

Abstract
Huntington disease (HD) is a dominantly inherited neurodegenerative disorder characterized by dysregulation of various genes. Recently, microRNAs (miRNAs) have been reported to be involved in this dysregulation, suggesting that manipulation of appropriate miRNA regulation may have a therapeutic benefit. Here, we report the beneficial effects of miR-196a (miR196a) on HD in cell, transgenic mouse models, and human induced pluripotent stem cells derived from one individual with HD (HD-iPSCs). In the in vitro results, a reduction of mutant HTT and pathological aggregates, accompanying the overexpression of miR-196a, was observed in HD models of human embryonic kidney cells and mouse neuroblastoma cells. In the in vivo model, HD transgenic mice overexpressing miR-196a revealed the suppression of mutant HTT in the brain and also showed improvements in neuropathological progression, such as decreases of nuclear, intranuclear, and neuropil aggregates and late-stage behavioral phenotypes. Most importantly, miR-196a also decreased HTT expression and pathological aggregates when HD-iPSCs were differentiated into the neuronal stage. Mechanisms of miR-196a in HD might be through the alteration of ubiquitin-proteasome systems, gliosis, cAMP response element-binding protein pathway, and several neuronal regulatory pathways in vivo. Taken together, these results show that manipulating miR-196a provides beneficial effects in HD, suggesting the potential therapeutical role of miR-196a in HD.
Main Text
Huntington disease (HD [MIM 143100]) is an autosomal-dominant disease that is caused by a mutation of CAG trinucleotide repeats in the exon 1 region of the huntingtin (HTT [MIM 613004]) gene.1 In principle, the longer the CAG trinucleotide repeats, the earlier is onset of HD. Pathological characteristics of HD include nuclear, intranuclear, and neuropil aggregates and they lead to clinical symptoms such as cognitive dysfunction, emotional changes, mental deterioration, motor deficits, chorea, and dystonia.2 Effective treatment of HD remains a high-priority goal because effective therapies have not emerged despite considerable insight into pathogenic mechanisms.
Normally, endogenous HTT is involved in mechanisms of gene regulation, suggesting that mutant HTT disrupts physiological gene regulation in HD individuals.3 Recently, posttranscriptional regulations by noncoding RNAs, especially microRNAs (miRNAs), have been extensively studied in central nervous system and neuronal diseases,4,5 and there are indications that miRNAs might also affect HD; however, there are still relatively few studies investigating the relationship between miRNAs and HD. Because HD is characterized by the dysfunction of several mechanisms and one miRNA could target multiple pathways, this suggests that miRNA could be a potential treatment for HD. We identify here that miR-196a (MIM 608632) has a potential effect on HD and further confirm that increased expression of miR-196a ameliorates the molecular, pathological, and behavioral phenotypes both in vitro and in vivo.
Based on the results of a miRNA microarray study by Packer et al. in 20086 and our unpublished microarray data with the brain tissues of HD transgenic monkeys, miR-196a was identified as a possible candidate for involvement in the pathogenesis of HD. To study the effects of miR-196a on HD in vitro, we transfected miR-196a mimics or inhibitors (Genepharma) with mutant HTT into 293 FT (human embryonic kidney cells) and N2a (mouse neuroblastoma cells) cells. Because a direct degradation of mRNA through the complementary disruption between miR-196a and the coding region of our transgene is one possible mechanism leading to a lower expression of the mutant HTT transgene, we aligned sequences of miR-196a and the coding region of our transgene and confirmed that there was no complementary sequence (data not shown), suggesting that the expression of mutant HTT might not be through a direct degradation of mRNA. In the transfection experiments, we observed that miR-196a mimics suppressed the expression of mutant HTT compared to that of an unrelated control, as shown by immunoblotting with mEM48 against mutant HTT (Figure 1A). In addition, we also examined the RNA expression level of miR-196a (Figures S1A and S1C available online) and mutant HTT (Figures S1B and S1D) via real-time reverse-transcription quantitative PCR (qRT-PCR) and observed that a higher expression of miR-196a could significantly suppress the mRNA level of mutant HTT in two cell lines. These results suggest that miR-196a can suppress the mRNA and protein expression of mutant HTT in vitro and that the process is not through a direct degradation of mutant HTT mRNA.
Figure 1.
