

Abstract
The first Negishi nickel-catalyzed stereospecific cross-coupling reaction of secondary benzylic esters is reported. A series of traceless directing groups are evaluated for ability to promote cross-coupling with dimethylzinc. Esters with a chelating thioether derived from commercially-available 2-(methylthio)acetic acid are most effective. The products are formed in high yield and with excellent stereospecificity. A variety of functional groups are tolerated in the reaction including alkenes, alkynes, esters, amines, imides, and O-, S-, and N-heterocycles. The utility of this transformation is highlighted in the enantioselective synthesis of a retinoic acid receptor (RAR) agonist and a fatty acid amide hydrolase (FAAH) inhibitor.
INTRODUCTION
In medicinal chemistry, incorporation of a methyl group at a key position is a common strategy for improving drug potency.1 Inserting a “magic” methyl group can improve efficacy by multiple mechanisms: increased binding affinity, improved pharmacokinetics, and greater specificity within a family of targets. For instance, methyl incorporation can provide additional surfaces for hydrophobic interactions, resulting in enhanced binding to the target protein. This effect increases the potency of Lipitor, which features two benzylic methyl groups.2 In addition, replacing a hydrogen with a methyl group can force a conformational change and lower the enthalpic cost of binding.3,4 When methyl placement affords both increased hydrophobic interactions and a more favorable conformation, the potency has been observed to increase by 200-fold.4 Additionally, methyl substituents can prevent formation of toxic by products by providing an alternative soft site for predictable metabolic oxidation.1,5 When oxidation of a benzylic methylene is problematic, incorporation of a tertiary benzylic site can improve pharmacokinetics by slowing degradation and drug clearance.1,6 Due to the above reasons, a tertiary benzylic stereocenter bearing a methyl group is a common motif in medicinal agents. For example, the compounds shown in Figure 1 possess biological activity against a variety of targets, with potential applications ranging from treatment of autoimmune disorders and inflammation to cancer and obesity.7 Stereoselective methods for the introduction of methyl groups8 are necessary because stereoisomers typically exhibit different biological activity. For instance, the enantiomers of MCHR1 antagonist 1 and FAAH inhibitor 3 show at least an order of magnitude difference in activity (Figure 1).7a,d
Figure 1.

Bioactive small molecules containing a tertiary benzylic stereocenter.
Previously, we developed a stereospecific nickelcatalyzed Kumada cross-coupling of benzylic ethers for the formation of 3° benzylic stereocenters.9 Unlike enantioselective methods that employ a chiral catalyst with racemic or achiral starting materials, stereospecific methods use an achiral catalyst to translate the stereochemistry of the starting material to the product. For example, during the course of our Kumada coupling, a readily prepared enantioenriched secondary ether10 is transformed to a tertiary benzylic stereocenter with overall inversion of configuration. While this transformation is highly stereoselective and has been utilized in synthesis of medicinal agents with activity against breast cancer and insomnia, the use of Grignard reagents as the coupling partner precludes synthesis of highly functionalized compounds such as 2 and 3 without recourse to lengthy protecting group strategies.
Recently, we have turned our attention to milder coupling partners in an effort to expand the scope of our reaction.11,12 We chose to focus on organozinc reagents because Negishi reactions are highly functional group tolerant.13 Stereoselective sp2–sp3 cross-coupling has been shown for α-chiral organozinc reagents, which undergo stereospecific Negishi reactions with aryl and vinyl halides.14 Additionally, the Fu group has developed enantioselective alkyl-alkyl cross-coupling reactions using chiral nickel catalysts.15 However, stereospecific sp3–sp3 cross-coupling of alkylzinc reagents with enantioenriched electrophiles has not yet been reported.
While Kumada reactions benefit from the high nucleophilicity of Grignard reagents, and proceed with simple benzylic ethers, we postulated that cross-coupling of alcohol derivatives with the less nucleophilic organozinc reagents would require a more reactive electrophile. Directing groups have been used in organic synthesis to promote transformations of otherwise unreactive substrates.16,17 While incorporation of the directing group into the body of the substrate is a common strategy, it can limit the scope of the transformation. A traceless approach places the directing functionality on the leaving group. Over the course of the reaction the directing group is cleaved, affording products that bear no trace of the directing functionality. Liebeskind demonstrated that traceless directing groups promote the cross-coupling of thioethers by accelerating the transmetallation step (Scheme 1a).18 The pendant carboxylic acid complexes zinc and promotes dissociation of the leaving group, providing an open coordination site on the nickel center for transmetallation. Inspired by this work, we designed traceless directing groups to promote the oxidative addition of sluggish electrophiles for Kumada couplings (Scheme 1b).9b,19,20 Magnesium coordination to the 2-methoxyethyl ether moiety is proposed to weaken the benzylic C-O bond, facilitating oxidative addition. We postulated that a similar strategy could accelerate cross-coupling reactions with dimethylzinc. A leaving group bearing a pendant ligand could serve two functions (Scheme 1c). Coordination to a zinc reagent could activate the substrate for oxidative addition and facilitate the subsequent transmetallation step. We anticipated that tuning the properties of the X and L groups would provide a synergistic enhancement of reactivity.
Scheme 1.

Traceless directing group strategy for promoting challenging organometallic transformations.
