

Abstract
The current model of apoptosis holds that upstream signals lead to activation of downstream effector caspases. We generated mice deficient in the two effectors, caspase 3 and caspase 7, which died immediately after birth with defects in cardiac development. Fibroblasts lacking both enzymes were highly resistant to both mitochondrial and death receptor–mediated apoptosis, displayed preservation of mitochondrial membrane potential, and had defective nuclear translocation of apoptosis-inducing factor (AIF). Furthermore, the early apoptotic events of Bax translocation and cytochrome c release were also delayed. We conclude that caspases 3 and 7 are critical mediators of mitochondrial events of apoptosis.
Mitochondria play a central role in apoptosis. Mitochondrial outer membrane permeabilization (MOMP) leads to release of proapoptotic factors such as cytochrome c and AIF (1). Furthermore, loss of mitochondrial membrane potential (Δψm) is thought to contribute to cell death by disruption of normal mitochondrial function (2, 3). Interaction of members of the Bcl-2 family of proteins regulates MOMP, the key event of cytochrome c release into the cytoplasm (3, 4). What is less clear, however, is the precise role of caspase proteases in mitochondrial events of apoptosis. Although upstream caspases, such as caspase 2 and caspase 8, affect mitochondrial events in both death-receptor and mitochondrial pathways of apoptosis, either directly or through interaction with Bcl-2 family members, the role of presumed downstream “effector” caspases in this process is less clear (5, 6). Therefore, we studied the two highly related effectors, caspase 3 and caspase 7, to elucidate their functions in apoptosis.
We generated caspase 7−/− mice (fig. S1), which were born in ratios consistent with Mendelian inheritance. They had normal appearance, organ morphology, and lymphoid development. When caspase 7−/− mouse embryonic fibroblasts (MEFs) were treated with inducers of apoptosis, they exhibited a slight survival advantage as compared with wild-type MEFs. Apoptosis caused by a range of insults in other caspase 7−/− cells proceeded normally, however, including the death of activated T cells following stimulation of the T cell receptor, thymocyte apoptosis, Fas-mediated death of B cells, and Fas-mediated death of hepatocytes (fig. S2).
Caspase 3, which is structurally similar to caspase 7, might compensate for the lack of caspase 7, which would lead to this relatively mild antiapoptotic phenotype (7, 8). Thus, we bred caspase 7−/− mice to caspase 3−/− mice previously described by our laboratory (9). The embryonic stem cells containing the mutation were from the 129/SvJ genetic background. Mice derived from these embryonic stem cells were backcrossed six generations onto the C57BL/6 background. We obtained no live caspase 3−/−/caspase 7−/− double-knockout (DKO) mice when progeny were genotyped at an age of 10 to 14 days. DKO mice were present at normal Mendelian numbers through embryonic day 20 (E20), but died rapidly after birth. A small percentage (~10%) of both caspase 3−/−/caspase 7+/− and DKO embryos displayed exencephaly, likely due to the absence of caspase 3 in combination with residual genes from the 129/SvJ background (10). The majority of E20 DKO embryos had a grossly normal appearance. This suggests that neither caspase 3 nor caspase 7 are important for brain development on the C57BL/6 genetic background.
Histologic examination of DKO hearts revealed dilation of the atria (Fig. 1, A and B) and disorganization and noncompaction of the ventricular musculature (Fig. 1, C through F). This noncompaction is similar to that of mice deficient in the death receptor–signaling molecules caspase 8, FADD, and c-FLIP (caspase 8–related protein) (11-13), although all of these mice exhibit additional developmental abnormalities and die in mid-gestation. Thus, caspases 3 and 7 together are important for proper cardiac development, and noncompaction may occur because they act downstream of death receptor signaling. Other aspects of death receptor–mediated development, however, leading to earlier lethality in caspase 8, FADD, and c-FLIP knockout mice, likely proceed through alternative pathways.
Fig. 1.

(A to F) Defective cardiac development in DKO embryos. Hematoxylin-and-eosin staining of transverse sections of caspase 3+/−/caspase 7+/− and DKO E20 embryo hearts. Higher magnification of the right ventricular free wall (E and F). Scale bars: (A and B), 500 μm; (C and D), 200 μm; and (E and F), 100 μm. Abbreviations: A, aorta; PT, pulmonary trunk; RA, right atrium; LA, left atrium; RSVC, right superior vena cava; LSVC, left superior vena cava; LV, left ventricle; RV, right ventricle; S, septum; TV, tricuspid valve; RVFW, right ventricular free wall.