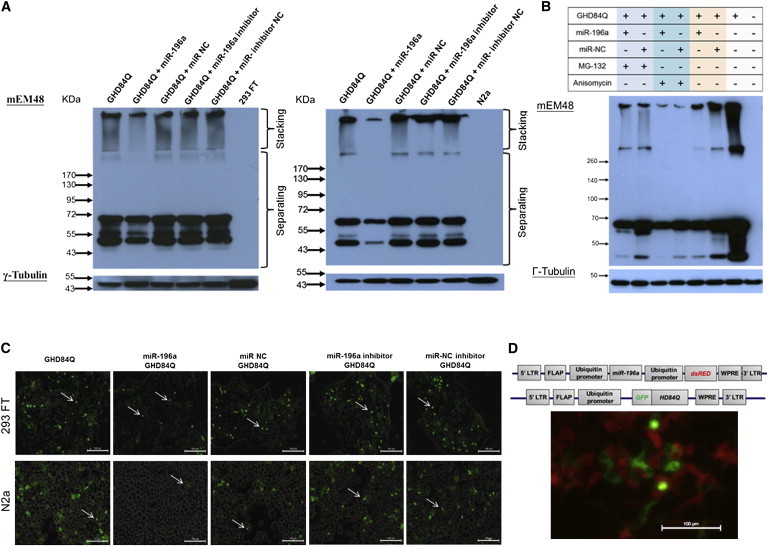
miR-196a Suppressed the Expression of Mutant HTT In Vitro
293 FT and N2a cells were transfected with miR-196a mimics, miR-196a inhibitors, unrelated controls (NC), and mutant HTT (GHD) as indicated in different graphs. Expression of mutant HTT was analyzed 48 hr after transfection.
(A) Immunoblotting by mEM48 showed lower expression of mutant HTT at either high (stacking gel) or low (separating gel) molecular weight in 293 FT (left) and N2a (right) cells transfected with miR-196a and GHD. γ-tubulin (Sigma) was used as an internal control.
(B) Immunoblotting results showed that transfected 293 FT cells with MG-132 (Enzo Life Sciences) treatment did not obviously alter expression profiles of mutant HTT, whereas anisomycin (Enzo Life Sciences) suppressed the expression of aggregated HTT, suggesting that miR-196 suppressed the expression of mutant HTT through the suppression of protein synthesis predominantly and protein degradation partly.
(C) Fluorescent signals of mutant HTT were detected via GFP fusion proteins in 293 FT (top) and N2a (bottom) cells. Under the same exposure condition, cells transfected with miR-196a and GHD showed lower fluorescence signal. These images were merged with transmission light, and arrows indicate intranuclear aggregates.
(D) Top panel shows the miR-196a construct containing the ubiquitin promoter to drive miR-196a precursor and red fluorescence protein (dsRED), respectively, and the mutant HTT construct containing the ubiquitin promoter to drive HD84Q fused with GFP. Abbreviations are as follows: WPRE, woodchunk hepatitis virus posttranscriptional regulatory element; FLAP, human immunodeficiency virus-1 falp element; LTR, long terminal repeat. Bottom panel shows the expression of mutant HTT (green) and the expression of miR-196a (red), suggesting the suppression of mutant HTT.
However, because we used human ubiquitin promoter to drive the mutant HTT gene,7,8 there remains a question of whether miR-196a affected the ubiquitin promoter rather than the expression of mutant HTT. Therefore, different promoters, including cytomegalovirus (CMV) and endogenous human HTT promoters, were constructed to explore this question (Figure S2A), and the results also showed repression of mutant HTT (Figures S2B and S2C). This suggests that miR-196a suppressed the expression of mutant HTT, not the expression of an ubiquitin promoter.
The next question is whether miR-196a functions through the inhibition of mutant HTT synthesis or through the acceleration of degradation. We therefore treated 293 FT cells with either 1 μM MG-132 (a proteasome inhibitor) or 0.1 μM anisomycin (a protein synthesis inhibitor) when miR-196a and mutant HTT were cotransfected. Upon treatment with MG-132, a stronger signal was observed in the miR-196a group compared to that of miR-NC, whereas no aggregated protein was observed in either group after anisomycin treatment (Figure 1B). This result suggests that miR-196 suppressed the expression of mutant HTT predominantly through the suppression of protein synthesis and partly through protein degradation. Furthermore, because a fusion protein, green fluorescent protein (GFP), was constructed in the 5′ end of mutant HTT in this study, fluorescent signals could be used to indicate the expression of mutant HTT. Under the same exposure conditions, weaker fluorescent signals were detected in 293 FT and N2a cells treated with miR-196a mimics (Figure 1C). In addition, different fluorescent reporter genes were cloned into the vector system (Figure 1D, top), and the results showed a reduction of mutant HTT along with a higher expression of miR-196a (Figure 1D, bottom). These in vitro results taken together suggested a suppression ability of miR-196a on HTT in vitro.