RESULTS AND DISCUSSION
Identification of traceless directing group for Negishi coupling
To test our hypothesis we examined a range of activating groups to promote the cross-coupling of benzylic electrophiles with dimethylzinc (Figure 2). As anticipated, simple benzylic ether 4 was unreactive. Next, we employed a thioether with the thought that formation of the zinc-sulfur bond would provide a strong thermodynamic driving force for the reaction.21 While substrate 5 was more reactive, elimination to provide styrene 23 was the major pathway. We reasoned that if thioether 5 underwent oxidative addition, sluggish transmetallation could have resulted in β-hydride elimination to give alkene 23 as the major product. To promote transmetallation over β-hydride elimination, we examined ethers and thioethers bearing a second ligand (Group 2). While acetal 6 and 2-methoxyethyl ether 8 remained unreactive, hydroxyethyl thioether 7 afforded the desired cross-coupled product 22 as the major species, albeit with low enantiospecificity (es).22 To increase the yield and enantiospecificity of the transformation, we increased the cooridinating ability of the directing group by switching to a pendant pyridyl ligand. Pyridyl ether 10 was the first of the oxygen series to afford an appreciable yield of desired product with good es. In contrast, pyridyl thioether 11, afforded lower yields than 7, with significant erosion of enantiomeric excess. Carboxylic acids 12 and 13 afforded the desired product in moderate yield, but with less than satisfactory es. We reasoned that in order to achieve higher reactivity and high es we could invert the carboxylic acid to an isomeric ester. These compounds would be less likely to undergo radical racemization, which is more likely for thioethers than ethers, improving the es. In addition, maintaining the thiol functionality would allow for strong coordination of zinc to the leaving group. Indeed, a series of isomeric ester leaving groups provided the desired product in both synthetically useful yields and high es (Group 3). Although the ester leaving groups addressed the issue of chirality transfer, their synthesis necessitated employing protecting groups to mask the free thiol, which added a step to the synthetic sequence (see SI for details). Additionally, free thiols are not optimal substrates because they are susceptible to oxidative decomposition. We postulated that utilizing 2-(methylthio)ester 18 instead would simplify substrate synthesis and prevent oxidative decomposition of the starting material. This directing group is particularly convenient since (methylthio)acetic acid is commercially available and can be easily appended onto the benzylic alcohol through a DCC coupling.23 Functionalized with the thioether directing group, (R)-18 cross-coupled to afford (S)-22 in 81% and excellent es with overall inversion of configuration (Figure 2 and Table 1, entry 1).24
Figure 2.

Evaluation of leaving groups for cross-coupling with dimethylzinc.a
aNickel sources used in this reaction included NiCl2•DME, Ni(acac)2, or Ni(COD)2. Ligands included DPEphos, rac-BINAP, or Xantphos. Solvents included PhMe, THF, or Et2O. Reaction temperatures ranged between rt and 50 °C. The optimal results for each directing group are shown. For a complete list of reaction conditions for each experiment, please refer to the SI. Yields were determined either by 1H NMR based on comparison with PhTMS as internal standard or by GC analysis with dodecane as internal standard. Enantiospecificity (es) = (eeproduct/eesubstrate) × 100%; determined by chiral SFC chromatography. bDPEphos as ligand. cXantphos as ligand.
Table 1.
Scope of cross-coupling in the presence of a range of functional groups
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Product | Yield (%)a |
SM ee (%)b |
ee (%)b |
es (%)c |
|
| 1d,e | 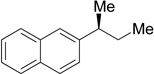 |
(S)-22 | 81 | 98 | 98 | >99 |
| 2f,h | 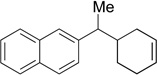 |
24 | 71 | N/A | N/A | N/A |
| 3f | 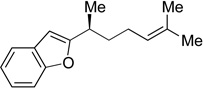 |
25 | 86 | 93 | 93 | >99 |
| 4g | 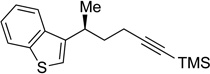 |
26 | 81 | 97 | 96 | 99 |
| 5f | 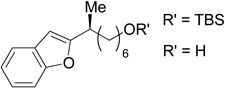 |
27 | 75 | 90 | 88 | 98 |
| 6f | 28 | 77 | 92 | 90 | 98 | |
| 7d,f | 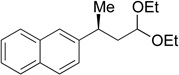 |
(S)-29 | 81 | 89 | 89 | >99 |
| 8d,e | 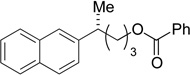 |
(R)-30 | 81 | 97 | 97 | >99 |
| 9f,h | 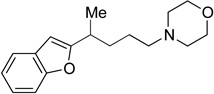 |
31 | 68 | N/A | N/A | N/A |
| 10g,i | 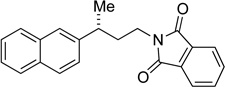 |
32 | 84 | 94 | 94 | >99 |
Isolated yield after column chromatography.
Determined by supercritical fluid chromatography.
Enantiospecificity (es) = (eeproduct/eesubstrate) × 100%.
Absolute configuration determined by comparison of optical rotations with literature values. Please see SI for details.
Approximately 20% styrene byproduct arising form β-hydride elimination was observed in these reactions.
10–15% styrene byproduct arising form β-hydride elimination was observed in these reactions.
Less than 10% styrene byproduct arising form β-hydride elimination was observed in these reactions.
Prepared from racemic substrate.
Reaction ran with the following amounts of reagents: NiCl2•DME (13 mol %), DPEphos (26 mol %), ZnMe2 (3.8 equiv).
