (m, 5H, Ar-H), 7.85 (s, 1H, CH=CCH3) ppm; 13C-NMR (100 MHz, CDCl3) δ 1 3.7, 127.6, 128.4, 128.7, 129.8, 135.6, 141.1, 174.3 ppm; MS (ESI) m/z 163 (M+H+).
5.1.38. 2-Benzylpropanoic acid (48a)
Compound 48a [25] was synthesized according to the general procedure B. Colorless oil, yield: 100%. IR (film): νmax 3401, 2977, 2664, 1705, 1455, 1292, 1240 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.17 (d, J = 6.9 Hz, 3H, CHCH3), 2.66 (dd, J = 13.3, 8.0 Hz, 1H, CH2CH), 2.71–2.81 (ddq, J = 8.0, 6.3, 6.9 Hz, 1H, CHCH3), 3.07 (dd, J = 13.3, 6.3 Hz, 1H, CH2CH), 7.16–7.31 (m, 5H, Ar-H), 11.03 (br s, 1H, COOH) ppm; 13C NMR (100 MHz, CDCl3) δ 16.4, 39.3, 41.3, 126.4, 128.4, 129.0, 139.0, 182.6 ppm; MS (ESI) m/z 165 (M+H+).
6.1.39. 2,3-Dihydro-2-methylinden-1-one (49a)
Compound 49a [26] was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). Yellow oil, yield: 83%. IR (film): νmax 3072, 2929, 1709, 1605, 1462, 1288, 1201 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 7.3 Hz, 3H, CHCH3), 2.66–2.76 (m, 2H, CH2CH+CH2CH), 3.40 (dd, J = 17.9, 8.8 Hz, 1H, CH2CH), 7.33–7.78 (m, 4H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.2, 34.9, 41.9, 123.9, 126.5, 127.3, 134.6, 136.3, 153.4, 209.4 ppm; MS (ESI) m/z 169 (M+Na+).
5.1.40. Ethyl 2-(2-methyl-1H-inden-3-yl) acetate (50a)
Compound 50a [26] was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Yellow oil, yield: 72%. IR (film): νmax 2976, 2903, 1732, 1610, 1470, 1394, 1366, 1308, 1257, 1156, 1037 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.14 (s, 3H, C=CCH3), 3.35 (s, 2H, ArCH2C=C), 3.54 (s, 2H, CH2COOEt), 4.15 (q, J = 7.1 Hz, 2H, OCH2CH3), 7.11–7.16 (m, 1H, Ar-H), 7.24–7.32 (m, 2H, Ar-H), 7.36–7.40 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.13, 14.16, 31.6, 42.7, 60.7, 118.4, 123.1, 123.9, 126.1, 129.8, 141.95, 142.0, 145.9, 171.0 ppm; MS (ESI) m/z 239.1 (M+Na+, 100%).
5.1.41. (Z)-2-(1-(4-Isopropylbenzylidene)-2-methyl-1H-inden-3-yl) acetic acid (23)
Compound 23 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 78%. M.p. 133–134 °C (hexane/EtOAc); IR (film): νmax 3420, 2964, 1705, 1601, 1452, 1409, 1299, 1217, 1162, 1052, 1016 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.20 (s, 3H, C=CCH3), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.63 (s, 2H, CH2COOH), 6.89–6.95 (m, 1H, Ar-H), 7.14–7.19 (m, 2H, Ar-H), 7.21 (s, 1H, vinyl-H), 7.27–7.31 (m, 2H, Ar-H), 7.45–7.51 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.4, 23.9, 31.3, 34.0, 117.9, 122.8, 124.5, 126.4, 127.7, 129.4, 130.6, 131.0, 134.1, 136.9, 140.7, 143.8, 149.1, 176.1 ppm; MS (ESI) m/z 341.1 (M+Na+, 100%); HRMS (ESI) calcd for C22H22NaO2+ [M+Na+]: 341.1512; found: 341.1512.
5.1.42. 2-Methyl-3-p-tolylacrylic acid (47b)
Compound 47b was synthesized according to the general procedure A. White solid, yield: 30%. M.p. 163–164 °C (hexane/EtOAc); IR (film): νmax 3410, 2924, 1662, 1601, 1412, 1318, 1263, 1208, 1122 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.16 (s, 3H, C=CCH3), 2.39 (s, 3H, Ar-CH3), 7.19–7.40 (m, 4H, Ar-H), 7.82 (s, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.8, 21.4, 126.6, 129.2, 130.0, 132.8, 139.0, 141.2, 174.4 ppm; MS (ESI) m/z 175.1 (M−H+); HRMS (ESI) calcd for C11H11O2− [M−H+]: 175.0765; found: 175.0762.
5.1.43. 2-Methyl-3-p-tolylpropanoic acid (48b)
Compound 48b was synthesized according to the general procedure B. Colorless oil yield: 98%. IR (film): νmax 3400, 2979, 1702, 1516, 1461, 1412, 1290, 1241, 1198, 1119, 1037 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.18 (d, J = 6.9 Hz, 3H, CHCH3), 2.33 (s, 3H, Ar-CH3), 2.65 (dd, J = 13.4, 8.0 Hz, 1H, CHCH2), 2.74 (ddq, J = 8.0, 6.4, 6.9 Hz, 1H, CHCH3), 3.04 (dd, J = 13.4, 6.4 1H, CHCH2), 7.06–7.13 (m, 4H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.4, 21.0, 38.9, 41.3, 128.9, 129.1, 135.87, 135.92, 182.7 ppm; MS (ESI) m/z 177.1 (M−H+); HRMS (ESI) calcd for C11H13O2− [M−H+]: 177.0921; found: 177.0919.
5.1.44. 2,6-Dimethyl-2,3-dihydro-1H-inden-1-one (49b)
Compound 49b was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). Yellow oil yield: 87%. IR (film): νmax 2960, 2924, 2869, 1710, 1610, 1494, 1281, 1150, 1116, 1034 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.30 (d, J = 7.3 Hz, 3H, CHCH3), 2.40 (s, 3H, Ar-CH3), 2.67 (dd, J = 17.0, 3.9 Hz, 1H, CHCH2), 2.67–2.75 (m, 1H, CHCH3), 3.34 (dd, J = 17.0, 7.9 Hz, 1H, CHCH2), 7.30–7.35 (m, 1H, Ar-H), 7.38–7.42 (m, 1H, Ar-H), 7.54–7.57 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.3, 21.1, 34.6, 42.3, 123.9, 126.2, 135.9, 136.5, 137.2, 150.8, 209.6 ppm; MS (ESI) m/z 183.1 (M+Na+, 100%); HRMS (ESI) calcd for C11H12NaO+ [M+Na+]: 183.0780; found: 183.0772.