In order to further study the effects of miR-196a in vivo, miR-196a transgenic mice (196a) were generated via lentiviral transgenesis (Figure 2A, top)7 and then bred with GFP-HTT (GHD) transgenic mice8 (Figure 2A, top) to obtain four groups of mice: double transgenic mice (D-Tg) carrying mutant HTT and miR-196a, GHD transgenic mice, 196a transgenic mice, and wild-type (WT) mice. All animal procedures performed in this study were approved by the Institutional Animal Care and Use Committees at National Cheng Kung University. Tests for HTT mRNA and protein in brain samples of D-Tg and GHD transgenic mice at 1 month of age showed lower expression levels of mutant HTT in D-Tg transgenic mice compared to those of GHD transgenic mice (Figure 2A, bottom). We also confirmed the significantly higher expression of miR-196a in D-Tg transgenic mice compared to that of GHD transgenic mice (Figure S3). Because HD is a progressive disease resulting from the continuous accumulation of mutant HTT,2 the suppression ability of miR-196a at different stages was examined. Immunoblotting results from brain samples taken from D-Tg and GHD transgenic mice at 1 and 12 months of age showed an increase of aggregated mutant HTT in GHD mice at 12 months of age, whereas D-Tg mice displayed much fewer mutant HTT at either 1 or 12 months of age (Figure 2B), implying that miR-196a can inhibit the expression of mutant HTT in vivo.
Figure 2.
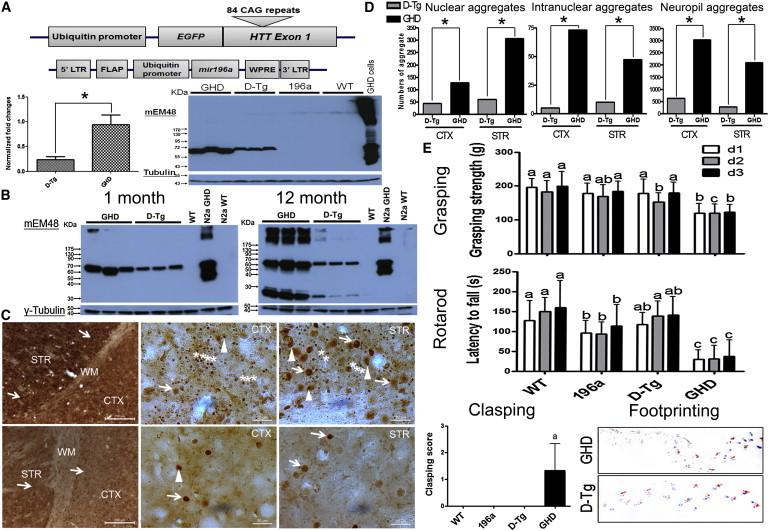
miR-196a Suppressed the Expression of Mutant HTT in HD Transgenic Mouse Model
(A) Top panel shows the construct of mutant HTT and miR-196a used for the generation of GHD transgenic mice. Bottom left shows the suppression of mutant HTT mRNA determined by qRT-PCR (Applied Biosystems) using brain samples of different transgenic mice at 1 month of age (n = 6–8 for each group). Bottom right shows the immunoblotting, with suppression of mutant HTT in the brains of D-Tg transgenic mice at 1 month of age compared to those of GHD transgenic mice. mEM48 was used to detect mutant HTT, and γ-tubulin was used as the internal control. Asterisk represents significant difference between two groups.
(B) Immunoblotting shows the suppression of mutant HTT in the brains of D-Tg transgenic mice at 1 (left) and 12 (right) months of age. One brain sample of a wild-type mouse (WT) was used as a negative control. N2a cells transfected with mutant HTT (N2a GHD) were used as a positive control, and wild-type N2a cells (N2a WT) were used as a negative control of cells.