Simple esters were also evaluated to determine the importance of a pendant ligand in these transformations (Figure 2, Group 4). Both acetyl and pivaloyl esters provided either high yield or high es depending on the reaction conditions (see SI for details). For example, acetate 19 could be cross-coupled to provide the desired product in 84% yield and 87% es or 45% yield with no loss of chirality. The same trend was observed for pivaloyl ester 21. While both are viable alternatives to 18, the presence of the thioether ligand is necessary for obtaining optimal yields of highly enantioenriched product.
Scope of reaction
With the optimal leaving group in hand, we prepared a range of enantioenriched substrates for cross-coupling by the general strategies outlined in Scheme 2. Synthesis of the chiral alcohol intermediates was accomplished by CBS reduction of the corresponding ketone25 or enantioselective arylation of the requisite aldehyde.26,27 Absolute configuration of the intermediate alcohols was assigned using the accepted models for selectivity for each reaction.28 The absolute configuration was then confirmed by the Competing Enantioselective Conversion (CEC) Method.29 DCC coupling appended the necessary leaving group with no loss of ee, providing the starting materials for the transformation.23
Scheme 2.

Synthesis of enantioenriched benzylic alcohol derivatives for Negishi cross-coupling.
A variety of functional groups are well tolerated under our optimized reaction conditions (Tables 1 and 2). For example, products containing internal alkenes, 24 and 25, are formed in high yield and in the case of 25, with high ee (Table 1, entries 2 and 3). Additionally, the increased steric bulk of 24 does not significantly slow down the reaction. The presence of a TMS-protected alkyne is also compatible with the reaction conditions and 26 is formed in 81% yield and 99% es (entry 4). TMS-alkynes are easily deprotected to the free terminal alkyne, which provides a convenient functional handle for further elaboration. Oxygenation is also well tolerated under reaction conditions. Substrates containing a silyl ether or a free alcohol form 27 and 28 in good yield and with high es (entries 5 and 6). Furthermore, the use of zinc reagents allows for cross-coupling of substrates containing sensitive functionality such as acetals (entry 7) as well as electrophilic fragments such as esters (entry 8). We did not observe decomposition of the acetal or addition to the ester under our reaction conditions. With these promising results we moved to nitrogenated substrate classes.
Table 2.
Substrate scope of diarylethanes
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Product | Yield (%)a |
SM ee (%)b |
ee (%)b |
es (%)c |
|
| 1 |
|
35 | 87 | 99 | 98 | 99 |
| 2d | (R)-36 | 89 | 96 | 93 | 97 | |
| 3 | 37 | 78 | 97 | 91 | 94 | |
| 4 | 38 | 93 | 95 | 95 | 99 | |
| 5 | 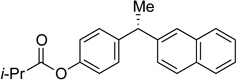 |
39 | 76 | 98 | 97 | 99 |
| 6 | 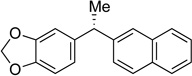 |
40 | 98 | 95 | 90 | 95 |
Isolated yield after column chromatography.
Determined by supercritical fluid chromatography.
Enantiospecificity (es) = (eeproduct/eesubstrate) × 100%.
Absolute configuration determined by comparison of optical rotations with literature values. Please see SI for details.
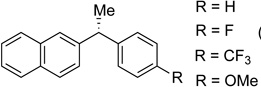
N-Heterocycles, amines, and imides are common functional groups in biologically active molecules (Figure 1, compounds 1 and 3). Since nitrogen-based ligands are often employed in nickel-catalyzed cross-coupling reactions, we anticipated that this class of substrates could be problematic. Initially, we synthesized a morpholine-containing substrate. Morpholine is a common motif in many pharmaceuticals, including the antibiotic linezolid, anticancer agent gefitinib, and analgesic dextromoramide.30 We were pleased to see that the morpholino ring was well tolerated in our cross-coupling and 31 was formed in 68% yield. Amides are also well tolerated in the reaction; 32 was formed in 84% yield with excellent es (entry 10). Phthalimides are interesting because they are readily deprotected to reveal primary amines. Encouraged by these results, we next designed an indole substrate. This class of substrates is particularly challenging since the indole moiety stabilizes carbocation intermediates, which, if formed, would afford racemic product and increased levels of byproducts resulting from elimination (Scheme 3a). In prior studies, we obsereved that cross-coupling of indole substrates under our original Kumada coupling conditions afforded only racemic product.31 We found, however, that under our Negishi conditions, when dimethylzinc is used, 33 couples to form 34 in 91% yield and with excellent es (Scheme 3b).
Scheme 3.

Cross-coupling of indole electrophiles.
Stereospecific cross-coupling of diaryl electrophiles is challenging because this substrate class is prone to racemization through pathways involving carbocation intermediates. As predicted, erosion of enantiospecificity was observed at ambient temperatures; however, upon cooling to 0 °C, excellent transfer of chirality was observed (Table 2, entry 1). Both electron-poor ((R)-36 and 37) and electron-rich (38 and 40) products were formed in good yield and es (entries 2, 3, 4, and 6 respectively). To probe the functional group compatibility of the reaction, we evaluated a substrate that included an isobutyric acid ester (entry 5). Esters are common masking groups used in prodrugs as they are readily hydrolyzed in vivo by non-specific esterases to reveal the active metabolite bearing a hydroxyl group.32 For example, the antimuscarinic 1,1-diarylalkane fesoterodine contains an aryl isobutyric acid ester.33 Our reaction conditions tolerate the isobutyric acid ester moiety nicely: product 39 was formed selectively in 76% yield and 99% es, with no competitive cross-coupling of the aryl ester.