5.1.45. Ethyl 2-(2,5-dimethyl-1H-inden-3-yl) acetate (50b)
Compound 50b was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Colorless oil yield: 79%. IR (film) νmax 2982, 2915, 1735, 1616, 1479, 1366, 1253, 1153, 1037 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.12 (s, 3H, C=CCH3), 2.39 (s, 3H, Ar-CH3), 3.30 (s, 2H, CH2C=C), 3.52 (s, 2H, CH2COOEt), 4.15 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.92–6.97 (m, 1H, Ar-H), 7.09–7.13 (m, 1H, Ar-H), 7.23–7.27 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 21.5, 31.6, 42.3, 60.7, 119.2, 122.8, 124.6, 129.7, 135.7, 139.1, 142.2, 146.1, 171.1 ppm; MS (ESI) m/z 253.1 (M+Na+, 100%); HRMS (ESI) calcd for C15H18NaO2+ [M+Na+]: 253.1199; found: 253.1199.
5.1.46. (Z)-2-(1-(4-iso-Propylbenzylidene)-2,5-dimethyl-1H-inden-3-yl) acetic acid (24)
Compound 24 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 70%. M.p. 143–144 °C (hexane/EtOAc); IR (film): νmax 3416, 2961, 2924, 1705, 1607, 1467, 1415, 1311, 1159, 1046 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.19 (s, 3H, C=CCH3), 2.33 (s, 3H, Ar-CH3), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.61 (s, 2H, CH2COOH), 6.71–6.76 (m, 1H, Ar-H), 6.97–7.02 (m, 1H, Ar-H), 7.14 (s, 1H, vinyl-H), 7.27 (m, 2H, Ar-H), 7.34–7.39 (m, 1H, Ar-H), 7.44–7.50 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 21.7, 23.9, 31.3, 34.0, 118.8, 122.6, 125.1, 126.4, 129.5, 130.0, 130.4, 131.5, 134.3, 137.2, 137.7, 140.6, 144.1, 148.9, 176.3 ppm; MS (ESI) m/z 355.2 (M+Na+, 100%); HRMS (ESI) calcd for C23H24NaO2+ [M+Na+]: 355.1699; found: 355.1664.
5.1.47. 3-(4-Methoxyphenyl)-2-methylacrylic acid (47c)
Compound 47c [25] was synthesized according to the general procedure A. White solid, yield: 32%. M.p. 154–155 °C (hexane/EtOAc); IR (film): νmax 3390, 2945, 2836. 1662, 1598, 1513, 1424, 1281, 1257, 1177, 1037 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.16 (s, 3H, C=CCH3), 3.85 (s, 3H, OCH3), 6.90–7.00 (m, 2H, Ar-H), 7.40–7.48 (m, 2H, Ar-H), 7.79 (s, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.7, 55.3, 113.9, 125.1, 128.2, 131.7, 140.8, 160.0, 174.5 ppm; MS (ESI) m/z 215.1 (M+Na+, 100%).
5.1.48. 3-(4-Methoxyphenyl)-2-methylpropanoic acid (48c)
Compound 48c [25] was synthesized according to the general procedure B. Colorless oil, yield: 99%. IR (film): νmax 3380, 2933, 1711, 1613, 1513, 1461, 1247, 1299, 1183, 1116, 1037 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.17 (d, J = 6.9 Hz, 3H, CHCH3), 2.63 (dd, J = 13.4, 7.9 Hz, 1H, CHCH2), 2.72 (ddq, J = 7.9, 6.3, 6.9 Hz, 1H, CHCH3), 3.02 (dd, J = 13.4, 6.3 1H, CHCH2), 3.79 (s, 3H, OCH3), 6.81–6.87 (m, 2H, Ar-H), 7.08–7.14 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.4, 38.4, 41.5, 55.2, 113.8, 129.9, 131.1, 158.1, 182.6 ppm; MS (ESI) m/z 217.1 (M+Na+, 100%).
5.1.49. 6-Methoxy-2-methyl-2,3-dihydro-1H-inden-1-one (49c)
Compound 49c [27] was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). Colorless oil, yield: 50%. IR (film): νmax 2961, 2927, 1708, 1619, 1488, 1436, 1278, 1244, 1171, 1028 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.30 (d, J = 7.4 Hz, 3H, CHCH3), 2.64 (dd, J = 16.6, 3.6 Hz, 1H, CHCH2), 2.68–2.78 (m, 1H, CHCH3), 3.32 (dd, J = 16.6, 7.6 Hz, 1H, CHCH2), 3.83 (s, 3H, OCH3), 7.15–7.20 (m, 2H, Ar-H), 7.31–7.35 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.3, 34.3, 42.8, 55.6, 105.1, 124.1, 127.2, 137.4, 146.2, 159.4, 209.5 ppm; MS (ESI) m/z 199.1 (M+Na+, 100%).
5.1.50. Ethyl 2-(5-methoxy-2-methyl-1H-inden-3-yl) acetate (50c)
Compound 50c [27] was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). White solid, yield: 75%. M.p. 36–37 °C (hexane/EtOAc); IR (film): νmax 2985, 2933, 2908, 1729, 1619, 1583, 1479, 1284, 1244, 1202, 1153, 1092, 1040 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.12 (s, 3H, C=CCH3), 2.29 (s, 2H, ArCH2C=C), 3.50 (s, 2H, CH2COOEt), 3.83 (s, 3H, OCH3), 4.14 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.66–6.71 (m, 1H, Ar-H), 6.85–6.88 (m, 1H, Ar-H), 7.22–7.26 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.19, 14.27, 31.6, 42.0, 55.5, 60.8, 104.5, 109.4, 123.5, 129.7, 134.1, 143.6, 147.4, 158.9, 171.0 ppm; MS (ESI) m/z 269.1 (M+Na+, 100%).