(C) Improvement of neuropathological phenotypes in brains of D-Tg mice at 12 months of age via a mEM48 antibody (n = 3–5 mice). Top three images show significant neuropil (stars), nuclear (arrows), and intranuclear (arrowheads) aggregates in different brain regions from one representative GHD transgenic mouse, including striatum (STR), cortex (CTX), and white matter (WM), via a 3, 3′-diaminobezidine (DAB) method (Vector Laboratories). Bottom three images show brain pathology from one representative D-Tg transgenic mouse.
(D) Quantitation figures show accumulated number of nuclear, intranuclear, and neuropil aggregates via the MetaMorph software. Accumulated number of aggregates was counted in five random 120 × 120 μM2 regions. Asterisk represents significant difference between two groups.
(E) Improvement of behavioral phenotypes in D-Tg mice at 12 months of age. Clasping, grasping (Bioseb), rotarod (Singa), and footprinting tests were performed at 12 months of age in four groups (n = 6–9 mice for each group). Data represent mean ± SD, and different characters on different bars indicate statistically significant difference.
Accumulation of mutant HTT leads to neuropathological phenotypes, such as neuropil aggregates, nuclear aggregates, and intranuclear aggregates, in the brains of HD models.2,8 Therefore, pathological features in these mice were examined. In GHD transgenic mice at 12 months of age, severe pathological aggregates were observed in different brain regions, such as the cortex and striatum (Figure 2C, top), whereas much fewer aggregates were observed in D-Tg transgenic mice (Figure 2C, bottom). These results were also confirmed through computational quantitation (Figure 2D), revealing that miR-196a had significantly alleviated pathological phenotypes in vivo.
Gradual deterioration of behavioral phenotypes is an important symptom for HD, especially motor functions.2 Therefore, four longitudinal examinations, including clasping, grasping, rotarod, and footprinting tests, were performed in mice. At 4 and 8 months of age, comparable behavioral phenotypes were observed (Figure S4); however, deteriorated performance was observed in GHD mice at 12 months of age, whereas D-Tg mice showed a similar performance to that of 196a and WT mice (Figure 2E). Taken together, these studies in mice indicate that miR-196a can alleviate molecular, pathological, and behavioral phenotypes in vivo.
Because the above HD transgenic mice constitute an overexpression model, we were curious about the effects of miR-196a on human endogenous HTT. The model of HD individual-derived induced pluripotent stem cells (HD-iPSCs) is a unique platform to study endogenous HTT in HD research.9,10 Therefore, we used HD-iPSCs (GM23225) purchased and permitted from the Coriell Institute in this study and differentiated these cells into neuronal cells. As HD-iPSCs were differentiated into neuronal cells in N3 medium, miR-196a and empty lentiviruses were used to infect these differentiated cells (Figure 3A). Because it is difficult to induce pathological phenotypes in HD-iPSCs, 10 μM MG-132 (a proteasome inhibitor) was added to accelerate the accumulation of mutant HTT. First, we confirmed the characteristics of HD-iPSCs at different stages, demonstrating that these cells carrying 72 CAG repeats could successfully differentiate into neuronal cells (Figures 3B and 3C). Immunostaining via a mEM48 antibody was performed 30 hr after MG-132 treatments, revealing certain signals of aggregates in empty vector group compared to that in miR-196a group (Figure 3D). Immunoblotting also revealed a lower expression level of mutant HTT in the miR-196a group via mEM48 and MAB2166 antibodies (Figure 3E), suggesting that miR-196a could suppress the expression of mutant HTT and alleviate the pathological phenotypes in human samples.
Figure 3.
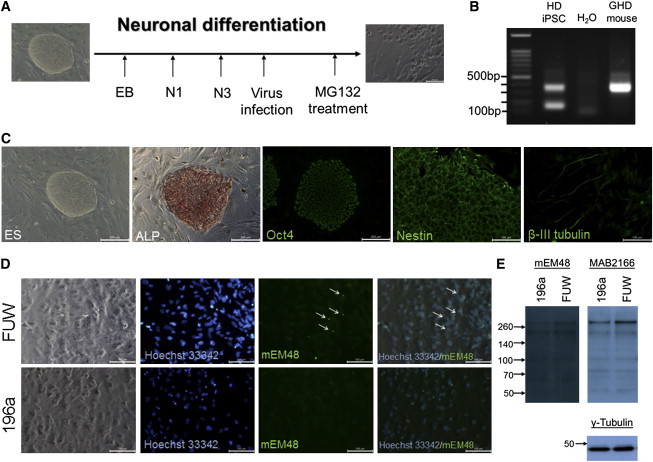
miR-196a Suppressed Pathological Aggregates in HD-iPSCs
(A) Schematic of experimental design for differentiating HD-iPSCs into neuronal cells.