Cross-coupling with diethylzinc
The cross-coupling reaction can be utilized with longer-chain alkylzinc reagents such as diethylzinc. Reactions employing such reagents are more complex as additional competitive reaction pathways are possible: in addition to undesired β-hydride elimination to afford byproduct 23, hydrogenolysis to provide 42 is also possible. Indeed, in initial studies 2-(methylthio)ester 18 gave only a modest yield of the desired ethylated product 41 and significant amounts of both byproducts 23 and 42 (Table 3, entry 1). This result is in direct contrast to cross-coupling with dimethylzinc, where the thiomethyl ether was found to be the ideal traceless directing group (Figure 2). To suppress these undesired pathways, we once again turned to tuning the directing group. During our earlier investigation of leaving groups in reactions with dimethylzinc, we identified thiols 17 and 15 as promising leads (Figure 2). When thiol 17 was coupled with with diethylzinc, the yield of 41 improved and formation of both 23 and 42 decreased; however, we observed formation of free alcohol 43 (Table 3, entry 2). We hypothesized that increased steric bulk at the α-position would slow addition to the ester; directing group 15 further improved the yield of desired product to 55% (entry 3).
Table 3.
Cross-coupling of diethylzinc
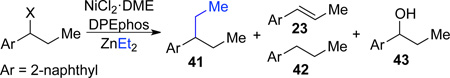 | ||||||
|---|---|---|---|---|---|---|
| Entry | Leaving Group | (X) | Yieldc 41 (%) |
Yieldc 23 (%) |
Yieldc 42 (%) |
Yieldc 43 (%) |
| 1a | 18 | 38 | 29 | 18 | 0 | |
| 2b | 17 | 49 | 7 | 10 | 27 | |
| 3b | 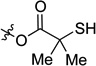 |
15 | 55 | 14 | 6 | 18 |
Conditions used: NiCl2•DME (10 mol %), DPEphos (20 mol %), ZnEt2 (3.0 equiv), PhMe, rt, 24 h.
THF used instead of PhMe.
Determined by 1H NMR based on comparison with PhTMS as internal standard.
To determine the stereospecificity of the cross-coupling reaction with diethylzinc, substrate 44, equipped with the thiol directing group, was subjected to cross-coupling conditions. Despite the more challenging nature of this transformation, product 45 was formed in excellent es (eq 1).
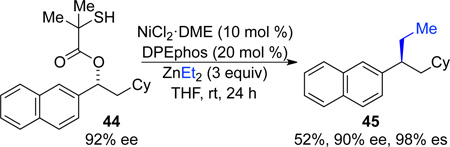 |
(1) |
Synthesis of biologically active arylethanes: retinoic acid receptor (RAR) agonist and fatty acid amide hydrolase (FAAH) inhibitor
The retinoic acid receptors (RAR) are implicated in several disease states. RARβ is targeted in treatment of certain cancers;34 modulating RARγ activity could enable treatment of skin diseases such as psoriasis,35 melanoma,36 and acne.37 Compounds containing the diaryl moiety are isoform selective ligands for RARβ and γ. 1,1-Diarylethane 2 and BMS 184394, which is the saponified ester 50, are RARγ agonists.38 RARγ is known to be sensitive to stereochemistry at the benzylic position. The (S)-enantiomer of BMS 184394 is 10-fold more potent than the (R)-enantiomer.7c While racemic 1,1-diarylethane 2 has comparable activity to (S)-BMS184394, activities of the two enantiomers of 2 have not been reported. We envisioned using our cross-coupling strategy as a key step in the synthesis of enantioenriched acid 2. We began our synthesis with bromide 46, which was converted to boronic acid 47 from the corresponding Grignard reagent by quenching with trimethoxyborane followed by hydrolysis.39 Enantioselective arylation of aldehyde 48 according to the Bolm strategy furnished the desired enantioenriched alcohol (R)-50 in 94% yield and 94% ee.27b The traceless directing group was then installed by DCC coupling23 and the substrate was subjected to standard cross-coupling conditions. Diarylethane 53 was obtained in 92% yield with excellent transfer of stereochemical information (97% es). We confirmed that the cross-coupling reaction proceeded with inversion at the benzylic center by preparation of a crystalline derivative that was subjected to X-ray crystallographic analysis (see Supporting Information for details).40 Saponification with 1 N NaOH provided the desired retinoic acid receptor ligand (S)-2 in high yield.7c
We next applied our methodology to the synthesis of fatty acid amide hydrolase (FAAH) inhibitor 3. FAAH is a membrane bound serine hydrolase, which has recently gained interest for pain treatment as an alternative target to cannabinoid receptor 1 (CB1). Inactivation of FAAH produces analgesic and anti-inflammatory effects in rodents without the weight gain that commonly accompanies CB1 agonists.7d The first reported synthesis of 3 featured a sodium borohydride reduction followed by chiral chromatography to separate the enantiomers. While the authors demonstrated that the two enantiomers exhibited a 20-fold difference in activity, they did not report the absolute configuration of the more active stereoisomer.7d Since our cross-coupling proceeds with net inversion, the configuration of the product can be readily assigned when the configuration of the enantioenriched alcohol is known.29,41 We began our synthesis with addition of aryl lithiate 54 to 1-Boc-piperidine-4-carboxaldehyde to afford racemic 56, followed by oxidation to ketone 55.42 Subsequent CBS reduction afforded the requisite enantioenriched alcohol intermediate in 93% ee.25b,c We assigned the absolute configuration of alcohol 56 as R based on the accepted model for selectivity in CBS reductions,28a which was then confirmed by the Competing Enantioselective Conversion (CEC) method.29 The directing group was installed by a DCC coupling without loss of ee.23 Cross-coupling of 57 under our optimized reaction conditions afforded the methyl-bearing benzylic stereocenter in 87% yield with 99% es.