To examine the combined functions of caspases 3 and 7 in apoptosis, we tested the sensitivity of MEFs to two inducers of mitochondrially mediated apoptosis—ultraviolet (UV) irradiation and staurosporine, and two activators of the death receptor pathway—Fas ligand (FasL) and tumor necrosis factor–α (TNFα). DAPI (4′,6-diamidinole-2-phenolindole) staining of the nuclei of UV irradiated cells revealed typical morphologic characteristics of cell death in caspase 3+/−/caspase 7+/− (Fig. 2A) and caspase 3+/−/caspase 7−/− (Fig. 2B) cells. Caspase 3−/−/caspase 7+/− MEFs displayed distorted chromatin condensation (Fig. 2C), and nuclear fragmentation was absent (14). In contrast, UV-irradiated DKO MEFs maintained a normal nuclear morphology (Fig. 2D), even 24 hours after treatment (Fig. 2, E and F). At 24 hours after irradiation, caspase 3+/−/caspase 7+/− (Fig. 2G) and caspase 3+/−/caspase 7−/− (Fig. 2H) MEFs underwent complete loss of cellular morphology, and many cells detached from the plate. Some caspase 3−/−/caspase 7+/− MEFs partially retained gross morphology (Fig. 2I), but their cytoplasm was contracted. DKO MEFs, on the other hand, appeared completely normal (Fig. 2J), with flattened, attached cell bodies.
Fig. 2.

Apoptosis resistance in caspase 3– and caspase 7–deficient MEFs. (A) Caspase 3+/−/caspase 7+/−, (B) caspase 3+/−/caspase 7−/−, (C) caspase 3−/−/caspase 7+/− and (D) DKO MEFs were treated with UV irradiation at 30 J/m2, fixed after 6 hours, then stained with DAPI to visualize chromatin condensation (arrows) and nuclear fragmentation (arrowhead). Partial chromatin condensation was seen in caspase 3−/−/caspase 7+/− MEFs (asterisks). (E) Caspase 3+/−/caspase 7+/− and (F) DKO MEFs, 24 hours after UV irradiation. Light microscopy at 24 hours after UV in (G) caspase 3+/−/caspase 7+/−, (H) caspase 3+/−/caspase 7−/−, (I) caspase 3−/−/caspase 7+/− (some with partial preservation of morphology, arrows), and (J) DKO MEFs. (K) Improved survival in DKO MEFs. MEFs were treated as indicated, FasL and TNF-α each supplemented with cyclohexamide (10 μg/ml). After 24 hours, survival was determined using a live/dead cytotoxicity/viability assay. Data are means ± SD from a single experiment in triplicate, representative of at least three separate experiments. P < 0.01 for pairwise comparison of differences between DKO and each of the other six genotypes. (L) DNA cleavage is associated with caspase 3 alone. Cells were treated with UV and harvested after 3 hours, then nucleosome ELISA was done to measure DNA fragmentation. Data are means ± SD from a single experiment in duplicate, representative of at least three separate experiments. (M) PARP cleavage is a function of caspase 3. Cells were treated with UV (20 J/m2) and harvested at indicated time points, then Western blots were performed on whole-cell lysates.
Consistent with this, DKO MEFs displayed increased viability after stimulation of the death-receptor pathway (Fig. 2K) as compared with that of other genotypes. Caspase 3−/−/caspase 7+/− and caspase 3+/−/caspase 7−/− cells showed an intermediate effect. Interestingly, caspase 7−/− MEFs showed a consistently greater viability than caspase 3−/− MEFs. In contrast, when we measured DNA fragmentation by nucleosome enzyme-linked immunosorbent assay (ELISA) (Fig. 2L) caspase 3−/− MEFs showed complete absence of DNA fragmentation, whereas both caspase 7−/− and caspase 3+/−/caspase 7−/− cells showed DNA fragmentation comparable to that of the wild type. Furthermore, cleavage of the caspase substrate poly(adenosine diphosphate–ribose) polymerase was entirely dependent on caspase 3 and not caspase 7 (Fig. 2M). These results show that caspases 3 and 7 have some overlapping, but also some distinct, roles in apoptosis. Caspase 3 controls DNA fragmentation and morphologic changes of apoptosis, whereas caspase 7 plays little role in these processes. In contrast, caspase 7 appears to be more important to the loss of cellular viability, although the combined role of both caspases is crucial in this area.