(B) PCR was used to confirm 72 and 17 CAG repeats inside HD-iPSCs, showing 309 and 144 bp of amplicons.
(C) Immunostaining indicated that undifferentiated HD-iPSCs (ALP [Bio SB] and Oct4 [GeneTex]) differentiated into neuronal precursor stage (Nestin; GeneTex) and neuronal stage (β-III tubulin; Millipore).
(D) Differentiated HD-iPSCs treated with control viruses (FUW; top) displayed pathological aggregates (arrows), whereas cells treated with miR-196a viruses (196a; bottom) did not.
(E) Immunoblotting by mEM48 and Huntingtin (Chemicon; MAB2166) antibodies showed weaker HTT fragments in the 196a cells compared to those of the FUW group. γ-tubulin was used as an internal control.
Because miR-196a can suppress the expression of mutant HTT both in vitro and in vivo, the detailed mechanisms should therefore be addressed. Based on online bioinformatic analysis (TargetScan 6.2), HTT is not the target gene of miR-196a, suggesting that miR-196a influences HD through other indirect pathways. The impairment of protein clearance pathways, such as the ubiquitin-proteasome system (UPS), in HD11 led us to examine the UPS in brain samples, which showed a clear decrease of high-molecular-weight unibiquitin protein in D-Tg transgenic mice compared to that of GHD transgenic mice (Figure 4A). Also, because reactive gliosis is an important manifestation of damage to the central nervous system in brains of HD,12 we therefore assessed the profiling of glial fibrillary acidic protein (GFAP; a marker representing reactive gliosis) in these mice, and the results showed a decrease of GFAP (Figure 4A) and less-reactive gliosis in D-Tg brains (Figure 4B). Furthermore, a decrease of calmodulin kinase II (CaMKII) activity has been reported in HD models, leading to an impairment of the cAMP response element-binding protein (CREB) pathway.13,14 Therefore, we also determined the total CaMKII and phosphorylated CaMKII (pCaMKII) in the brain samples, and the results showed that D-Tg transgenic mice reverted to higher expression levels of pCaMKII (Figure 4C). However, UPS and CaMKII results in infected HD-iPSCs were to some extent different to the in vivo mouse model (Figure S5). This might be due to the difference between in vitro and in vivo systems and might also be due to the different severity of pathological aggregates in these two models.
Figure 4.
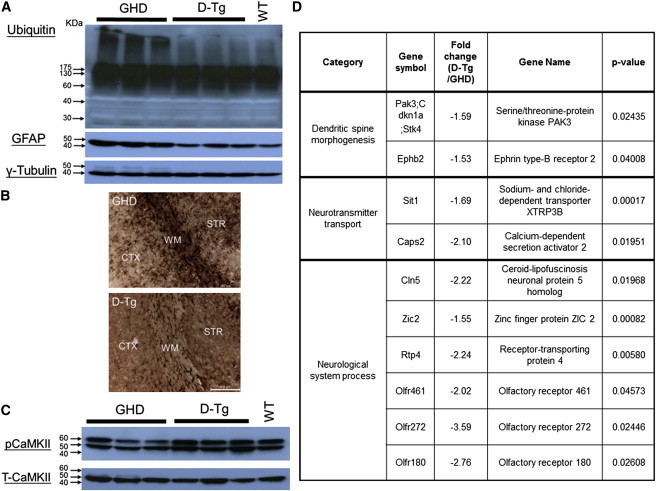
Improvement of D-Tg Transgenic Mice Was through Several Different Mechanisms
Brain samples from GHD and D-Tg transgenic mice at 12 months of age were subjected to immunoblotting, immunohistochemical staining, and microarray analyses.
(A) Higher expression level of ubiquitin (DakoCytomation) and GFAP (Sigma) proteins in GHD transgenic mice compared to those of D-Tg transgenic mice upon immunoblotting.
(B) Immunohistochemical staining showed ameliorated gliosis in the different brain regions, including striatum (STR), cortex (CTX), and white matter (WM), of D-Tg transgenic mice (bottom) compared to those of GHD transgenic mice (top).
(C) Immunoblotting showed a higher expression level of pCaMKII (Abcam) in D-Tg transgenic mice compared to that in GHD transgenic mice.