Subsequent elaboration of key intermediate 58 to FAAH inhibitor 3 was accomplished in four steps. To introduce the requisite methyl substituent on the benzothiophene ring, we chose to employ a second cross-coupling reaction. Bromination at the 3-position of the benzothiophene provided 59,43 which was then subjected to cross-coupling conditions. A Suzuki cross-coupling with methylboronic acid required elevated temperatures and did not proceed to conversion, resulting in an inseparable mixture of desired product, starting material, and several byproducts. In contrast, Negishi cross-coupling provided 60 in 75% at ambient temperature. Subsequent deprotection and treatment with pyridine-3-isocyanate 61 provided the FAAH inhibitor 3.44 Since the absolute configuration of 56 was readily assigned as R, our synthesis afforded enantioenriched (S)-3. In the event that both enantiomers were required for testing, synthesis of (R)-3 via (S)-56 could be achieved using the other enantiomer of the CBS catalyst. Therefore, our stereospecific Negishi cross-coupling methodology can be used to construct both enantiomers of the product for biological testing.
CONCLUSIONS
In summary, we have developed a stereospecific nickelcatalyzed Negishi-type alkyl–alkyl cross-coupling for the introduction of benzylic methyl substitutents found in biologically active small molecules. Reactions proceed with high levels of chirality transfer. The mild reaction conditions are compatible with a variety of functional groups including alkenes, protected alkynes, acetals, and esters. Heterocycles, amines and imides are also well tolerated in these reactions. Benzylic alcohols are activated for cross-coupling using a traceless directing group that is easily installed by DCC coupling with commercially available (methylthio)acetic acid. Using this methodology, we report enantioselective synthesis of two bioactive molecules. This strategy enables biological testing of such compounds in their enantioenriched form, allowing for a more complete evaluation of their activity.
Supplementary Material
Scheme 4.

Enantioselective synthesis of retinoic acid receptor ligand 2.
(a) Mg, THF, rt, 2 h; (b) B(OMe)3, Et2O, 0 °C, 1 h, 94%; (c) 1 M HCl; (d) i) Et2Zn (6.0 equiv), PhMe, 65 °C, 24 h, ii) 48, 49 (0.10 equiv), PhMe, 0 °C, 24 h; (e) 51, DCC, DMAP, CH2Cl2, rt, 12 h, 90%; (f) NiCl2•DME (10 mol%), Xantphos (20 mol%), ZnMe2 (3.0 equiv), PhMe, 0 °C, 24 h; (g) 1 N NaOH, THF:MeOH (1:1), 60 °C, 2 h, 92%.
Scheme 5.

Synthesis of FAAH inhibitor 3.
(a) 54 (1.0 equiv), THF, −78 °C to rt, 1 h, 75%; (b) MnO2 (20 equiv), CH2Cl2, rt, 9 h, 72%; (c) (S)-Me-CBS (10 mol%), H3B•SMe2 (2.0 equiv), THF, 0 °C, 16 h, 99%, 67% ee, recrystallized to 95% ee; (d) 51, DCC, DMAP, CH2Cl2, rt, 12 h, 83%; (e) NiCl2•DME (10 mol %), DPEphos (20 mol %), ZnMe2 (3.0 equiv), PhMe, rt, 24 h; (f) Br2, THF, 0 °C, 6 h, 24%; (e) NiCl2•DME (10 mol %), DPEphos (20 mol %), ZnMe2 (3.0 equiv), PhMe, rt, 24 h, 75%; (g) TFA, CH2Cl2, rt, 30 min; (h) 61, CH2Cl2, rt, 20 h 68% over 2 steps.
ACKNOWLEDGMENT
Funding Sources
This work was supported by NIH NIGMS (R01GM100212) and the University of California Cancer Research Coordinating Committee. Prof. Scott D. Rychnovsky and Alexander J. Wagner are acknowledged for confirming absolute configuration of enantioenriched alcohol intermediates by their CEC method (see Supporting Information for details). Justin A. Hilf is acknowledged for synthesis of two racemic substrates (Table 1, entries 9 and 11). Dr. John Greaves is acknowledged for mass spectrometry data.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
No competing financial interests have been declared.