5.1.51. (Z)-2-(1-(4-Isopropylbenzylidene)-5-methyl-2-methyl-1H-inden-3-yl) acetic acid (25)
Compound 25 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 76%. M.p. 117–119 °C (hexane/EtOAc); IR (film) νmax 3411, 2957, 1705, 1598, 1473, 1214, 1162, 1037 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.19 (s, 3H, C=CCH3), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.60 (s, 2H, CH2COOH), 3.79 (s, 3H, OCH3), 6.41–6.47 (m, 1H, Ar-H), 6.74–6.77 (m, 1H, Ar-H), 7.10 (s, 1H, vinyl-H), 7.26–7.29 (m, 2H, Ar-H), 7.37–7.41 (m, 1H, Ar-H), 7.44–7.49 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.6, 23.9, 31.3, 34.0, 55.4, 104.7, 108.9, 123.6, 126.4, 126.9, 129.1, 129.5, 130.1, 134.3, 138.4, 140.2, 145.8, 148.9, 160.0, 175.7 ppm; MS (ESI) m/z 371.2 (M+Na+, 100%); HRMS (ESI) calcd for C23H24NaO3+ [M+Na+]: 371.1618; found: 371.1620.
5.1.52. 3-(4-Ethylphenyl)-2-methylacrylic acid (47d)
Compound 47d was synthesized according to the general procedure A. White solid, yield: 41%. M.p. 126–127 °C (hexane/EtOAc); IR (film): νmax 3411, 2957, 1671, 1607, 1424, 1314, 1269, 1180, 1132 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.28 (t, J = 7.6 Hz, 3H, CH2CH3), 2.18 (s, 3H, C=CCH3), 2.70 (q, J = 7.6 Hz, 2H, CH2CH3), 7.23–7.30 (m, 2H, Ar-H), 7.37–7.44 (m, 2H, Ar-H), 7.85 (s, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.7, 15.3, 28.7, 126.6, 128.0, 130.1, 133.0, 141.2, 145.2, 174.6 ppm; MS (ESI) m/z 189.1 (M−H+); HRMS (ESI) calcd for C12H13O2− [M−H+]: 189.0921; found: 189.0919.
5.1.53. 3-(4-Ethylphenyl)-2-methylpropanoic acid (48d)
Compound 48d was synthesized according to the general procedure B. Colorless oil, yield: 100%. IR (film): νmax 3401, 2967, 2933, 1708, 1595, 1516, 1464, 1418, 1293, 1238, 1196, 1122 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.18 (d, J = 6.9 Hz, 3H, CHCH3), 1.23 (t, J = 7.6 Hz, 3H, CH2CH3), 2.63 (q, J = 7.6 Hz, 2H, CH2CH3), 2.65 (dd, J = 13.4, 8.2 Hz, 1H, CHCH2), 2.74 (ddq, J = 8.2, 6.3, 6.9 Hz, 1H, CHCH3), 3.06 (dd, J = 13.4, 6.3 1H, CHCH2), 7.08–7.16 (m, 4H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 15.5, 16.5, 28.4, 38.9, 41.3, 127.9, 128.9, 136.2, 142.3, 182.4 ppm; MS (ESI) m/z 191.1 (M−H+); HRMS (ESI) calcd for C12H15O2− [M−H+]: 191.1078; found: 191.1074.
5.1.54. 6-Ethyl-2-methyl-2,3-dihydro-1H-inden-1-one (49d)
Compound 49d [28] was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). Yellow oil, yield: 91%. IR (film): νmax 2967, 2930, 2872, 1711, 1613, 1494, 1452, 1275, 1153, 1122 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.25 (t, J = 7.6 Hz, 3H, CH2CH3), 1.30 (d, J = 7.3 Hz, 3H, CHCH3), 2.64–2.76 (m, 4H, CHCH3, CHCH2, CH2CH3), 3.35 (dd, J = 17.2, 8.0 Hz, 1H, CHCH2), 7.33–7.39 (m, 1H, Ar-H), 7.40–7.46 (m, 1H, Ar-H), 7.56–7.61 (s, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ: 15.5, 16.3, 28.5, 34.6, 42.3, 122.6, 126.3, 135.0, 136.5, 143.7, 151.1, 209.6 ppm; MS (ESI) m/z 197.1 (M+Na+, 100%).
5.1.55. Ethyl 2-(5-ethyl-2-methyl-1H-inden-3-yl) acetate (50d)
Compound 50d was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Colorless oil, yield: 84%. IR (film): νmax 2957, 2924, 1732, 1613, 1479, 1366, 1305, 1263, 1144, 1034 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.26 (t, J = 7.6 Hz, 3H, CH2CH3), 2.12 (s, 3H, C=CCH3), 2.69 (q, J = 7.6 Hz, 2H, CH2CH3), 3.30 (s, 2H, ArCH2C=C), 3.52 (s, 2H, CH2COOEt), 4.15 (q, J = 7.1 Hz, 2H, OCH2CH3), 6.94–7.00 (m, 1H, Ar-H), 7.11–7.16 (m, 1H, Ar-H), 7.26–7.30 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.2 (2C), 16.2, 29.1, 31.7, 42.4, 60.7, 118.0, 123.0, 123.6, 129.8, 139.4, 142.2, 142.4, 146.2, 171.1 ppm; MS (ESI) m/z 267.1 (M+Na+, 100%); HRMS (ESI) calcd for C16H20NaO2+ [M+Na+]: 267.1356; found: 267.1352.
5.1.56. (Z)-2-(5-Ethyl-1-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl) acetic acid (26)
Compound 26 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 76%. M.p. 111–112 °C (hexane/EtOAc); IR (film): νmax 3413, 2957, 2933, 2872, 1705, 1604, 1506, 1467, 1409, 1293, 1055 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.23 (t, J = 7.6 Hz, 3H, CH2CH3), 1.32 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.20 (s, 3H, C=CCH3), 2.64 (q, J = 7.6 Hz, 2H, CH2CH3), 2.98 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.63 (s, 2H, CH2COOH), 6.75–6.80 (m, 1H, Ar-H), 7.01–7.06 (m, 1H, Ar-H), 7.16 (s, 1H, vinyl-H), 7.27–7.31 (m, 2H, Ar-H), 7.38–7.43 (m, 1H, Ar-H), 7.46–7.51 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ: 10.5, 15.7, 23.9, 29.1, 31.4, 34.0, 117.6, 122.7, 124.0, 126.4, 129.5, 130.0, 130.5, 131.7, 134.3, 137.2, 140.7, 144.1, 144.2, 148.9, 176.9 ppm; MS (ESI) m/z 369.2 (M+Na+, 100%); HRMS (ESI) calcd for C24H26NaO2+ [M+Na+]: 369.1825; found: 369.1818.