(D) Candidate genes showing significant alternation between D-Tg and GHD transgenic mice were obtained from microarray analyses.
In addition, microarray analyses (Phalanx Biotech Group) of the striatum from D-Tg and GHD transgenic mice via MetaCore (GeneGo) revealed that several genes related to neuronal functions, such as dendritic spine morphogenesis, neurotransmitter transport, and neurological system processes, had been altered (Figure 4D). We also analyzed genes from both TargetScan 6.2 and our microarray with the MetaCore software and observed that several genes from the two data sets are involved in CREB, gliosis, and UPS pathways (Table S1), suggesting that miR-196a might function through these TargetScan 6.2-predicted genes to affect these three pathways. In addition, we observed that three predicted genes, E3 ubiquitin-protein ligase NEDD4-like (MIM 606384), E3 ubiquitin-protein ligase RNF5 (MIM 602677), and Ubiquitin carboxyl-terminal hydrolase 15 (MIM 604731), were downregulated in D-Tg mice in the microarray data (Table S1), suggesting that miR-196a might function through destabilizing the mRNAs of these three genes and subsequently affect the UPS pathway. However, further evidence is needed. Taken together, these results suggest that miR-196a might alleviate phenotypes of HD in vivo via improvement of UPS, gliosis, CREB pathway, and several neuronal regulatory pathways.
A higher expression of miR-196a in HD models has been reported in the microarray database;6,15 however, overexpression of miR-196a can alleviate HD both in vitro and in vivo in our study, implying that a higher expression of miR-196a might be caused by HD and that the expression level of miR-196a in the above HD models might be too low to provide protective functions. In several disease models, miR-196a has offered beneficial effects, including hepatitis C (MIM 609532)16 and spinal and bulbar muscular atrophy (SBMA [MIM 313200]).17 Moreover, miR-196a also plays critical roles such as in gene regulation, nuclear architecture, antiapoptosis, and calcium binding.18 Therefore, whether miR-196a provides neuroprotective effects on HD through similar regulatory mechanisms should be further investigated.
Although miR-196a has multiple predicted targets, HTT is not one of them,19 suggesting that miR-196a does not alleviate HD directly through targeting mutant HTT. One direct target, CUGBP Elav-like family member 2 (CELF2 [MIM 602538]), has attracted our attention because CELF2 can be regulated by miR-196a in SBMA, another neurodegenerative disease characterized by the expansion of CAG repeats in the mutant androgen receptor (AR [MIM 313700]) gene.17 Because of the binding of CELF2 to the mRNA of expanded CAG repeats in SBMA,17 we strongly suspected a role for CELF2 in HD. However, we did not observe a lower expression level of CELF2 either in HD 293 FT cells transfected with miR-196a or in our mouse model (Figure S6A). In addition, we also tested whether CELF2 bound to the mRNA of mutant HTT via RNA immunoprecipitation and observed that precipitated CELF2 did not bind more mutant HTT mRNA compared to that of the IgG control (Figure S6B). This suggests that CELF2 might not be involved in the mechanism of miR-196a on HD, and so other candidates should be screened. Of particular interest for further research is the question of whether miR-196a could also have beneficial effects in other polyglutamine diseases.
In summary, a higher expression of miR-196a not only reduced the expression of mutant HTT in vitro but also improved molecular, pathological, and behavioral phenotypes in a HD transgenic mouse model. Most importantly, miR-196a ameliorated the formation of pathological aggregates in HD-iPSCs differentiating into the neuronal stage. We further show that the effects of miR-196a might be through the alteration of the ubiquitin-proteasome systems, gliosis, and the CREB pathway. This study suggests the important role of miR-196a in HD and provides an insight into a therapeutical strategy for HD.
Acknowledgments
We thank Jonathan Courtenay for critical reading of the manuscript, Chauying Jen and Pi-Hsueh Shirley Li for providing equipment, and Xiao-Jiang Li for providing mEM48 antibodies. This work was supported by National Science Council grants (NSC 99-2320-B-006-026-MY3 and NSC 101-2627-B-006) and in part by a grant of the Ministry of Education, Taiwan, Republic of China, under the ATU plan. Computational analyses and data mining were done with the system provided by the Bioinformatics Core at National Cheng Kung University, supported by a National Science Council grant (NSC 97-3122-B-006-011).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
miRBase, http://www.mirbase.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
TargetScan 6.2, http://www.targetscan.org/
Accession Numbers
The GEO accession number for our microarray data reported in this paper is GSE47500. The miRBase accession number for the miR-196a sequence reported in this paper is MI0000279.