REFERENCES
- 1.Barreiro EJ, Kümmerle AE, Fraga CAM. Chem. Rev. 2011;111:5215. doi: 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]
- 2.(a) Istvan ES, Deisenhofer J. Science. 2001;292:1160. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]; (b) Roth BD, Blankley CJ, Chucholowski AW, Ferguson E, Hoefle ML, Ortwine EF, Newton RS, Sekerke CS, Sliskovic DR, Stratton CD, Wilson M. J. Med. Chem. 1991;34:357. doi: 10.1021/jm00105a056. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zimmerman J, Buchdunger E, Mett H, Meyer T, Lydon NB, Traxler P. Bioorg. Med. Chem. Lett. 1996;6:1221. [Google Scholar]; (b) Zimmerman J, Furet P, Buchdunger E. ACS Symp. Ser. 2001;796:245. [Google Scholar]
- 4.Leung CS, Leung SSF, Tirado-Rives J, Jorgensen WL. J. Med. Chem. 2012;55:4489. doi: 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For examples of methyl substitution to improve metabolic stability of heterocycles, see the following references: Obach RS, Kalgutkar AS, Ryder TF, Walker GS. Chem. Res. Toxicol. 2008;21:1890. doi: 10.1021/tx800185b. Talley JJ, Bertenshaw SR, Brown DL, Carter JS, Graneto MJ, Kellogg MS, Koboldt CM, Yuan J, Zhang YY, Seibert K. J. Med. Chem. 2000;43:1661. doi: 10.1021/jm000069h. Talley JJ, Brown DL, Carter JS, Graneto MJ, Koboldt CM, Masferrer JL, Perkins WE, Rogers RS, Shaffer AF, Zhang YY, Zweifel BS, Seibert K. J. Med. Chem. 2000;43:775. doi: 10.1021/jm990577v.
- 6.for an example of methyl substitution at the benzylic position as part of a strategy to slow down metabolic oxidation, see: Palani A, Shapiro S, Josien H, Bara T, Clader JW, Greenlee WJ, Cox K, Strizki JM, Baroudy BM. J. Med. Chem. 2002;45:3143. doi: 10.1021/jm0200815.
- 7.(a) Mihalic JT, Chen X, Fan P, Chen X, Fu Y, Liang L, Reed M, Tang L, Chen J-L, Jaen J, Li L, Dai K. Bioorg. Med. Chem. Lett. 2011;21:7001. doi: 10.1016/j.bmcl.2011.09.110. [DOI] [PubMed] [Google Scholar]; (b) Pereira AR, Strangman WK, Marion F, Feldberg L, Roll D, Mallon R, Hollander I, Andersen RJ. J. Med. Chem. 2010;53:8523. doi: 10.1021/jm100531u. [DOI] [PubMed] [Google Scholar]; (c) Yu K-L, Spinazze P, Ostrowski J, Currier SJ, Pack EJ, Hammer L, Roalsvig T, Honeyman JA, Tortolani DR, Raczek PR, Mansuri MM, Starrett JE., Jr J. Med. Chem. 1996;39:2411. doi: 10.1021/jm9502293. [DOI] [PubMed] [Google Scholar]; (d) Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, Fay L, Gregory T, Stiff C, Dunbar JB, Jr, Liimatta M, Beidler D, Smith S, Nomanbhoy TK, Cravatt BF. Bioorg. Med. Chem. Lett. 2009;19:2865. doi: 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For methods to construct methyl-bearing tertiary benzylic stereocenters, see: Hatanaka Y, Hiyama T. J. Am. Chem. Soc. 1990;112:7793. Imao D, Glasspoole BW, Laberge VS, Crudden CM. J. Am. Chem. Soc. 2009;131:5024. doi: 10.1021/ja8094075. Paquin J-F, Defieber C, Stephenson CRJ, Carreira EM. J. Am. Chem. Soc. 2005;127:10850. doi: 10.1021/ja053270w. Nave S, Sonawane RP, Elford TG, Aggarwal VK. J. Am. Chem. Soc. 2010;132:17096. doi: 10.1021/ja1084207. Podhajsky SM, Iwai Y, Cook-Sneathen A, Sigman MS. Tetrahedron. 2011;67:4435. doi: 10.1016/j.tet.2011.02.027. Pámies O, Andersson PG, Diéguez M. Chem.-Eur. J. 2010;16:14232. doi: 10.1002/chem.201001909. Wang X, Guram A, Caille S, Hu J, Preston JP, Ronk M, Walker S. Org. Lett. 2011;13:1881. doi: 10.1021/ol200422p. Mazuela J, Norrby P-O, Andersson PG, Pámies O, Diéguez M. J. Am. Chem. Soc. 2011;133:13634. doi: 10.1021/ja204948k.
- 9.(a) Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J. Am. Chem. Soc. 2011;133:389. doi: 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]; (b) Greene MA, Yonova IM, Williams FJ, Jarvo ER. Org. Lett. 2012;14:4293. doi: 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]
- 10.For representative strategies, see the following references: Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Vols 1–3. Berlin: Springer; 1990. Corey EJ, Bakshi RK, Shibata S. J. Am. Chem. Soc. 1987;109:5551. Kitamura M, Suga S, Kawai K, Noyori R. J. Am. Chem. Soc. 1986;108:6071. doi: 10.1021/ja00279a083. Kitamura M, Ohkuma T, Inoue S, Sayo N, Kumobayashi H, Akutagawa S, Ohta T, Takaya H, Noyori R. J. Am. Chem. Soc. 1988;110:629. Fu GC. Acc. Chem. Res. 2000;33:412. doi: 10.1021/ar990077w. Copeland GT, Miller SJ. J. Am. Chem. Soc. 2001;123:6496. doi: 10.1021/ja0108584. Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2001;123:7725. doi: 10.1021/ja015791z. Jensen DR, Pugsley JS, Sigman MS. J. Am. Chem. Soc. 2001;123:7475. doi: 10.1021/ja015827n. Zhang F-Y, Yip C-W, Cao R, Chan ASC. Tetrahedron: Asymmetry. 1997;8:585.