5.1.57. 3-(4-Ethoxyphenyl)-2-methylacrylic acid (47e)
Compound 47e was synthesized according to the general procedure A. White solid, yield: 30%. M.p. 166–167 °C (hexane/EtOAc); IR (film): νmax 3385, 2930, 1671, 1601, 1509, 1424, 1256, 1180, 1122, 1043 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.44 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.16 (d, J = 1.3 Hz, 3H, C=CCH3), 4.07 (q, J = 7.0 Hz, 2H, OCH2CH3), 6.91–6.95 (m, 2H, Ar-H), 7.39–7.45 (m, 2H, Ar-H), 7.78 (q, J = 1.3 Hz, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.8, 14.8, 63.5, 114.4, 124.9, 128.1, 131.8, 140.8, 159.4, 174.0 ppm; MS (ESI) m/z 205.1 ([M−H+); HRMS (ESI) calcd for C12H13O3−[M−H+]: 205.0870; found: 205.0866.
5.1.58. 3-(4-Ethoxyphenyl)-2-methylpropanoic acid (48e)
Compound 48e was synthesized according to the general procedure B. White solid, yield: 98%. M.p. 56–57 °C (hexane/EtOAc); IR (film) νmax 3378, 2973, 2933, 1705, 1610, 1509, 1378, 1244, 1177, 1119, 1046 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.17 (d, J = 6.9 Hz, 3H, CHCH3), 1.41 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.62 (dd, J = 13.4, 7.9 Hz, 1H, CHCH2), 2.72 (ddq, J = 7.9, 6.4, 6.9 Hz, 1H, CHCH3), 3.02 (dd, J = 13.4, 6.4, 1H, CHCH2), 4.02 (q, J = 7.0 Hz, 2H, OCH2CH3), 6.80–6.87 (m, 2H, Ar-H), 7.06–7.13 (m, 2H, Ar-H), 10.33 (br s, 1H, COOH) ppm; 13C NMR (100 MHz, CDCl3) δ 14.8, 16.4, 38.5, 41.5, 63.3, 114.4, 129.9, 130.9, 157.5, 182.6 ppm; MS (ESI) m/z 207.1 (M−H+); HRMS (ESI) calcd for C12H15O3− [M−H+]: 207.1027; found: 207.1023.
5.1.59. 6-Ethoxy-2-methyl-2,3-dihydro-1H-inden-1-one (49e)
Compound 49e was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). White solid, yield: 50%. M.p. 49–50 °C (hexane/EtOAc); IR (film): νmax 2969, 2927, 1705, 1616, 1491, 1446, 1278, 1241, 1174, 1116, 1040 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.30 (d, J = 7.4 Hz, 3H, CHCH3), 1.41 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.64 (dd, J = 16.6, 3.6 Hz, 1H, CHCH2), 2.67–2.78 (m, 1H, CHCH3), 3.31 (dd, J = 16.6, 7.6 Hz, 1H, CHCH2), 4.05 (q, J = 7.0 Hz, 2H, OCH2CH3), 7.14–7.19 (m, 2H, Ar-H), 7.29–7.34 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.7, 16.3, 34.2, 42.7, 63.8, 105.7, 124.5, 127.2, 137.4, 146.1, 158.7, 209.5 ppm; MS (ESI) m/z 213.1 (M+Na+, 100%); HRMS (ESI) calcd for C12H14NaO2+[M+Na+]: 213.0886; found: 213.0886.
5.1.60. Ethyl 2-(5-ethoxy-2-methyl-1H-inden-3-yl) acetate (50e)
Compound 50e was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). White solid, yield: 82%. M.p. 37–38 °C (hexane/EtOAc); IR (film): νmax 2979, 2909, 1732, 1607, 1467, 1394, 1259, 1208, 1153, 1086, 1034 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.24 (t, J = 7.1 Hz, 3H, COOCH2CH3), 1.42 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.12 (s, 3H, C=CCH3), 3.27 (s, 2H, ArCH2C=C), 3.49 (s, 2H, CH2COOEt), 4.05 (q, J = 7.0 Hz, 2H, OCH2CH3), 4.14 (q, J = 7.1 Hz, 2H, COOCH2CH3), 6.65–6.69 (m, 1H, Ar-H), 6.84–6.87 (m, 1H, Ar-H), 7.21–7.24 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.19, 14.27, 14.9, 31.6, 42.0, 60.7, 63.6, 105.3, 110.1, 123.5, 129.8, 134.1, 143.5, 147.3, 158.2, 171.0 ppm; MS (ESI) m/z 283.1 (M+Na+, 100%); HRMS (ESI) calcd for C16H20NaO3+ [M+Na+]: 283.1305; found: 283.1303.
5.1.61. (Z)-2-(5-Ethoxy-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl) acetic acid (27)
Compound 27 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 67%. M.p. 146–147 °C (hexane/EtOAc); IR (film) νmax 3410, 2957, 2930, 1705, 1610, 1464, 1385, 1211, 1156, 1119, 1040 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.39 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.19 (s, 3H, C=CCH3), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.59 (s, 2H, CH2COOH), 4.02 (q, J = 7.0 Hz, 2H, OCH2CH3), 6.40–6.46 (m, 1H, Ar-H), 6.72–6.78 (m, 1H, Ar-H), 7.09 (s, 1H, vinyl-H), 7.24–7.30 (m, 2H, Ar-H), 7.36–7.40 (m, 1H, Ar-H), 7.43–7.49 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 14.8, 23.9, 31.3, 34.0, 63.5, 105.3, 109.5, 123.6, 126.4, 126.7, 128.9, 129.5, 130.1, 134.3, 138.3, 140.2, 145.7, 148.8, 159.3, 176.1 ppm; MS (ESI) m/z 385.2 (M+Na+, 100%); HRMS (ESI) calcd for C24H26NaO3+ [M+Na+]: 385.1774; found: 385.1777.
5.1.62. 3-(4-iso-Propylphenyl)-2-methylacrylic acid (47f)
Compound 47f was synthesized according to the general procedure A. White solid, yield: 30%. M.p. 86–87 °C (hexane/EtOAc); IR (film) νmax 3380, 2961, 1677, 1619, 1421, 1360, 1272, 1217, 1122, 1046 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.28 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.17 (d, J = 1.3 Hz, 3H, C=CCH3), 2.95 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 7.26–7.31 (m, 2H, Ar-H), 7.38–7.43 (m, 2H, Ar-H), 7.83 (q, J = 1.3 Hz, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ: 13.8, 23.8, 34.0, 126.6, 130.1, 130.4, 133.1, 141.1, 149.8, 174.4 ppm; MS (ESI) m/z 203.1 (M−H+); HRMS (ESI) calcd for C13H15O2−[M−H+]: 203.1078; found: 203.1075.