References
- 1.The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Yang S.H., Chan A.W. Transgenic animal models of Huntington’s disease. Current Topics in Behavioral Neurosciences. 2011;7:61–85. doi: 10.1007/7854_2010_105. [DOI] [PubMed] [Google Scholar]
- 3.Li S.H., Li X.J. Huntingtin and its role in neuronal degeneration. Neuroscientist. 2004;10:467–475. doi: 10.1177/1073858404266777. [DOI] [PubMed] [Google Scholar]
- 4.Junn E., Mouradian M.M. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 2012;133:142–150. doi: 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu J.Y., Chung K.H., Deo M., Thompson R.C., Turner D.L. MicroRNA miR-124 regulates neurite outgrowth during neuronal differentiation. Exp. Cell Res. 2008;314:2618–2633. doi: 10.1016/j.yexcr.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Packer A.N., Xing Y., Harper S.Q., Jones L., Davidson B.L. The bifunctional microRNA miR-9/miR-9∗ regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008;28:14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang S.H., Cheng P.H., Banta H., Piotrowska-Nitsche K., Yang J.J., Cheng E.C., Snyder B., Larkin K., Liu J., Orkin J. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature. 2008;453:921–924. doi: 10.1038/nature06975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng P.H., Li C.L., Her L.S., Chang Y.F., Chan A.W., Chen C.M., Yang S.H. Significantly differential diffusion of neuropathological aggregates in the brain of transgenic mice carrying N-terminal mutant huntingtin fused with green fluorescent protein. Brain Struct. Funct. 2013;218:283–294. doi: 10.1007/s00429-012-0401-x. [DOI] [PubMed] [Google Scholar]
- 9.Park I.H., Arora N., Huo H., Maherali N., Ahfeldt T., Shimamura A., Lensch M.W., Cowan C., Hochedlinger K., Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeon I., Lee N., Li J.Y., Park I.H., Park K.S., Moon J., Shim S.H., Choi C., Chang D.J., Kwon J. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30:2054–2062. doi: 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- 11.Chiang M.C., Chen C.M., Lee M.R., Chen H.W., Chen H.M., Wu Y.S., Hung C.H., Kang J.J., Chang C.P., Chang C. Modulation of energy deficiency in Huntington’s disease via activation of the peroxisome proliferator-activated receptor gamma. Hum. Mol. Genet. 2010;19:4043–4058. doi: 10.1093/hmg/ddq322. [DOI] [PubMed] [Google Scholar]
- 12.Lin C.H., Tallaksen-Greene S., Chien W.M., Cearley J.A., Jackson W.S., Crouse A.B., Ren S., Li X.J., Albin R.L., Detloff P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 13.Sugars K.L., Brown R., Cook L.J., Swartz J., Rubinsztein D.C. Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington’s disease that contributes to polyglutamine pathogenesis. J. Biol. Chem. 2004;279:4988–4999. doi: 10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- 14.Marambaud P., Dreses-Werringloer U., Vingtdeux V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009;4:20. doi: 10.1186/1750-1326-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaughwin P.M., Ciesla M., Lahiri N., Tabrizi S.J., Brundin P., Björkqvist M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 2011;20:2225–2237. doi: 10.1093/hmg/ddr111. [DOI] [PubMed] [Google Scholar]
- 16.Hou W., Tian Q., Zheng J., Bonkovsky H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology. 2010;51:1494–1504. doi: 10.1002/hep.23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyazaki Y., Adachi H., Katsuno M., Minamiyama M., Jiang Y.M., Huang Z., Doi H., Matsumoto S., Kondo N., Iida M. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat. Med. 2012;18:1136–1141. doi: 10.1038/nm.2791. [DOI] [PubMed] [Google Scholar]
- 18.Chen C., Zhang Y., Zhang L., Weakley S.M., Yao Q. MicroRNA-196: critical roles and clinical applications in development and cancer. J. Cell. Mol. Med. 2011;15:14–23. doi: 10.1111/j.1582-4934.2010.01219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinha M., Ghose J., Bhattarcharyya N.P. Micro RNA -214,-150,-146a and-125b target Huntingtin gene. RNA Biol. 2011;8:1005–1021. doi: 10.4161/rna.8.6.16035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.