- 11.Stereospecific Suzuki cross-coupling to form triarylmethanes:Harris MR, Hanna LE, Greene MA, Moore CE, Jarvo ER. J. Am. Chem. Soc. 2013;135:3303. doi: 10.1021/ja311783k.
- 12.For a closely related transformation, see: Zhou Q, Srinivas HD, Dasgupta S, Watson MP. J. Am. Chem. Soc. 2013;135:3307. doi: 10.1021/ja312087x.
- 13.(a) Knochel P, Leuser H, Gong L-Z, Perrone S, Kneisel FF. Polyfunctional Zinc Organometallics for Organic Synthesis. In: Knochel P, editor. Handbook of Functionalized Organometallics: Applications in Synthesis. Weinheim: Wiley; 2005. p. 251. [Google Scholar]; (b) Phapale VB, Cárdenas DJ. Chem. Soc. Rev. 2009;38:1598. doi: 10.1039/b805648j. [DOI] [PubMed] [Google Scholar]; (c) Giovannini R, Stüdemann T, Devasagayaraj A, Dussin G, Knochel P. J. Org. Chem. 1999;64:3544. doi: 10.1021/jo982317b. [DOI] [PubMed] [Google Scholar]; (d) Piber M, Jensen AE, Rottländer M, Knochel P. Org. Lett. 1999;1:1323. [Google Scholar]; (e) Jensen AE, Knochel P. J. Org. Chem. 2002;67:79. doi: 10.1021/jo0105787. [DOI] [PubMed] [Google Scholar]
- 14.Campos KR, Klapars A, Waldman JH, Dromer PG, Chen C-y. J. Am. Chem. Soc. 2006;128:3538. doi: 10.1021/ja0605265. Beng TK, Gawley RE. Org. Lett. 2011;13:394. doi: 10.1021/ol102682r. For a review of general asymmetric cross-coupling strategies, see: Swift EC, Jarvo ER. Asymmetric transition metal-catalyzed cross-coupling reactions for the construction of tertiary stereocenters. [accessed May 22, 2013];Tetrahedron. doi: 10.1016/j.tet.2013.05.001. [Online early access]. http://authors.elsevier.com/sd/article/S0040402013007084 DOI: 10.1016/j.tet.2013.05.001. Published Online: May 6 2013.
- 15.(a) Lundin PM, Esquivias J, Fu GC. Angew. Chem., Int. Ed. 2009;121:160. doi: 10.1002/anie.200804888. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fischer C, Fu GC. J. Am. Chem. Soc. 2005;127:4594. doi: 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]; (c) Smith SW, Fu GC. J. Am. Chem. Soc. 2008;130:12645. doi: 10.1021/ja805165y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Arp FO, Fu GC. J. Am. Chem. Soc. 2005;127:10482. doi: 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]; (e) Oelke AJ, Sun J, Fu GC. J. Am. Chem. Soc. 2012;134:2966. doi: 10.1021/ja300031w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Choi J, Fu GC. J. Am. Chem. Soc. 2012;134:9102. doi: 10.1021/ja303442q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Binder JT, Cordier CJ, Fu GC. J. Am. Chem. Soc. 2012;134:17003. doi: 10.1021/ja308460z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For lead references on general directing group strategies, see: Rousseau G, Breit B. Angew. Chem., Int. Ed. 2011;50:2450. doi: 10.1002/anie.201006139. Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307.
- 17.For representative examples of the use of directing group in nickel catalysis, see: Devasagayaraj A, Stüdemann T, Knochel P. Angew. Chem., Int. Ed. Engl. 1995;34:2723. Didiuk MT, Morken JP, Hoveyda AH. Tetrahedron. 1998;54:1117. Wilsily A, Tramutola F, Owston NA, Fu GC. J. Am. Chem. Soc. 2012;134:5794. doi: 10.1021/ja301612y.
- 18.Srogl J, Liu W, Marshall D, Liebeskind LS. J. Am. Chem. Soc. 1999;121:9449. [Google Scholar]
- 19.Taylor BLH, Harris MR, Jarvo ER. Angew. Chem., Int. Ed. 2012;51:7790. doi: 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]
- 20.For acceleration of oxidative addition by pendant thiols during cross-coupling of thioacetals, see:Ni Z-J, Mei N-W, Shi X, Tzeng Y-L, Wang MC, Luh T-Y. J. Org. Chem. 1991;56:4035.
- 21.For a discussion see ref. 18
- 22.For es, see: Denmark SE, Vogler T. Chem.—Eur. J. 2009;15:11737. doi: 10.1002/chem.200901377.
- 23.Stayshich RM, Meyer TY. J. Polym. Sci., Part A: Polym Chem. 2008;46:4704. [Google Scholar]
- 24.The stereochemical course of the reaction was determined by comparison to the reported optical rotations of the intermediate alcohol and cross-coupling product. See Supporting Information for details.
- 25.(a) Lee AS, Norman AW, Okamura WH. J. Org. Chem. 1992;57:3846. [Google Scholar]; (b) Li JJ, Limberakis C, Pflum DA. Modern Organic Synthesis in the Laboratory: A Collection of Standard Experimental Procedures. New York: Oxford University Press; 2007. Reductions; p. 96. [Google Scholar]; Dakin LA, Panek JS. Org. Lett. 2003;5:(c). 3995. [Google Scholar]
- 26.For enantioselective arylation of alkyl aldehydes, see:Wu K-H, Zhou S, Chen C-A, Yang M-C, Chiang R-T, Chen C-R, Gau H-M. Chem. Commun. 2011;47:11668. doi: 10.1039/c1cc15059f.