5.1.63. 3-(4-iso-Propylphenyl)-2-methylpanoic acid (48f)
Compound 48f was synthesized according to the general procedure B. Colorless oil, yield: 97%. IR (film): νmax 3365, 2957, 1705, 1513, 1461, 1418, 1284, 1238, 1193, 1116, 1052 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.19 (d, J = 6.9 Hz, 3H, CHCH3), 1.25 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.64 (dd, J = 13.4, 7.1 Hz, 1H, CHCH2), 2.76 (ddq, J = 7.1, 6.2, 6.9 Hz, 1H, CHCH3), 2.89 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.08 (dd, J = 13.4, 6.2, 1H, CHCH2), 7.10–7.19 (m, 4H, Ar-H), 9.70 (br s, 1H, COOH) ppm; 13C NMR (100 MHz, CDCl3) δ 16.5, 24.0, 33.7, 38.9, 41.2, 126.4, 128.9, 136.3, 146.9, 182.6 ppm; MS (ESI) m/z 205.1 (M−H+); HRMS (ESI) calcd for C13H17O2− [M−H+]: 205.1234; found: 205.1230.
5.1.64. 6-iso-Propyl-2-methyl-2,3-dihydro-1H-inden-1-one (49f)
Compound 49f was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). Yellow oil, yield: 87%. IR (film): νmax 2960, 2927, 2866, 1708, 1622, 1494, 1436, 1259, 1174, 1119 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.26 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.30 (d, J = 7.3 Hz, 3H, CHCH3), 2.64–2.76 (m, 2H, CHCH2, CHCH3), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.35 (dd, J = 17.0, 7.9 Hz, 1H, CHCH2), 7.34–7.39 (m, 1H, Ar-H), 7.45–7.49 (m, 1H, Ar-H), 7.61–7.64 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.3, 23.9, 33.9, 34.6, 42.3, 121.1, 126.3, 133.8, 136.5, 148.5, 151.3, 209.7 ppm; MS (ESI) m/z 211.1 (M+Na+, 100%). HRMS (ESI) calcd for C13H16NaO+ [M+Na+]: 211.1093; found: 211.1095.
5.1.65. Ethyl 2-(5-isopropyl-2-methyl-1H-inden-3-yl) acetate (50f)
Compound 50f was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Colorless oil, yield: 82%. IR (film): νmax 2954, 1741, 1610, 1482, 1366, 1302, 1253, 1156, 1031 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.28 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.32 (d, J = 6.8 Hz, 6H, CH(CH3)2), 2.16 (s, 3H, C=CCH3), 2.99 (sept, J = 6.8 Hz, 1H, CH(CH3)2), 3.33 (s, 2H, ArCH2C=C), 3.57 (s, 2H, CH2COOEt), 4.18 (q, J = 7.2 Hz, 2H, OCH2CH3), 7.02–7.07 (m, 1H, Ar-H), 7.18–7.24 (m, 1H, Ar-H), 7.30–7.36 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.2, 24.3 (2C), 31.7, 34.3, 42.3, 60.7, 116.5, 122.1, 122.9, 129.8, 139.6, 142.2, 146.1, 147.0, 171.1 ppm; MS (ESI) m/z 281.2 (M+Na+, 100%); HRMS (ESI) calcd for C17H22NaO2+ [M+Na+]: 281.1512; found: 281.1517.
5.1.66. (Z)-2-(5-iso-Propyl-1-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl) acetic acid (28)
Compound 28 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 78%. M.p. 125–126 °C (hexane/EtOAc); IR (film) νmax 3405, 2954, 1714, 1607, 1507, 1467, 1406, 1299, 1213, 1052, 1013 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.24 (d, J = 6.9 Hz, 6H, CH(CH3)2), 1.32 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.19 (s, 3H, C=CCH3), 2.89 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.63 (s, 2H, CH2COOH), 6.78–6.83 (m, 1H, Ar-H), 7.03–7.08 (m, 1H, Ar-H), 7.15 (s, 1H, vinyl-H), 7.26–7.31 (m, 2H, Ar-H), 7.38–7.42 (m, 1H, Ar-H), 7.45–7.50 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 23.9, 24.0, 31.4, 34.0, 34.3, 116.3, 122.4, 122.7, 126.4, 129.4, 130.0, 130.6, 131.9, 134.3, 137.1, 140.7, 144.1, 148.88, 148.92, 176.6 ppm; MS (ESI) m/z 383.2 (M+Na+, 100%). HRMS (ESI) calcd for C25H28NaO2+ [M+Na+]: 383.1982; found: 383.1982.
5.1.67. 3-(4-Chlorophenyl)-2-methylacrylic acid (47g)
Compound 47g [25] was synthesized according to the general procedure A. White solid, yield: 55%. M.p. 164–165 °C (hexane/EtOAc); IR (film): νmax 3360, 2957, 2826, 1671, 1491, 1446, 1424, 1308, 1287, 1263, 1214, 1132, 1092, 1012 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.13 (d, J = 1.4 Hz, C=CCH3), 7.35–7.41 (m, 4H, Ar-H), 7.79 (q, J = 1.4 Hz, 1H, vinyl-H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.7, 128.1, 128.7, 131.1, 134.0, 134.7, 139.7, 173.7 ppm; MS (ESI) m/z 197.0 (M+Na+, 100%).
5.1.68. 3-(4-Chlorophenyl)-2-methylpropanoic acid (48g)
A mixture of the acrylic acid 47g (1.5 g, 7.63 mmol) and 10 % Pd/C (150 mg) in EtOAc (30 mL) was hydrogenated under 3 atm of hydrogen for 12 h. The catalyst was filtered off and the filtrate concentrated to afford compound 48g as a white solid (1.46 g, 96%). M.p. 52–53 °C (hexane/EtOAc); IR (film): νmax 3351, 2976, 2933, 1698, 1494, 1461, 1406, 1296, 1232, 1196, 1089, 1019 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.19 (d, J = 6.8 Hz, 3H, CHCH3), 2.66 (dd, J = 13.3, 7.6 Hz, 1H, CHCH2), 2.74 (ddq, J = 7.6, 6.5, 6.8 Hz, 1H, CHCH3), 3.02 (dd, J = 13.3, 6.5, 1H, CHCH2), 7.09–7.14 (m, 2H, Ar-H), 7.24–7.28 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ: 16.5, 38.6, 41.1, 128.5, 130.3, 132.3, 137.4, 182.2 ppm; MS (ESI) m/z 221.0 (M+Na+, 100%).