We transferred fetal liver cells into RAG-deficient mice and saw a normal distribution of CD4+, CD8+, and double-positive thymocytes in the DKO (fig. S3). We subjected these thymocytes to apoptotic stimuli. Determination of the fraction of cells with hypodiploid (subG0) DNA content (Fig. 3A), reflecting the nuclear changes seen with apoptosis, showed a complete resistance in DKO and a slightly smaller degree of resistance in caspase 3−/−/caspase 7+/− thymocytes. When analyzed for viability (Fig. 3B), however, only DKO thymocytes were resistant to apoptosis mediated by the mitochondrial pathway, although they were susceptible to death receptor–mediated apoptosis. These data imply that caspases 3 and 7 are not necessary for positive and negative selection of thymocytes. They further support a primary role for caspase 3 in DNA fragmentation, and a combined role of caspases 3 and 7 in viability. Because DKO thymocytes die normally from death-receptor stimuli, some cells, such as thymocytes, may have alternative pathways to loss of viability that bypass caspases 3 and 7.
Fig. 3.
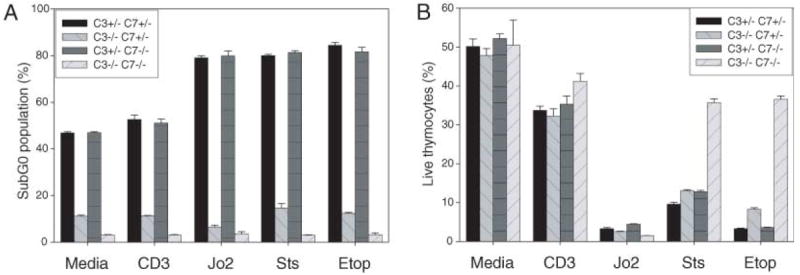
Thymocyte apoptosis. (A) Reduced subG0 population in caspase 3−/−/caspase 7+/− and DKO thymocytes. Thymocytes from RAG−/− chimeras were plated in media alone or treated with plate-bound antibodies against CD3 and CD28 (20 μg/ml each), the Fas-specific antibody Jo2 (1 μg/ml) + cyclohexamide (10 μg/ml), staurosporine (0.1 μM), or etoposide (25 μM). Cells were fixed at 24 hours and stained with propidium iodide. SubG0 population was identified by using flow cytometry in the FL-2 channel and was expressed as a percentage of total cells. Data are means ± SD from a single experiment in triplicate, representative of at least three separate experiments. For each treatment, P < 0.001 for the difference between DKO and C3KO. (B) Reduced mitochondrially mediated cell death in DKO thymocytes. Thymocytes with above treatments were analyzed at 24 hours using a live-dead cytotoxicity-viability assay, and viable cells were expressed as a percentage of total cells analyzed. Data are means ± SD from a single experiment in triplicate, representative of at least three separate experiments.
The central role of mitochondria in cell death is highlighted by the importance of Δψm. This gradient is critical for normal mitochondrial function, and its loss is associated with apoptosis (3). Inhibition of caspase activity protects dying cells from loss of Δψm, at least in part through cleavage of respiratory chain proteins, but the identity of the caspase(s) involved has been unclear (15-17). We examined Δψm after treatment of MEFs with UV irradiation (Fig. 4A). In DKO MEFs, Δψm was preserved, whereas the other genotypes showed a decrease in the magnitude of Δψm. These results identify caspases 3 and 7 as key mediators of the loss of Δψm that occurs during apoptosis.
Fig. 4.

(A) Preservation of Δψm in DKO MEFs. MEFs were treated with UV irradiation at 30 J/m2 and harvested at indicated time points. Δψm was assessed by incubating with JC-1 reagent and by fluorescence-activated cell sorting (FACS). JC-1 enters cells and gives a green fluorescent signal (FL-1, x axis), and is processed to give an additional red fluorescent signal (FL-2, y axis) only in mitochondria with preserved Δψm. Cells with preservation of Δψm are both FL-1 high, FL-2 high (top right in each plot); cells with loss of Δψm are FL-1 high, FL-2 low (bottom right in each plot). FACS data plots are shown, with percentage of total events indicated. Data are from a single experiment, representative of at least three independent experiments. (B and C) Control of release of mitochondrial apoptotic factors by caspases 3 and 7. (B) Delayed Bax translocation and cytochrome c release in DKO MEFs. MEFs treated with UV irradiation at 30 J/m2, mitochondrial (top) and cytoplasmic (bottom) fractions were separated at indicated time points. Western blots were probed with indicated antibodies. (C) Absent AIF nuclear translocation in DKO MEFs. MEFs were treated with UV irradiation at 30 J/m2 and fixed after 6 hours. Cells with AIF nuclear translocation were counted in five random fields and expressed as a percentage of total cells (minimum 120 total cells counted per genotype). Data are means ± SD from a single experiment, representative of two separate experiments.