- 27.For enantioselective arylation of aryl aldehydes, see Bolm C, Rudolph J. J. Am. Chem. Soc. 2002;124:14850. doi: 10.1021/ja028518l. Braga AL, Paixão MW, Westermann B, Schneider PH, Wessjohann LA. J. Org. Chem. 2008;73:2879. doi: 10.1021/jo702413n.
- 28.Absolute configuration of known alcohols was assigned by optical rotation (see Supporting Information). Absolute configuration of new alcohols was assigned as follows: (a) Alcohols prepared by the CBS reduction were assigned based on the predictive model described in Corey EJ, Helal CJ. Angew. Chem., Int. Ed. 1986;37 doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. (b) Singly benzylic alcohols were assigned by analogy to similar substrates prepared by the same method as ref. 26 (c) Benzydryl alcohols were assigned by predictive model described in ref. 27b.
- 29.(a) Wagner AJ, David JG, Rychnovsky SD. Org. Lett. 2011;13:4470. doi: 10.1021/ol201902y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wagner AJ, Rychnovsky SD. J. Org. Chem. 2013;78:4594. doi: 10.1021/jo400432q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) For linezolid see: Brickner SJ, Hutchinson DK, Barbachyn MR, Manninen PR, Ulanowicz DA, Garmon SA, Graga KC, Hendges SK, Toops DS, Ford CW, Zurenko GE. J. Med. Chem. 1996;39:673. doi: 10.1021/jm9509556. (b) For gefitinib see: Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. Science. 2004;304:1497. doi: 10.1126/science.1099314.(c) For dextromoramide see: Lasagna L, De Kornfeld T, Safar P. J. Chron. Dis. 1958;8:689. doi: 10.1016/0021-9681(58)90124-3.
- 31.Yonova IM, Jarvo ER. Unpublished results [Google Scholar]
- 32.Smith FT, Clark CR. Prodrugs and Drug Latentiation. In: Block JH, Beale JM Jr., editors. Organic Medicinal and Pharmaceutical Chemistry. 11th ed. Baltimore: 2004. p. 142. [Google Scholar]
- 33.Malhotra B, Gandelman K, Sachse R, Wood N, Michel MC. Curr. Med. Chem. 2009;16:4481. doi: 10.2174/092986709789712835. [DOI] [PubMed] [Google Scholar]
- 34.Nagpal S, Chandraratna RAS. Curr. Pharm. Design. 1996;2:295. doi: 10.2174/1381612003400146. [DOI] [PubMed] [Google Scholar]
- 35.Nagpal S, Thatcher SM, Patel S, Friant S, Malhotra M, Shafer J, Krasinski G, Asano AT, Teng M, Duvic M, Chandraratna RAS. Cell Growth Differ. 1996;7:1783. [PubMed] [Google Scholar]
- 36.Schadendorf D, Worm M, Jurgovsky K, Dippel E, Michel S, Charpentier B, Bernardon JM, Reichert U, Czarnetzki BM. Int. J. Oncol. 1994;5:1325. doi: 10.3892/ijo.5.6.1325. [DOI] [PubMed] [Google Scholar]
- 37.Fisher GJ, Voorhees JJ. FASEB J. 1996;10:1002. doi: 10.1096/fasebj.10.9.8801161. [DOI] [PubMed] [Google Scholar]
- 38.Graupner G, Malle G, Maignan J, Lang G, Prunieráes M, Pfahl M. Biochem. Biophys. Res. Commun. 1991;179:1554. doi: 10.1016/0006-291x(91)91750-7. See also ref. 7c. [DOI] [PubMed] [Google Scholar]
- 39.Frohn H-J, Adonin NY, Bardin VV, Starichenko VFZ. Anorg. Allg. Chem. 2002;628:2827. [Google Scholar]
- 40.See Supporting Information for full details. CCDC-940966 contains the supplemental crystallographic data for this structure. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif
- 41.For assignment of absolute configuration of alcohols, see: Hoye TR, Jeffrey CS, Shao F. Nat. Protoc. 2007;2:2451. doi: 10.1038/nprot.2007.354. Dale JA, Dull DL, Mosher HS. J. Org. Chem. 1969;34:2543. Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512. Ohtani I, Kusumi T, Ishitsuka MO, Kakisawa H. Tetrahedron Lett. 1989;30:3147. Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Org. Chem. 1991;56:1296. Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Am. Chem. Soc. 1991;113:4092.
- 42.(a) Li JJ, Limberakis C, Pflum DA. Modern Organic Synthesis in the Laboratory: A Collection of Standard Experimental Procedures. New York: Oxford University Press; 2007. Oxidation; p. 55. [Google Scholar]; (b) Wipf P, Xu W. J. Org. Chem. 1996;61:6556. doi: 10.1021/jo960845m. [DOI] [PubMed] [Google Scholar]
- 43.Kose M. J. Photochem. Photobiol., A. 2004;165:97. [Google Scholar]
- 44.Swanson DM, Dubin AE, Shah C, Nasser N, Chang L, Dax SL, Jetter M, Breitenbucher JG, Liu C, Mazur C, Lord B, Gonzales L, Hoey K, Rizzolio M, Bogenstaetter M, Codd EE, Lee DH, Zhang S-P, Chaplan SR, Carruthers NI. J. Med. Chem. 2005;48:1857. doi: 10.1021/jm0495071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.