5.1.69. 6-Chloro-2-methyl-2,3-dihydro-1H-inden-1-one (49g)
Compound 49g was synthesized according to the general procedure C, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:40). White solid, yield: 78%. M.p. 62–64 °C (hexane/EtOAc); IR (film): νmax 3079, 2967, 1930, 1774, 1704, 1604, 1455, 1433, 1256, 1235, 1193, 1104, 1064 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.30 (d, J = 7.3 Hz, 3H, CHCH3), 2.68 (dd, J = 16.8, 3.9 Hz, 1H, CHCH2), 2.71–2.79 (m, 1H, CHCH3), 3.36 (dd, J = 16.8, 7.6, 1H, CHCH2), 7.35–7.40 (m, 1H, Ar-H), 7.50–7.55 (m, 1H, Ar-H), 7.67–7.71 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 16.2, 34.5, 42.6, 123.8, 127.8, 133.7, 134.7, 137.9, 151.5, 208.0 ppm; MS (ESI) m/z 203.0 (M+Na+, 100%).
5.1.70. Ethyl 2-(5-chloro-2-methyl-1H-inden-3-yl) acetate (50g)
Compound 50g was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Colorless oil, yield: 67%. IR (film): νmax 2976, 2906, 1732, 1601, 1464, 1363, 1305, 1256, 1156, 1098, 1074, 1034 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.14 (s, 3H, C=CCH3), 3.32 (s, 2H, ArCH2C=C), 3.50 (s, 2H, CH2COOEt), 4.17 (q, J = 7.1 Hz, 2H, OCH2CH3), 7.08–7.13 (m, 1H, Ar-H), 7.24–7.29 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.15, 14.2, 31.4, 42.3, 60.9, 118.8, 123.7, 124.0, 129.4, 132.2, 140.2, 144.1, 147.7, 170.7 ppm; MS (ESI) m/z 273.1 (M+Na+, 100%); HRMS (ESI) calcd for C14H15ClNaO2+ [M+Na+]: 273.0653; found: 273.0659.
5.1.71. (Z)-2-(5-Chloro-1-(4-isopropylbenzylidene)-2-methyl-1H-inden-3-yl) acetic acid (29)
Compound 29 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 77%. M.p. 182–183 °C (hexane/EtOAc); IR (film): νmax 3392, 2961, 2930, 1705, 1595, 1452, 1412, 1308, 1217, 1086, 1022 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.21 (s, 3H, C=CCH3), 2.97 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.60 (s, 2H, CH2COOH), 6.86–6.90 (m, 1H, Ar-H), 7.14–7.16 (m, 1H, Ar-H), 7.23 (s, 1H, vinyl-H), 7.27–7.31 (m, 2H, Ar-H), 7.35–7.39 (m, 1H, Ar-H), 7.42–7.47 (m, 2H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 10.5, 23.9, 31.2, 34.0, 118.3, 123.6, 124.1, 126.6, 129.4, 129.7, 131.9, 132.3, 133.5, 133.7, 138.6, 139.7, 145.6, 149.4, 176.0 ppm; MS (ESI) m/z 375.1 (M+Na+, 100%); HRMS (ESI) calcd for C22H21ClNaO2+ [M+Na+]: 375.1122; found: 375.1125.
5.1.72. Ethyl 2-(5-Fluoro-1H-inden-3-yl)acetate (52)
Compound 52 was synthesized according to the general procedure D, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:50). Colorless oil, yield: 55%. IR (film): νmax 3054, 2982, 2931, 1704, 1636, 1486, 1446, 1369, 1345, 1288, 1276 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.28 (t, J = 7.2 Hz, 3H, CH2CH3), 3.35 (s, 2H, CH2CO), 3.56 (m, 2H, CH2CH), 4.19 (q, J = 7.2 Hz, 2H, CH2CH3), 6.52 (s, 1H, C=CH), 6.87–6.94 (m, 1H, Ar-H), 7.03–7.09 (m, 1H, Ar-H), 7.33–7.39 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 14.2, 34.1, 37.4, 61.0, 106.5 (d, JC-F = 24.0 Hz), 111.5 (d, JC-F = 23.0 Hz), 124.4 (d, JC-F = 9.0 Hz), 134.1, 136.36 (d, JC-F = 3.0 Hz), 139.24 (d, JC-F = 2.0 Hz), 146.34 (d, JC-F = 8.0 Hz), 162.3 (d, JC-F = 241.0 Hz), 170.7 ppm; MS (ESI) m/z 243.1 (M+Na+); Anal. Calcd for C13H13FO2: C, 70.90; H, 5.95. Found: C, 71.30; H, 6.23.
5.1.73. (E)-2-(5-Fluoro-1-(4-isopropylbenzylidene)-1H-inden-3-yl)acetic acid (30)
Compound 30 was synthesized according to the general procedure E, and purified by flash column chromatography on silica gel (eluent: ethyl acetate: petroleum ether = 1:10). Yellow solid, yield: 78%. M.p. 128–129 °C (hexane/EtOAc); IR (film): νmax 3426, 2960, 1708, 1604, 1513, 1461, 1418, 1247, 1159, 1049 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.29 (d, J = 6.9 Hz, 6H, CH(CH3)2), 2.96 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 3.69 (s, 2H, CH2COO), 6.90–6.98 (m, 1H, Ar-H), 7.01–7.06 (m, 1H, Ar-H), 7.09 (s, 1H, CH2C=CH), 7.27–7.32 (m, 2H, Ar-H), 7.40 (s, 1H, vinyl-H), 7.50–7.55 (m, 2H, Ar-H), 7.59–7.64 (m, 1H, Ar-H) ppm; 13C NMR (100 MHz, CDCl3) δ 23.8, 33.8, 34.0, 106.5 (d, JC-F = 23.0 Hz), 111.96 (d, JC-F = 23.0 Hz), 119.97 (d, JC-F = 9.0 Hz), 126.9 (2C), 127.0, 129.2, 130.2 (2C), 133.6, 134.2, 137.0, 137.6, 142.96 (d, JC-F = 9.0 Hz), 149.7, 162.9 (d, JC-F = 243.0 Hz), 176.1 ppm; MS (ESI) m/z 345.1 (M+Na+, 100%); HRMS (ESI) calcd for C21H19FNaO2+ [M+Na+]: 345.1261; found: 345.1262.