Mitochondria in cells undergoing stress-induced apoptosis may act as amplifiers of caspase activity, much as they do in death receptor–mediated apoptosis (18, 19). We investigated whether caspases may act as amplifiers of mitochondrial events. In DKO cells, early mitochondrial events—Bax translocation to the mitochondria and cytochrome c release into the cytoplasm—were both delayed (Fig. 4B). Similar findings were also made at early time points for caspase 9−/− MEFs (fig. S4). Notably, caspase 9 processing was normal in DKO MEFs, whereas caspases 2 and 8 were not processed in either wild-type or DKO MEFs (fig. S4). This suggests a pathway from caspase 9 through caspases 3 and 7 that promotes cytochrome c release at early time points. These events did occur ultimately, as living, healthy-appearing DKO MEFs displayed Bax translocation and cytochrome c release at later time points (fig. S5).
Translocation of the mitochondrial flavo-protein AIF to the nucleus is associated with chromatin condensation and DNA cleavage during apoptosis (20). Data using chemical inhibitors of caspase activity or cells deficient in single caspases, have suggested that AIF release may be either caspase-dependent or caspase-independent (21). Given the profound absence of chromatin condensation in DKO cells, we examined AIF release with immunocytochemistry (fig. S6). DKO MEFs completely lacked AIF translocation (Fig. 4C), whereas caspase 3−/−/caspase 7+/− MEFs had a partial deficiency. These data are consistent with a role for AIF in chromatin condensation and DNA cleavage and indicate that, under these conditions, AIF release is fully dependent on the combined action of caspases 3 and 7.
The data presented here show that caspases 3 and 7 are crucial for apoptosis and contribute to some mitochondrial events once thought to lie upstream of effector caspases. They control the loss of Δψm and AIF release. They may also serve to amplify the initial death signal by helping to promote further cytochrome c release. Nevertheless, as Bax translocation and cytochrome c release occur eventually in DKO cells, there are likely multiple routes to regulate mitochondrial function in apoptosis.
Supplementary Material
Acknowledgments
The authors wish to thank Y. Lazebnik (Cold Spring Harbor) for caspase 7 antibody and Biogen Idec for their contribution of interleukin-2; Yale Liver Center Core Facilities for hepatocyte preparation; A. Ferrandino and A. Mohapatra for technical assistance; and F. Manzo for assistance with manuscript preparation. This work was supported in part by NIH grants K12 HD00850, 5 K12 HD01401, 1 K08 HD044580 (S.A.L.); Liver Center Grant NIDDK P30-34989 (R.A.F.); R.A.F. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/311/5762/847/DC1
Materials and Methods
Figs. S1 to S6
References and Notes
References and Notes
- 1.van Loo G, et al. Cell Death Differ. 2002;9:1031. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- 2.Halestrap AP, McStay GP, Clarke SJ. Biochimie. 2002;84:153. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 3.Green DR, Kroemer G. Science. 2004;305:626. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 4.Wei MC, et al. Science. 2001;292:727. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. J Biol Chem. 2002;277:13430. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Zhu H, Xu CJ, Yuan J. Cell. 1998;94:491. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 7.Lippke JA, Gu Y, Sarnecki C, Caron PR, Su MS. J Biol Chem. 1996;271:1825. doi: 10.1074/jbc.271.4.1825. [DOI] [PubMed] [Google Scholar]
- 8.Zheng TS, et al. Nat Med. 2000;6:1241. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]
- 9.Kuida K, et al. Nature. 1996;384:368. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 10.Leonard JR, Klocke BJ, D’Sa C, Flavell RA, Roth KA. J Neuropathol Exp Neurol. 2002;61:673. doi: 10.1093/jnen/61.8.673. [DOI] [PubMed] [Google Scholar]
- 11.Varfolomeev EE, et al. Immunity. 1998;9:267. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 12.Yeh WC, et al. Science. 1998;279:1954. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 13.Yeh WC, et al. Immunity. 2000;12:633. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 14.Woo M, et al. Genes Dev. 1998;12:806. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bossy-Wetzel E, Newmeyer DD, Green DR. EMBO J. 1998;17:37. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricci JE, Gottlieb RA, Green DR. J Cell Biol. 2003;160:65. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricci JE, et al. Cell. 2004;117:773. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Hausmann G, et al. J Cell Biol. 2000;149:623. doi: 10.1083/jcb.149.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marsden VS, et al. Nature. 2002;419:634. doi: 10.1038/nature01101. [DOI] [PubMed] [Google Scholar]
- 20.Susin SA, et al. Nature. 1999;397:441. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 21.Cande C, Vahsen N, Garrido C, Kroemer G. Cell Death Differ. 2004;11:591. doi: 10.1038/sj.cdd.4401400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.