5.2. Biological assays
5.2.1. Ligand-binding Competition Assay
The GST-tagged human RXRα-LBD (223–462) was incubated with unlabeled 9-cis-RA or different concentrations of compounds in 200 μL binding buffer [0.15 M KCl, 10 mMTris·HCl (pH7.4), 8% glycerol, and 0.5% CHAPS detergent] at 4°C for 1 h. [3H]-9-cis-RA was added to the final concentration of 7.5 nM and final volume of 300 μL and incubated overnight at 4°C. The RXRα-LBD was captured by Glutathione sepharose beads. Bound [3H]-9-cis-RA was quantitated by liquid scintillation counting [29, 30].
5.2.2. Cell Culture and Transfection
HeLa cervix cancer, HCT-116 colon cancer and A549 lung cancer cells were cultured in Dulbecco modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS). PC3 prostate cancer and ZR-75-1 breast cancer cells were grown in RPMI1640 medium containing 10% FBS. The cells were maintained at 5% CO2 at 37°C.
5.2.3. MTT assay
Cells cultured in 96-well dishes were treated with various concentrations of compound 30 for 24 hr. The cells were then incubated with 2 mg/mL MTT for 4 hr at 37°C and dissolved by addition of 150 μL DMSO each well. Absorbance was measured at 570 nm [30].
5.2.4 Western Blotting
Cells were lysed and equal proteins were electrophoresed on 10% SDS-PAGE gels and transferred onto PVDF membranes (Millipore). The membranes were blocked in 5% skimmed milk in TBST [50 mmol/L Tris-HCl (pH7.4),150 mmol/L NaCl and 0.1% Tween20] for 1 hr, then incubated with primary antibodies and secondary antibodies and detected using ECL system(Thermo). The dilutions of the primary antibodies were anti-RXRα(N197, Santa Cruz) in 1:1000, anti-PARP(H-250, Santa Cruz) in 1:3000, anti-p-AKT (D9E, Cell Signaling Technology) in 1:1000, anti-AKT1/2/3 (H-136, Santa Cruz) in 1:1000, anti-β-actin (Sigma) in 1:5000.
5.2.5. Transient transfection and reporter assay
HCT-116 colon cancer cells were transfected with pG5 luciferase reporter vector (50 ng/well) and pGAL-4-RXRα-LBD expression vector (50ng/well) for 24hr. Cells were incubated with varied concentrations of compounds for another 12 hr. Luciferase activities were measured using the Dual-Luciferase Assay System Kit (Promega).
5.2.6. RXRα siRNA and transfection
RXRα siRNA used in the experiments were obtained from Dharmacon Research, Inc. A 2.5-μl aliquot of 20 μmol of siRNA/well was transfected into cells grown in 12-well plates by using oligofectamine reagent (Invitrogen) according to the manufacturer’s recommendations. Two days after transfection, the cells were harvested for Western blotting [18].
5.3. Ligand Docking
Schrodinger’s (Portland, OR) (www.schrodinger.com) GLIDE[21], a grid-based docking program, was used for docking studies of the small molecule ligands to the protein. The crystal structure of RXRα LBD in complex with antagonist LG100754 (Protein Data Bank code 3A9E) was used. The GLIDE GScore was used as docking score to rank the docking results. Visual inspection was done to pick the docked pose from the ranked results. Schrödinger’s Maestro 6.5 was used to prepare Figure 5.
Highlights.
28 new Sulindac analogs as modulators of tRXRa-dependent AKT activation were synthesized.
Binding assay was used to test the 28 compounds for SAR study.
A new scaffold was found with improved biological properties.
Acknowledgments
This work was supported by Grants from the U.S. Army Medical Research and Material Command (W81XWH-11-1-0677), the National Institutes of Health (CA140980, GM089927), the 985 Project from Xiamen University, the National Natural Science Foundation of China (NSFC-91129302 and 20832005).
Abbreviations
- RXRα
retinoid X receptor alpha
- tRXRα
N-terminally-truncated retinoid X receptor alpha
- PI3K
phosphatidylinositol-3-OH kinase
- NR
nuclear receptor
- LBD
ligand-binding domain
- LBP
ligand-binding pocket
- NSAID
nonsteroidal antiinflammatory drug
- TNFα
Tumor necrosis factor-α
- LDA
lithium diisopropylamide
- TMS
tetramethylsilane
- LHMDS
bis(trimethylsilyl)amine lithium
- THF
tetrahydrofuran
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- p-TsOH
p-toluenesulfonic acid
- HOBt
N-Hydroxybenzotriazole
- EDCI
1-Ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride
- SAR
structure–activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Germain P, Chambon P, Eichele G, Evans RM, Lazar MA, Leid M, De Lera AR, Lotan R, Mangelsdorf DJ, Gronemeyer H. International Union of Pharmacology. LXIII. Retinoid X Receptors. Pharmacological Reviews. 2006;58:760–772. doi: 10.1124/pr.58.4.7. [DOI] [PubMed] [Google Scholar]
- 2.Szanto A, Narkar V, Shen Q, Uray IP, Davies PJA, Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11:S126–S143. doi: 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre P, Benomar Y, Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends in Endocrinology & Metabolism. 2010;21:676–683. doi: 10.1016/j.tem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Perez E, Bourguet W, Gronemeyer H, de Lera AR. Modulation of RXR function through ligand design. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2012;1821:57–69. doi: 10.1016/j.bbalip.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Dawson MI, Xia Z. The retinoid X receptors and their ligands. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2012;1821:21–56. doi: 10.1016/j.bbalip.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu K, Kim HT, Rodriquez JL, Hilsenbeck SG, Mohsin SK, Xu XC, Lamph WW, Kuhn JG, Green JE, Brown PH. Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiol Biomarkers Prev. 2002;11:467–474. [PubMed] [Google Scholar]
- 7.Bushue N, Wan YJY. Retinoid pathway and cancer therapeutics. Advanced Drug Delivery Reviews. 2010;62:1285–1298. doi: 10.1016/j.addr.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talpur R, Ward S, Apisarnthanarax N, Breuer-McHam J, Duvic M. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. Journal of the American Academy of Dermatology. 2002;47:672–684. doi: 10.1067/mjd.2002.124607. [DOI] [PubMed] [Google Scholar]
- 9.Dawson MI, Zhang XK. Discovery and design of retinoic acid receptor and retinoid X receptor class- and subtype-selective synthetic analogs of all-trans-retinoic acid and 9-cis-retinoic acid. Current Medicinal Chemistry. 2002;9:623–637. doi: 10.2174/0929867023370789. [DOI] [PubMed] [Google Scholar]
- 10.Esteva FJ, Glaspy J, Baidas S, Laufman L, Hutchins L, Dickler M, Tripathy D, Cohen R, DeMichele A, Yocum RC, Osborne CK, Hayes DF, Hortobagyi GN, Winer E, Demetri GD. Multicenter phase II study of oral bexarotene for patients with metastatic breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21:999–1006. doi: 10.1200/JCO.2003.05.068. [DOI] [PubMed] [Google Scholar]
- 11.Yen WC, Lamph WW. A selective retinoid X receptor agonist bexarotene (LGD1069, Targretin) prevents and overcomes multidrug resistance in advanced prostate cancer. The Prostate. 2006;66:305–316. doi: 10.1002/pros.20347. [DOI] [PubMed] [Google Scholar]
- 12.Ramlau R, Zatloukal P, Jassem J, Schwarzenberger P, Orlov SV, Gottfried M, Pereira JR, Temperley G, Negro-Vilar R, Rahal S, Zhang JK, Negro-Vilar A, Dziewanowska ZE. Randomized phase III trial comparing bexarotene (L1069-49)/cisplatin/vinorelbine with cisplatin/vinorelbine in chemotherapy-naive patients with advanced or metastatic non-small-cell lung cancer: SPIRIT I. J Clin Oncol. 2008;26:1886–1892. doi: 10.1200/JCO.2007.12.2614. [DOI] [PubMed] [Google Scholar]
- 13.Haanen C. Sulindac and its derivatives: a novel class of anticancer agents. Curr Opin Investig Drugs. 2001;2:677–683. [PubMed] [Google Scholar]
- 14.Yamamoto Y, Yin MJ, Lin KM, Gaynor RB. Sulindac inhibits activation of the NF-kappaB pathway. J Biol Chem. 1999;274:27307–27314. doi: 10.1074/jbc.274.38.27307. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 16.Davies NM, Watson MS. Clinical pharmacokinetics of sulindac. A dynamic old drug. Clin Pharmacokinet. 1997;32:437–459. doi: 10.2165/00003088-199732060-00002. [DOI] [PubMed] [Google Scholar]
- 17.Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006;98:736–747. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H, Liu W, Su Y, Wei Z, Liu J, Kolluri SK, Wu H, Cao Y, Chen J, Wu Y, Yan T, Cao X, Gao W, Molotkov A, Jiang F, Li WG, Lin B, Zhang HP, Yu J, Luo SP, Zeng JZ, Duester G, Huang PQ, Zhang XK. NSAID sulindac and its analog bind RXRalpha and inhibit RXRalpha-dependent AKT signaling. Cancer Cell. 2010;17:560–573. doi: 10.1016/j.ccr.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang GH, Jiang FQ, Duan YH, Zeng ZP, Chen F, Dai Y, Chen JB, Liu JX, Liu J, Zhou H, Chen HF, Zeng JZ, Su Y, Yao XS, Zhang XK. Targeting Truncated Retinoid X Receptor-alpha by CF31 Induces TNF-alpha-Dependent Apoptosis. Cancer research. 2012 doi: 10.1158/0008-5472.CAN-12-2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato Y, Ramalanjaona N, Huet T, Potier N, Osz J, Antony P, Peluso-Iltis C, Poussin-Courmontagne P, Ennifar E, Mely Y, Dejaegere A, Moras D, Rochel N. The “Phantom Effect” of the Rexinoid LG100754: structural and functional insights. PloS one. 2010;5:e15119. doi: 10.1371/journal.pone.0015119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. Journal of medicinal chemistry. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Zhou R, Li L, Chen J, Chen L, Li C, Ding H, Yu L, Hu L, Jiang H, Shen X. Danthron functions as a retinoic X receptor antagonist by stabilizing tetramers of the receptor. The Journal of biological chemistry. 2011;286:1868–1875. doi: 10.1074/jbc.M110.166215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Chen L, Chen J, Jiang H, Shen X. Structural basis for retinoic X receptor repression on the tetramer. The Journal of biological chemistry. 2011;286:24593–24598. doi: 10.1074/jbc.M111.245498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piazza GA, Rahm AL, Krutzsch M, Sperl G, Paranka NS, Gross PH, Brendel K, Burt RW, Alberts DS, Pamukcu R, et al. Antineoplastic Drugs Sulindac Sulfide and Sulfone Inhibit Cell Growth by Inducing Apoptosis. Cancer Res. 1995;55:3110–3116. [PubMed] [Google Scholar]
- 25.Verma RK, Singla R, Punniyakoti VT. A FACILE SYNTHESIS OF 2-BENZYLOXY/2-(4-ISOPROPYLBENZYLOXY)-2-METHYL-3-(4-SUBSTITUTED PHENYL)PROPANOIC ACID BASED INSULIN SNESITIZING AGENTS: RSR13-15 AND PKR13-15. Med Chem Res. 2004;13:660–676. [Google Scholar]
- 26.Sieckmann R. DE: 3720791, Patent. 1989 Jan 05;
- 27.Hagishita S, Yamada M, Shirahase K, Okada T, Murakami Y, Ito Y, Matsuura T, Wada M, Kato T, Ueno M, Chikazawa Y, Yamada K, Ono T, Teshirogi I, Ohtani M. Potent Inhibitors of Secretory Phospholipase A2: Synthesis and Inhibitory Activities of Indolizine and Indene Derivatives. Journal of Medicinal Chemistry. 1996;39:3636–3658. doi: 10.1021/jm960395q. [DOI] [PubMed] [Google Scholar]
- 28.GERHARD S, PAUL G, KLAUS B, PGA, RIFAT P. WO9747295, Patent. 1997 Dec 18;
- 29.Lu J, Dawson MI, Hu QY, Xia Z, Dambacher JD, Ye M, Zhang XK, Li E. The effect of antagonists on the conformational exchange of the retinoid X receptor alpha ligand-binding domain. Magnetic Resonance in Chemistry. 2009;47:1071–1080. doi: 10.1002/mrc.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu N, Liu J, Liu J, Zhang C, Jiang F, Wu H, Chen L, Zeng W, Cao X, Yan T, Wang G, Zhou H, Lin B, Yan X, Zhang XK, Zeng JZ. Antagonist effect of triptolide on AKT activation by truncated retinoid X receptor-alpha. PloS one. 2012;7:e35722. doi: 10.1371/journal.pone.0035722. [DOI] [PMC free article] [PubMed] [Google Scholar]